Asymmetric Diels–Alder reaction with >C=P– functionality of the 2-phosphaindolizine-η1-P-aluminium(O-menthoxy) dichloride complex: experimental and theoretical results
1,
2,
1,
2,
3 and
3
Rajendra K. Jangid
Department of Chemistry, University of Rajasthan, Jaipur 302004, India
1Department of Chemistry, University of Rajasthan, Jaipur 302004, India
2Department of Chemistry, IIS University, Jaipur 302020, India
3Fachbereich Chemie der Philipps Universität, D-35032 Marburg, Germany
Corresponding author email
Associate Editor: D. Y.-K. Chen Beilstein J. Org. Chem.2013,9, 392–400.https://doi.org/10.3762/bjoc.9.40 Received 14 Sep 2012,
Accepted 25 Jan 2013,
Published 18 Feb 2013
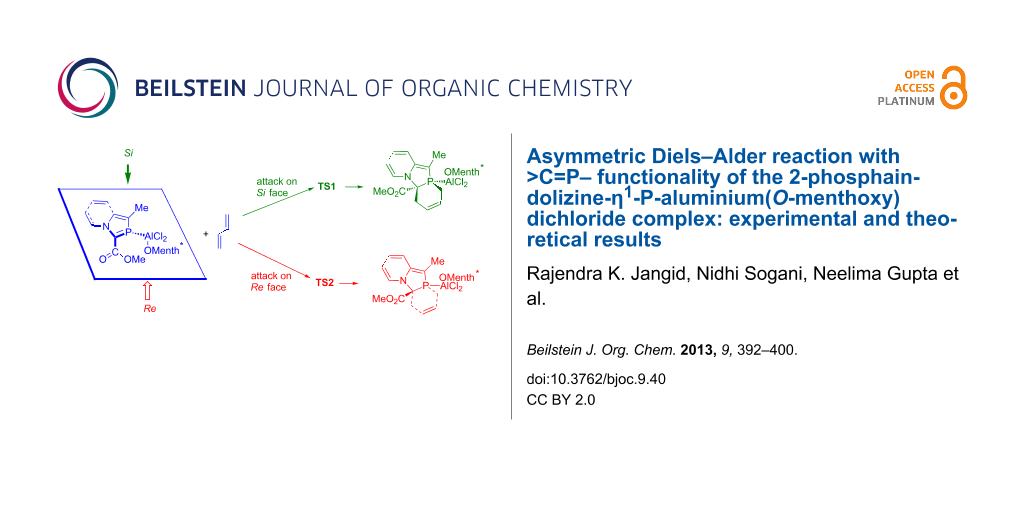
The Diels–Alder reaction of the 2-phosphaindolizine-η1-P-aluminium(O-menthoxy) dichloride complex with dimethylbutadiene was investigated experimentally and computationally. The >C=P– functionality of the complex reacts with 2,3-dimethylbutadiene with complete diastereoselectivity to afford [2 + 4] cycloadducts. Calculation of the model substrate, 3-methoxycarbonyl-1-methyl-2-phosphaindolizine-P-aluminium(O-menthoxy) dichloride (7a), at the DFT (B3LYP/6-31+G*) level reveals that the O-menthoxy moiety blocks the Re face of the >C=P– functionality, due to which the activation barrier of the Diels–Alder reaction of 7a with 1,3-butadiene, involving its attack from the Si face, is lower. It is found that in this case, the exo approach of the diene is slightly preferred over the endo approach.
There is an increasing emphasis on the synthesis of optically pure compounds, as far as possible, for environmental, economic and social reasons. Using chiral auxiliaries for changing enantiotopic faces into diastereotopic faces is a common approach in asymmetric synthesis, which is one of the most attractive methods from the atom-economy point of view [1] for producing single enantiomers selectively. Over the past three decades, a variety of reactions allowing the formation of C–H, C–C, C–N, C–O and other bonds enantioselectively have been developed [2]. The chiral pool continues to be an attractive and economic source of enantiomerically pure chiral auxiliaries (ligands or modifiers) for enantioselective synthesis [3]. Two naturally occurring enantiomers of menthol and synthetically prepared (1R)-(+)-8-phenylmenthol have often been used as chiral auxiliaries [3,4].
