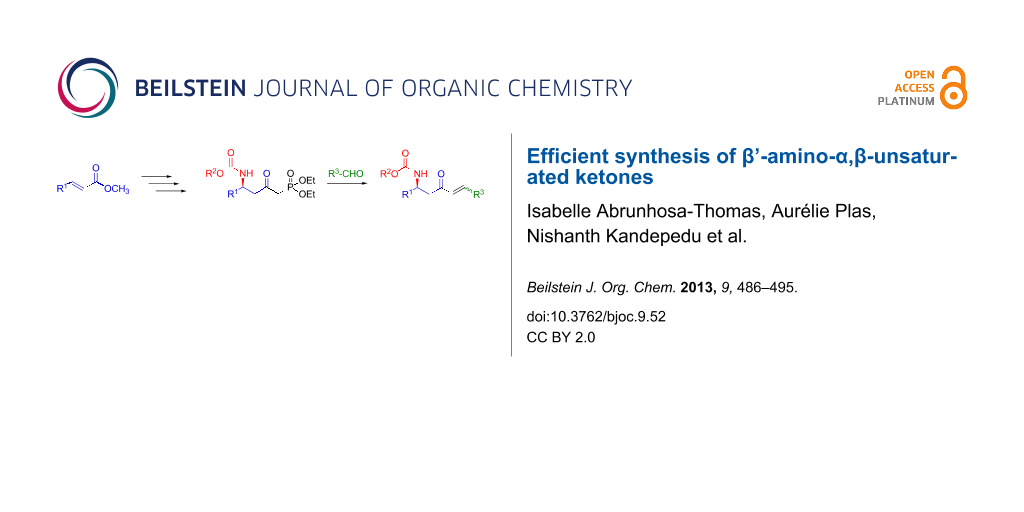
Abstract
A general and simple procedure to access chiral β'-amino-α,β-enones, in seven steps, from an α,β unsaturated ester has been described. The use of a Horner–Wadsworth–Emmons reaction as a key step for generating the β'-amino-α,β-enones, permits access to a range of substrates under mild conditions and in moderate to high yield.

Graphical Abstract
Introduction
Compounds incorporating β-amino ketone functionality are prevalent in many natural products of biological importance [1]. This versatile synthon has been extensively used in the construction of β-amino acids [2], β-amino alcohols [3], and homoallylic amines [4,5], and can serve as building blocks for the preparation of nitrogen-containing molecules often found in medicinal chemistry [6-10]. Thus, the development of efficient and stereoselective reactions for a useful approach to chiral β-amino ketones is still of importance. One of the most powerful approaches is the Mannich reaction, which can be conducted under different protocols in which the stereoselectivity of the reaction can be introduced through the use of a chiral catalyst [9,10] (Lewis acid, Brønsted acids, L-proline, Cinchona alkaloids derivatives, thioureas, etc.), or by the addition of chiral amines to α,β-unsaturated esters [11,12] or the reaction of chiral imines with enolates derived from Weinreb amides [13,14]. In previous work on the asymmetric synthesis of 2,6-disubstituted piperidines by C–N bond formation, we demonstrated that intramolecular aza-Michael ”type” cyclisation [15] using a β'-carbamate-α,β-unsaturated ketone predominantly induces the formation of a piperidine ring with the 2,6-trans configuration (Scheme 1).
![[1860-5397-9-52-i1]](/bjoc/content/inline/1860-5397-9-52-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Asymmetric synthesis of 2-methyl-6-phenyl piperidine.
Scheme 1: Asymmetric synthesis of 2-methyl-6-phenyl piperidine.
The relative stereochemistry of piperidine 2a was confirmed by further transformation to the known compound 3 [16,17] with 94% ee. In order to establish this new approach as a general method for the preparation of chiral 2,6-disubstituted piperidines, we wish to report here a facile synthetic route to various β’-carbamate-α,β-unsaturated ketones in good overall yields and good enantioselectivities.
Results and Discussion
In a preliminary approach, preparation of β-amino ketones was envisaged through a nucleophilic addition reaction of Grignard reagents to N-carbamoyl β-amino Weinreb amides (Scheme 2) [18].
![[1860-5397-9-52-i2]](/bjoc/content/inline/1860-5397-9-52-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: (a) Davies amine, BuLi, THF, −78 °C; dr ≥ 94% ; (b) H2, Pd(OH)2, MeOH; (c) Na2CO3, PhCH2CO2Cl, CH2Cl2/H2O; (d) NaOH 1 N, MeOH; (e) CDI, N,O-dimethylhydroxylamine·HCl, (f) Mg, 1-bromo-2-propene, THF.