Chiral phosphines constitute a very important group of ligands as their coordination compounds with transition metals have been extensively employed in asymmetric catalysis to convert achiral compounds into enantio-enriched products with high efficiency and enantioselectivity [5]. In many cases, chiral monophosphine ligands have been found to be more useful than chiral bisphosphines [6-8]. In view of this, efforts are always being made to obtain new chiral phosphines [9].
The first example of the Diels–Alder (DA) reaction with the >C=P– functionality of an azaphosphole was reported by Arbuzov and co-workers [10]. Subsequently, pioneering work by the research group of Appel established several interesting features associated with the DA reactions of phosphaalkenes [11,12]. Mathey and co-workers showed that 1H-phospholes underwent a 1,5-H shift followed by dimerization through a DA reaction [13]. The first DA reaction involving the –C=C–C=P– moiety of phosphinine as a diene was reported by Märkl and Lieb [14], while Mathey and Alcaraz showed that phosphinine could react as a dienophile as well, with the reaction taking place at the >C=P– functionality of phosphinine [15]. We recently compiled a review on the DA reactions involving the >C=P– functionality of various organophosphorus compounds wherein all these aspects have been discussed [16].
During the past few years, we have investigated the DA reaction with the >C=P– functionality of 1,3-azaphospholes theoretically as well as experimentally [16]. In this context, we found that 1,3-bis(alkoxycarbonyl)-2-phosphaindolizines (1a, Z = CO2R1) prepared through 1,5-electrocyclization of in situ generated bis(pyridinium ylidyl)phosphenium chlorides [17] lead to successful DA reaction [18,19], but 3-alkoxycarbonyl-2-phosphaindolizines having an electron-withdrawing group (EWG) only at the 3-position (1b, Z = Me) failed to undergo DA reaction even on heating under reflux in toluene alone or in the presence of sulfur [18] (Scheme 1).
Scheme 1:
Diels–Alder reaction of 2-phosphaindolizines.
Scheme 1:
Diels–Alder reaction of 2-phosphaindolizines.
It was demonstrated that the dienophilic reactivity of the >C=P– functionality of phosphinines could be enhanced by complexing the P-atom of phosphinine to a metal carbonyl, such as W(CO)5[15,20]. Thus, the phosphinine-η1-P-W(CO)5 complex reacted with 1,3-dienes to afford [2 + 4] cycloadducts. By following the same strategy, we recently reported our theoretical and experimental results of the DA reactions of 2-phosphaindolizine-η1-P-AlEtCl2 complexes [21]. Theoretical calculations at the DFT (B3LYP/6-31+G**) level indicated lowering of the activation barrier by 6 kcal mol–1 for the reaction of σ2,λ3-P-coordinated 2-phosphindolizine to methylaluminium dichloride with 1,3-butadiene as compared to that for the corresponding reaction of the uncomplexed 2-phosphaindolizine. The cycloadducts so obtained were well characterized by 1H, 31P and 27Al NMR data and, thus, confirmed the theoretical results.
Koga and co-workers [22] used, for the first time, chiral (−)-menthoxyaluminium dichloride, derived from the reaction of (–)-menthol with ethylaluminium dichloride, for the asymmetric catalytic DA reaction of methacrolein with cyclopentadiene leading to 66% ee. It led to the development of a variety of chiral aluminium and other organometallic catalysts for use in organic synthesis [23-25]. In view of this, it was considered interesting to prepare a 2-phosphindolizine-η1-P complex by using a chiral Lewis acid, (−)-menthoxyaluminium dichloride, and to investigate experimentally and theoretically the diastereoselectivity of its DA reaction. The results are described herein.
Results and Discussion
Experimental results
(2-Phosphaindolizine-η1-P)-Al(O-menthoxy)Cl2 (7) was generated in situ by reacting 2-phosphaindolizine with (O-menthoxy)aluminium dichloride (5); formation of the complex is confirmed by 31P NMR (δ 196.0–217.4 ppm). Coordination of the σ2,λ3-P atom of 2-phosphaindolizine to (O-menthoxy)aluminium dichloride causes a downfield shift in the 31P NMR signal by δ 34–55 ppm, which is in accordance with the previous results [26,27]. An attempt to isolate the complex was, however, unsuccessful. 2,3-Dimethylbutadiene was then added and the progress of the reaction was monitored by 31P NMR. The reaction proceeded with complete diastereoselectivity, and in each case, only one isomer (8) was formed, as shown by the 31P NMR of the reaction mixture (Scheme 2).
Scheme 2:
Diels–Alder reaction of 2-phosphaindolizine-η1-P-aluminium(O-menthoxy) dichloride with 2,3-dimethylbutadiene.
Scheme 2:
Diels–Alder reaction of 2-phosphaindolizine-η1-P-aluminium(O-menthoxy) dichloride with 2,3-dimethyl...
The cycloadducts 8 are pale yellow, fine, crystalline solids, sparingly soluble in methylene chloride and chloroform. Their structures have been confirmed on the basis of 31P, 27Al, 1H and 13C NMR studies. The upfield 31P NMR chemical shifts in the range of δ –9.4 to –14.3 ppm (Scheme 2) are in conformity with those reported for the cycloadducts resulting from the DA reactions of P-W(CO)5 complexes of λ3-phosphinines [15]. The 27Al NMR signal at δ 62.3 to 100.9 ppm (Scheme 2) indicates fourfold coordination of the aluminium atom [28]. In addition, a broad signal at δ 44.9 to 51.1 ppm (∆ν1/2 5707 to 8834 Hz) and the absence of 31P–27Al coupling may be due to exchange of the ligand [29]. 13C NMR studies have been used extensively in the characterization of azaphospholes and their [2 + 4] cycloadducts due to their characteristic 13C–31P coupling constants [30-32]. In view of this, the 13C NMR spectrum of a representative product 8a was recorded. The signals of the carbon atoms directly bonded to the phosphorus atom, namely C1 (δ = 132.7 ppm, 1JPC = 36.0 Hz), C3 (δ = 54.5 ppm, 1JPC = 31.7 Hz) and C9 (δ = 33.6 ppm, 1JPC = 44.5 Hz) are identified readily by large values of 1JPC[33,34]. The 13C NMR signals due to the O-menthoxy moiety were assigned on the basis of the reported results [35].
Mode of action of the catalyst
In the DA reactions catalysed by excess dialkylaluminium chloride, formation of the chelate complex cation 11 of the dienophile (Scheme 3) has been established experimentally [36-38], and the high reactivity of the dienophile in the presence of the organoaluminium catalyst was attributed to the formation of this cationic species.
Scheme 3:
Formation of the cationic 1:1 complex of the dienophile and dialkylaluminium.
Scheme 3:
Formation of the cationic 1:1 complex of the dienophile and dialkylaluminium.
Later, Tietze et al. [39] rationalized higher reactivity and observed stereoselectivity resulting from the formation of the cationic complex on the basis of computational calculations. Recently, Yu and co-workers [40] investigated theoretically and experimentally the InCl3-catalyzed cycloisomerisation of 1,6-enynes and demonstrated InCl2+ to be the actual catalytic species participating in the reaction. In this context, it has been emphasized that identifying the real catalytic species may be very challenging, because in many cases impurities in the catalysts act as the real catalytic species [41]. As one of the referees pointed out this possibility, we carefully checked for the formation of a chelate cationic complex 13 on addition of the catalyst. After adding 2-phosphaindolizine (1 equiv) solution to the previously generated (O-menthoxy)aluminium dichloride solution, 31P NMR of the resulting solution was performed, in which only one signal in the range of δ 196–211 ppm corresponding to the (2-phosphaindolizine-η1-P)-Al(O-menthoxy)Cl2 complex was observed, and no 31P NMR signal for the uncomplexed 2-phosphaindolizine was detected, thus ruling out formation of the cationic species 13 (Scheme 4).