Scheme 2: (a) Davies amine, BuLi, THF, −78 °C; dr ≥ 94% ; (b) H2, Pd(OH)2, MeOH; (c) Na2CO3, PhCH2CO2Cl, CH2Cl...
Conjugate addition of (R)-N-benzyl-N-methylbenzylamide to methyl cinnamate under basic conditions led to β–aminoester 5 with high diastereoselectivity (dr >94%) [11,12]. Subsequent transformation of the ester moiety to a Weinreb amide [18] followed by changing the nitrogen protecting group to a carbamate furnished the key intermediate 6, which could be further alkylated with Grignard reagents to give β’-amino protected α,β-enone 1 in good overall yield and high enantiomeric excess. As Grignard reagents did not allow the use of a wide range of functional groups and sometimes gave bad overall yields, we devised a general and simple method to easily access a variety of β’-amino-α,β-unsaturated ketones by a more convenient route using the Horner–Wadsworth–Emmons reaction [19,20] as the key step, as described in Scheme 3.
![[1860-5397-9-52-i3]](/bjoc/content/inline/1860-5397-9-52-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In order to gain access to phosphonates 13, in a general and convergent process, two ways were investigated (Scheme 4). In route A, we planned to obtain the desired compound by using a similar strategy to that which we have described previously (see above): chiral induction was obtained through the addition of Davies amine, furnishing 8. Hydrogenation of compound 8 followed by N-protection as a carbamate would furnish the β-amino ester precursor of the phosphonate 13. In route B, the most convergent, the preparation of the phosphonate was envisaged in the first step; then, hydrogenation would furnish amino phosphonate. In method A the last step to synthesize 13 would be a nucleophilic addition of diethyl methylphosphonate under basic conditions, whereas for method B, the final step would be the N-protection of the amino phosphonate as a carbamate.
![[1860-5397-9-52-i4]](/bjoc/content/inline/1860-5397-9-52-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Possible pathways to obtain phosphonate 13 (a) Davies amine, BuLi, THF, −78 °C; dr ≥ 95%; (b) H2, Pd(OH)2/C, MeOH, 60 psi; (c) Na2CO3, R2CO2Cl, CH2Cl2/H2O; (d) and (e) BuLi, (EtO)2P(O)Me, THF, −78 °C.
Scheme 4: Possible pathways to obtain phosphonate 13 (a) Davies amine, BuLi, THF, −78 °C; dr ≥ 95%; (b) H2, P...
The two routes were then tested, starting from methyl crotonate (7a, R1 = Me) as a model substrate (Scheme 4). While compounds 9 and 10a were obtained in reasonable yields (80 and 70%, respectively) in route A, the hydrogenation of 11 (obtained in a 74% yield from 8) to 12 did not proceed to provide the expected compound under various conditions (methanol in acid conditions, using either Pd(OH)2/C under H2 pressure (60 psi) or Pd/C under reflux in the presence of ammonium formate). Instead, the formation of 14 [21] was observed, resulting from β-elimination and reduction of the transient double bond (Scheme 5).
![[1860-5397-9-52-i5]](/bjoc/content/inline/1860-5397-9-52-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Thus, we focused on route A, and after optimization of the reaction conditions, we found that the transformation of 7a to 10a could be done without purification. Hence, the addition of enantiopure lithium N-benzyl-N-α-methylbenzylamide to α,β-unsaturated ester 7 followed by hydrogenation to the corresponding primary amine and further protection as a carbamate gave the β-amino methylester 10. At this stage, the ester function was transformed into the ketophosphonate 13 by treatment with 2.5 equivalents of the lithium anion of diethyl methylphosphonate [22-28] in THF at −78 °C, in moderate to good yields (Scheme 6, Table 1).
![[1860-5397-9-52-i6]](/bjoc/content/inline/1860-5397-9-52-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: General synthesis of compound 13 (a) Davies amine, BuLi, THF, −78 °C; (b) H2, Pd(OH)2/C, MeOH; (c) Na2CO3, R2CO2Cl, CH2Cl2/H2O; (d) BuLi, (EtO)2P(O)Me, THF, −78 °C.
Scheme 6: General synthesis of compound 13 (a) Davies amine, BuLi, THF, −78 °C; (b) H2, Pd(OH)2/C, MeOH; (c) ...
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-52-i8.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-52-i9.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-52-i10.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-52-i11.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-52-i12.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-52-i13.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-52-i14.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-52-i15.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-52-i16.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-52-i17.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-52-i18.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-52-i19.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-52-i20.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-52-i21.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-52-i22.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-52-i23.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-52-i24.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-52-i25.svg?max-width=637&scale=1.0)
Over the years, many examples of base-promoted Horner–Wadsworth–Emmons (HWE) reactions have been reported in the literature, and various combinations of bases and solvents (K2CO3/CH3CN[23], DBU/THF[25], NaH/THF[29], Et3N/LiCl/CH3CN[30] or Ba(OH)2/(THF/H2O)[31]) have been used. We subjected our substrate 13a to three of those mild sets of conditions (Scheme 7, Table 2), which, after reaction with the benzaldehyde, will furnish the chiral amino ketone 15a.
![[1860-5397-9-52-i7]](/bjoc/content/inline/1860-5397-9-52-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Optimization of conditions for the Horner–Wadsworth–Emmons reaction.
Scheme 7: Optimization of conditions for the Horner–Wadsworth–Emmons reaction.
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-52-i26.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-52-i27.svg?max-width=637&scale=1.0)
As illustrated in Table 2, we found that the use of 1.3 equiv of Ba(OH)2 THF/H2O (40/1) furnished the optimal yield of 95% with our model substrate. Those conditions were then applied to a wide range of functionalized aldehydes with phosphonate 13a–g, giving amino ketone 15a–z in good to excellent yields and high E/Z ratio (≥ 95%). The results are presented in Table 3.
Table 3: Formation of the β’-amino-α,β-unsaturated ketones 15 under Ba(OH)2 conditions.
| entry | phosphonate 13 | aldehyde | amino ketone 15 | yield (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-52-i28.svg?max-width=637&scale=1.0)
13a |
benzaldehyde |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-52-i29.svg?max-width=637&scale=1.0)
15a |
95 |
| 2 | 13a | o-nitrobenzaldehyde |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-52-i30.svg?max-width=637&scale=1.0)
15b |
89 |
| 3 | 13a | m-nitrobenzaldehyde |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-52-i31.svg?max-width=637&scale=1.0)
15c |
86 |
| 4 | 13a | p-nitrobenzaldehyde |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-52-i32.svg?max-width=637&scale=1.0)
15d |
91 |
| 5 | 13a | p-methoxybenzaldehyde |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-52-i33.svg?max-width=637&scale=1.0)
15e |
79 |
| 6 | 13a | o-bromobenzaldehyde |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-52-i34.svg?max-width=637&scale=1.0)
15f |
91 |
| 7 | 13a | p-bromobenzaldehyde |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-52-i35.svg?max-width=637&scale=1.0)
15g |
89 |
| 8 | 13a | 2-chloro-5-nitrobenzaldehyde |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-52-i36.svg?max-width=637&scale=1.0)
15h |
95 |
| 9 | 13a | pyridine-3-carboxaldehyde |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-52-i37.svg?max-width=637&scale=1.0)
15i |
87 |
| 10 | 13a | (E)-ethyl-4-oxo-2-butenoate |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-52-i38.svg?max-width=637&scale=1.0)
15j |
85 |
| 11 | 13a | ethylglyoxylate |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-52-i39.svg?max-width=637&scale=1.0)
15k |
53 |
| 12 | 13a | butanal |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-52-i40.svg?max-width=637&scale=1.0)
15l |
95 |