Scheme 4:
Disproportionation of the 1:1 complex of 2-phosphaindolizine and Al(O-menthoxy)Cl2.
Scheme 4:
Disproportionation of the 1:1 complex of 2-phosphaindolizine and Al(O-menthoxy)Cl2.
Furthermore, it has been established by X-ray crystal structure studies that Cr(CO)5 is coordinated to the phosphorus atom only, and no chelate complex involving the σ2,λ3-P atom and carbonyl oxygen atom is formed [27]. As reported recently, the DFT calculations reveal that the activation energy of the DA reaction is lowered only if the aluminium catalyst is coordinated to the phosphorus atom; when it is coordinated to the carbonyl oxygen atom, the activation energy barrier is rather high as compared to that for the DA reaction of the uncomplexed 2-phosphaindolizine [42]. Computational calculations also show that the conformation of 2-phosphaindolizine corresponding to the global minimum has phosphorous and carbonyl oxygen atoms in the antiperiplanar positions [35], thus reducing the possibility of chelate formation.
Theoretical results
We then investigated theoretically the mode of action of the chiral auxiliary in directing the complete diastereoselectivity of the DA reactions. The following model DA reactions (Scheme 5) were calculated at the DFT (B3LYP/6-31+G*) level.
Scheme 5:
Attack of 1,3-butadiene on Si and Re faces of >C=P– functionality of 2-phosphaindolizine complex.
Scheme 5:
Attack of 1,3-butadiene on Si and Re faces of >C=P– functionality of 2-phosphaindolizine complex.
It has been reported that for determining activation free energies and enthalpies of the pericyclic reactions, computational calculations at the B3LYP/6-31+G(d) level are very suitable [43-45]. Furthermore, the X-ray crystal investigation in one case confirmed the endo-structure of the resulting [4 + 2] cycloadduct [19]. In view of this, we also carried out computational calculations using the hybrid functional of Becke [46] and Lee, Yang and Parr [47]. Geometry optimizations of the reactants, the transition states and the cycloadducts were performed at the B3LYP/6-31+G* level. Stationary points were analysed by frequency calculations at the same level to confirm their character as local minima or transition structures. IRC calculations were performed in order to validate the connection of each transition state with the respective reactants and products. The solvent effect was computed by carrying out the single-point energy calculations of the gas-phase optimized geometries using the polarized continuum model (PCM). The Gaussian 03 program package [48] was used for all calculations.
Optimized geometries
Optimized geometries of (2-phosphaindolizine-η1-P)-Al(O-menth*)Cl2 (7a), the transition structures (TS1, TS2 and TS3 ), and the products (8a, 8a’ and 10) are shown in Figure 1.
Figure 1:
Geometries of 2-phosphaindolizine-η1-P-aluminium(O-menthoxy) dichloride, the transition structures, and the products optimized at the B3LYP/6-31+G* level in the gas-phase. The relative activation and reaction energies obtained in methylene chloride are given in parentheses.
Figure 1:
Geometries of 2-phosphaindolizine-η1-P-aluminium(O-menthoxy) dichloride, the transition structures,...
The optimized geometry of 7a (Figure 1) reveals that the menthol moiety shields the Re face of the >C=P– functionality in the coordinated 2-phosphaindolizine molecule. Attack of the 1,3-butadiene molecule from the less hindered Si face leads to the transition structures TS1 (endo) and TS2 (exo) and the products 8a and 8a’, respectively. On the other hand, attack of the diene from the sterically hindered Re face leads to the transition structure TS3 and the product 10.
Energetics
Ab initio investigations of the DA reaction of phosphaethene with 2H-phosphole [49] and with 1,3-butadiene [50-52] revealed low activation energies and a preference for the endo approach. In the present case, endo attack of the 1,3-butadiene molecule from the sterically more hindered Re face expectedly involves the higher-energy transition structure TS3. As regards the attack of the diene molecule from the sterically less hindered Si face, reactions involving both endo and exo approaches have been computed, and in contrast to the previous results, we find that the activation energy barrier for the exo approach involving TS2 is smaller than for the endo approach via TS1, by ca. 0.3 kcal mol−1. Presence of the bulky O-menthoxy moiety possibly makes the exo approach more preferable as compared to the endo approach. In methylene chloride, activation-energy barriers are increased by ca. 1 kcal mol−1. All the reactions are moderately exothermic, and exothermicity remains almost unaffected in methylene chloride.
Kinetics of the reactions
Standard-state entropies and entropy changes of different species, as well as the enthalpies and the Gibbs free energies of the computed reactions (as shown in Scheme 5) are given in Table 1.
Table 1:
Standard state entropies S0, entropy change ΔS, reaction enthalpies ΔH0 and reaction Gibbs free energies ΔG0.
Entry
Species
S0 (cal K−1 mol−1)
ΔS (cal K−1 mol−1)a
ΔH0 (kcal mol−1 )
ΔG0 (kcal mol−1 )
1
7a
211.1
–
2
9
66.1
–
3
TS1
231.4
−45.8
4
TS2
227.4
−49.8
5
TS3
231.9
−45.3
6
8a
225.7
−51.5
−3.70
+11.65
7
8a’
225.9
−51.3
−1.63
+13.67
8
10
225.8
−51.4
−2.24
+13.08
aThe relative entropy change; ΔS values have been obtained by subtracting the sum of the S0 values of 7a and 9 from the S0 value of the respective transition structure or the product.
The entropy effects have been found to play a major role in enzyme catalysis [53]. However, in the present case, the entropy difference between the TS1 and TS3 is negligible and does not appear to play significant role. On the other hand, entropy effects favour the endo approach over the exo approach from the Si face. Although the three reactions are endergonic, the reaction involving the endo approach of the diene from the less hindered Si face is preferred.
Π-Facial selectivity in the DA reactions has been investigated theoretically and the results have been found to be consistent with the experimentally observed results [54]. Origin of the diastereoselectivity observed in the cycloisomerisations of triynes has been correlated with the Gibbs free energies of the diastereomers calculated at the DFT B3LYP/TZV+P level; a difference of ca. 2 kcal mol−1 of Gibbs free energy corresponded to 84% diastereoselectivity [55]. In the present case also, the observed diastereoselectivity originates from the Re face being effectively blocked by the O-menthoxy moiety, thus making the diene attack the >C=P– functionality from the side of the Si face. In this case, the difference between the Gibbs free energies of 8a and 8a’ is found to be 2.02 kcal mol−1 in favour of the former.
The results suggest that the proposed mechanism involving the preferred attack of the diene from the Si face leading to the observed diastereoselectivity is valid, but the calculated absolute values for the energy barrier from this method are possibly too high.
Conclusion
The >C=P– functionality in 2-phosphaindolizines can be activated by coordinating the phosphorus atom to the Al(O-menthoxy)Cl2 moiety when it reacts with 2,3-dimethylbutadiene with complete diastereoselectivity. Computational calculations of the model DA reactions of (3-methoxycarbonyl-1-methyl-2-phosphaindolizine-η1-P)-Al(O-menth*)Cl2 with 1,3-butadiene reveal that the Re face is sterically hindered, and consequently, attack of the diene occurs preferentially from the Si face. Thermochemical data also support a preferential endo attack of the diene from the Si face. However, the absolute values for the energy barrier calculated for this method are possibly too high.
Experimental
Materials
Chemicals and solvents were purchased from Sigma-Aldrich. Solvents were dried according to the reported procedures. All the reactions were carried out in oxygen-free dry nitrogen under perfectly anhydrous conditions by using the Schlenk technique. 2-Phosphaindolizines (Scheme 2) were prepared by the [4 + 1] cyclocondensation method from the reaction of the respective 1-alkyl-2-ethylpyridinium bromide with phosphorus trichloride in the presence of triethylamine, as described in literature [56].
Analysis and characterisation of the products
Melting points were determined on a Tempo apparatus and are uncorrected. NMR spectra were recorded on a Jeol EX-300 MHz spectrometer: 31P NMR at a frequency of 121.50 MHz (using H3PO4 as the external reference), 1H NMR at a frequency of 300.40 MHz and 13C NMR at a frequency of 75.50 MHz (using TMS as the internal reference), and 27Al NMR at a frequency of 78.17 MHz (using Al(OiPr)3 as the external reference).
General method
A solution of (−)-menthoxyaluminium dichloride (5) (Scheme 2) was generated in situ [24] by adding ethylaluminium dichloride (4.6 mmol, 2.5 mL of 1 M solution in toluene) to a solution of (−)-menthol (4.6 mmol) (4) in CH2Cl2 under constant stirring at room temperature. This was followed by the addition of a solution of 2-phosphindolizine 6 (4.6 mmol) in CH2Cl2 (20mL) upon which an intense yellow colour developed (Scheme 2). After stirring for 30 minutes, the reaction mixture was cooled to −50 °C and a fivefold excess of 2,3-dimethylbutadiene (23 mmol, 1.8 g, 2.5 mL) was added under continuous stirring. The solution was then allowed to warm up to room temperature. After stirring of the reaction mixture overnight, completion of the reaction was revealed by the presence of only one signal (δ −9.4 to −14.3 ppm) in the 31P NMR spectrum. The solution was concentrated under vacuum to about 1/3 of its volume and left in a refrigerator after the addition of a few drops of hexane. Fine pale yellow crystals of the cycloadduct 8 deposited were separated, washed with hexane, and dried under vacuum.
Supporting Information File 1:
Cartesian coordinates of the geometries optimized (Table S1) and total energies of reactants, transition structures and products in the gas phase and in methylene chloride (Table S2) at the B3LYP/6-31+G* level.
Financial support from the Department of Science & Technology, New Delhi (Grant No. SR/SI/OC-71/2008) and the DFG, Bonn (Grant No. INT/FRG/DFG/P-22/2008) is gratefully acknowledged.
References
Jacobsen, E. N.; Pfaltz, A.; Yamamota, H., Eds. Comprehensive asymmetric catalysis I-III; Springer: Berlin, 1999.
Return to citation in text:
[1]
Collins, A. N.; Sheldrake, G. N.; Crosby, J., Eds. Chirality in industry-I & II; John Wiley: New York, 1992, 1996.
Return to citation in text:
[1]
Blaser, H. U. Chem. Rev.1992,92, 935–952. doi:10.1021/cr00013a009
Return to citation in text:
[1]
[2]
Oertling, H.; Reckziegel, A.; Surburg, H.; Bertram, H.-J. Chem. Rev.2007,107, 2136–2164. doi:10.1021/cr068409f
Return to citation in text:
[1]
Brunel, J. M.; Buono, G. New Chiral Organophosphorus Catalysts in Asymmetric Synthesis. In New aspects in phosphorus chemistry; Majoral, J., Ed.; Springer: Berlin, 2002; pp 79–105.
Return to citation in text:
[1]
Hayashi, T. Acc. Chem. Res.2000,33, 354–362. doi:10.1021/ar990080f
Return to citation in text:
[1]
Shin, J. H.; Parkin, G. J. Am. Chem. Soc.2002,124, 7652–7653. doi:10.1021/ja026059i
Return to citation in text:
[1]
Colby, E. A.; Jamison, T. F. J. Org. Chem.2003,68, 156–166. doi:10.1021/jo0264123
Return to citation in text:
[1]
Arbuzov, B. A.; Dianova, E. N.; Samitov, Y. Y. Dokl. Akad. Nauk SSSR1979,244, 1117.
Return to citation in text:
[1]
Appel, R.; Knoll, F.; Ruppert, I. Angew. Chem., Int. Ed. Engl.1981,20, 731–744. doi:10.1002/anie.198107311
Return to citation in text:
[1]
Appel, R. In Multiple Bonds and Low Coordination in Phosphorus Chemistry; Regitz, O.; Scherer, O. J., Eds.; Thieme: Stuttgart, Germany, 1990; p 157.
Return to citation in text:
[1]
Mathey, F.; Mercier, F.; Charrier, C.; Fischer, J.; Mitschler, A. J. Am. Chem. Soc.1981,103, 4595–4597. doi:10.1021/ja00405a058
Return to citation in text:
[1]
Märkl, G.; Lieb, F. Angew. Chem., Int. Ed. Engl.1968,7, 733. doi:10.1002/anie.196807331
Return to citation in text:
[1]
Bansal, R. K.; Kumawat, S. K. Tetrahedron2008,64, 10945–10976. doi:10.1016/j.tet.2008.09.037
Return to citation in text:
[1]
[2]
Bansal, R. K.; Surana, A.; Gupta, N. Tetrahedron Lett.1999,40, 1565–1568. doi:10.1016/S0040-4039(98)02645-8
Return to citation in text:
[1]
Bansal, R. K.; Gupta, N.; Kumawat, S. K.; Dixit, G. Tetrahedron2008,64, 6395–6401. doi:10.1016/j.tet.2008.04.080
Return to citation in text:
[1]
[2]
Bansal, R. K.; Jain, V. K.; Gupta, N.; Hemrajani, L.; Baweja, M.; Jones, P. G. Tetrahedron2002,58, 1573–1579. doi:10.1016/S0040-4020(02)00004-2
Return to citation in text:
[1]
[2]
Jangid, R. K.; Gupta, N.; Bansal, R. K.; von Hopffgarten, M.; Frenking, G. Tetrahedron Lett.2011,52, 1721–1724. doi:10.1016/j.tetlet.2011.02.007
Return to citation in text:
[1]
Hashimoto, S.; Komeshima, N.; Koga, K. J. Chem. Soc., Chem. Commun.1979, 437–438. doi:10.1039/C39790000437
Return to citation in text:
[1]
Wulff, W. D. In Lewis Acids in Organic Synthesis; Yamamota, H., Ed.; Wiley-VCH: Weinheim, Germany, 2000; Vol. 1, pp 301–354.
Return to citation in text:
[1]
Kagan, H. B.; Riant, O. Chem. Rev.1992,92, 1007–1019. doi:10.1021/cr00013a013
Return to citation in text:
[1]
[2]
Karaghiosoff, K. In Multiple Bonds and Low Coordination in Phosphorus Chemistry; Regitz, M.; Scherer, O. J., Eds.; Thieme: Stuttgart, Germany, 1990; p 463.
Return to citation in text:
[1]
Bansal, R. K.; Karaghiosoff, K.; Gupta, N.; Kabra, V.; Mahnot, R.; Sharma, D. C.; Munjal, R.; Kumawat, S. K. Z. Naturforsch.2005,60b, 7–14.
Return to citation in text:
[1]
Kwan, E. E.; Huang, S. G. Eur. J. Org. Chem.2008, 2671–2688. doi:10.1002/ejoc.200700966
Return to citation in text:
[1]
[2]
Lehmkuhl, H.; Kobs, H.-D. Justus Liebigs Ann. Chem.1969,719, 11–19. doi:10.1002/jlac.19687190103
Return to citation in text:
[1]
Evans, D. A.; Chapman, K. T.; Bisaha, J. J. Am. Chem. Soc.1988,110, 1238–1256. doi:10.1021/ja00212a037
Return to citation in text:
[1]
Castellino, S.; Dwight, W. J. J. Am. Chem. Soc.1993,115, 2986–2987. doi:10.1021/ja00060a061
Return to citation in text:
[1]
Tietze, L. F.; Schuffenhauer, A.; Schreiner, P. R. J. Am. Chem. Soc.1998,120, 7952–7958. doi:10.1021/ja980079w
Return to citation in text:
[1]
Zhuo, L.-G.; Zhang, J.-J.; Yu, Z.-X. J. Org. Chem.2012,77, 8527–8540. doi:10.1021/jo301471w
Return to citation in text:
[1]
Thomé, L.; Nijs, A.; Bolm, C. Chem. Soc. Rev.2012,41, 979–987. doi:10.1039/C2CS15249E
Return to citation in text:
[1]
Gupta, N.; Jangid, R. K.; Bansal, R. K.; von Hopffgarten, M. Curr. Catal.2012,1, 93–99. doi:10.2174/2211544711201020093
Return to citation in text:
[1]
Guner, V. A.; Khuong, K. S.; Houk, K. N.; Chuma, A.; Pulay, P. J. Phys. Chem. A2004,108, 2959–2965. doi:10.1021/jp0369286
Return to citation in text:
[1]
Guner, V. A.; Khuong, K. S.; Leach, A. G.; Lee, P. S.; Bartberger, M. D.; Houk, K. N. J. Phys. Chem. A2003,107, 11445–11459. doi:10.1021/jp035501w
Return to citation in text:
[1]
Chen, Y.; Ye, S.; Jiao, L.; Liang, Y.; Sinha-Mahapatra, D. K.; Herndon, J. W.; Yu, Z.-X. J. Am. Chem. Soc.2007,129, 10773–10784. doi:10.1021/ja072203u
Return to citation in text:
[1]
Becke, A. D. J. Chem. Phys.1993,98, 5648–5652. doi:10.1063/1.464913
Return to citation in text:
[1]
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B1988,37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text:
[1]
Gaussian 03, Revision B.05; Gaussian, Inc.: Wallingford, CT, 2004.
Return to citation in text:
[1]
Mulhearn, D. C.; Bachrach, S. M. Proceedings of the First Electronic Computational Chemistry Conference; Paper 11; Chem. Abstr.1994,124, 316229.
Return to citation in text:
[1]
Penkovsky, V.; Kharchenko, V.; Alexeiko, L. Phosphorus, Sulfur Silicon Relat. Elem.1993,77, 81–84. doi:10.1080/10426509308045624
Return to citation in text:
[1]
Wannere, C. S.; Bansal, R. K.; von Ragué Schleyer, P. J. Org. Chem.2002,67, 9162–9174. doi:10.1021/jo026284i
Return to citation in text:
[1]
Strajbl, M.; Sham, Y. Y.; Villà, J.; Chu, Z.-T.; Warchel, A. J. Phys. Chem. B2000,104, 4578–4584. doi:10.1021/jp0003095
Return to citation in text:
[1]
Marchand, A. P.; Chong, H.; Ganguly, B.; Coxon, J. M. Croat. Chem. Acta2000,73, 1027–1038.
Return to citation in text:
[1]
Sehnal, P.; Krausova, Z.; Teply, F.; Stara, I. G.; Stary, I.; Rulisek, L.; Saman, D.; Cisarova, I. J. Org. Chem.2008,73, 2074–2082. doi:10.1021/jo701997p
Return to citation in text:
[1]
Bansal, R. K.; Karaghiosoff, K.; Gandhi, N.; Schmidpeter, A. Synthesis1995, 361–369. doi:10.1055/s-1995-3935
Return to citation in text:
[1]
Brunel, J. M.; Buono, G. New Chiral Organophosphorus Catalysts in Asymmetric Synthesis. In New aspects in phosphorus chemistry; Majoral, J., Ed.; Springer: Berlin, 2002; pp 79–105.
Karaghiosoff, K. In Multiple Bonds and Low Coordination in Phosphorus Chemistry; Regitz, M.; Scherer, O. J., Eds.; Thieme: Stuttgart, Germany, 1990; p 463.
34.
Bansal, R. K.; Karaghiosoff, K.; Gupta, N.; Kabra, V.; Mahnot, R.; Sharma, D. C.; Munjal, R.; Kumawat, S. K. Z. Naturforsch.2005,60b, 7–14.


![[1860-5397-9-40-i1]](/bjoc/content/inline/1860-5397-9-40-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-40-i2]](/bjoc/content/inline/1860-5397-9-40-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-40-i3]](/bjoc/content/inline/1860-5397-9-40-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-40-i4]](/bjoc/content/inline/1860-5397-9-40-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-40-i5]](/bjoc/content/inline/1860-5397-9-40-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-40-1]](/bjoc/content/figures/1860-5397-9-40-1.png?scale=2.0&max-width=1024&background=FFFFFF)