| 13 | 13a | nonanal |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-52-i41.svg?max-width=637&scale=1.0)
15m |
88 |
| 14 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-52-i42.svg?max-width=637&scale=1.0)
13b |
benzaldehyde |
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-52-i43.svg?max-width=637&scale=1.0)
15n |
96 |
| 15 | 13b | decanal |
![[Graphic 37]](/bjoc/content/inline/1860-5397-9-52-i44.svg?max-width=637&scale=1.0)
15o |
78 |
| 16 | 13b | dodecanal |
![[Graphic 38]](/bjoc/content/inline/1860-5397-9-52-i45.svg?max-width=637&scale=1.0)
15p |
91 |
| 17 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-9-52-i46.svg?max-width=637&scale=1.0)
13c |
benzaldehyde |
![[Graphic 40]](/bjoc/content/inline/1860-5397-9-52-i47.svg?max-width=637&scale=1.0)
15q |
91 |
| 18 | 13c | p-nitrobenzaldehyde |
![[Graphic 41]](/bjoc/content/inline/1860-5397-9-52-i48.svg?max-width=637&scale=1.0)
15r |
84 |
| 19 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-9-52-i49.svg?max-width=637&scale=1.0)
13d |
benzaldehyde |
![[Graphic 43]](/bjoc/content/inline/1860-5397-9-52-i50.svg?max-width=637&scale=1.0)
15s |
94 |
| 20 | 13d | ethanal |
![[Graphic 44]](/bjoc/content/inline/1860-5397-9-52-i51.svg?max-width=637&scale=1.0)
15t |
78 |
| 21 | 13d | butanal |
![[Graphic 45]](/bjoc/content/inline/1860-5397-9-52-i52.svg?max-width=637&scale=1.0)
15u |
95 |
| 22 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-9-52-i53.svg?max-width=637&scale=1.0)
13e |
benzaldehyde |
![[Graphic 47]](/bjoc/content/inline/1860-5397-9-52-i54.svg?max-width=637&scale=1.0)
15v |
84 |
| 23 | 13e | ethanal |
![[Graphic 48]](/bjoc/content/inline/1860-5397-9-52-i55.svg?max-width=637&scale=1.0)
15w |
80 |
| 24 | 13e | butanal |
![[Graphic 49]](/bjoc/content/inline/1860-5397-9-52-i56.svg?max-width=637&scale=1.0)
15x |
95 |
| 25 | 13e | nonanal |
![[Graphic 50]](/bjoc/content/inline/1860-5397-9-52-i57.svg?max-width=637&scale=1.0)
15y |
89 |
| 26 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-9-52-i58.svg?max-width=637&scale=1.0)
13f |
ethanal |
![[Graphic 52]](/bjoc/content/inline/1860-5397-9-52-i59.svg?max-width=637&scale=1.0)
15z |
81 |
| 27 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-9-52-i60.svg?max-width=637&scale=1.0)
13g |
ethanal |
![[Graphic 54]](/bjoc/content/inline/1860-5397-9-52-i61.svg?max-width=637&scale=1.0)
1 |
76 |
Conclusion
In summary, a general methodology has been devised for the asymmetric synthesis of β’-amino-protected-α,β enones, a valuable intermediate for the synthesis of trans 2,6-disubstituted piperidines. The scope and limitation of the aza-Michael reaction were studied with a range of substrates. We are currently working on the application of this synthetic method to the preparation of piperidine natural products.
Experimental
Organic solutions were dried over MgSO4 or Na2SO4, and filtered. When anhydrous solvents were used, they were prepared as follows: tetrahydrofuran (THF) was distilled under N2 from sodium benzophenone ketyl and used immediately; anhydrous acetonitrile was freshly distilled from CaH2. All 1H and 13C NMR spectra were measured in CDCl3 or C6D6 and recorded on a Bruker 400 MHz (101 MHz for 13C) spectrometer with TMS as the internal standard. Chemical shifts are expressed in parts per million (ppm) and J-values are given in hertz. The following abbreviations are used: singlet (s), doublet (d), doublet of doublets (dd), triplet (t), multiplet (m). High-resolution mass spectroscopy (HRMS) was carried out in electrospray mode and was performed by CRMP (Clermont-Ferrand, France). Monitoring of the reactions was performed by using silica-gel TLC plates (silica Merck 60 F254). Spots were visualized by UV light at 254 nm. Flash chromatography was performed by using silica gel 60 (70–230 mesh) or RP18 (25–40 lM) from Merck Chimie SAS (France) on a Flash II apparatus (Armen Instrument, France).
General procedure for the synthesis of 10
(R)-Methyl 3-(ethoxycarbonylamino)butanoate 10a: To a cold solution (0 °C) of (+)-(R)-N-benzyl-N-α-methylbenzylamine (23.0 mL, 110 mmol, 1.1 equiv) in dry THF (280 mL) was added n-butyllithium (75.0 mL, 1.6 M in hexane, 120 mmol, 1.2 equiv) slowly under argon. The resultant pink solution of lithium amide was stirred for 30 min then cooled to −78 °C before dropwise addition of a solution of methyl crotonate (10.0 mL, 100 mmol, 1 equiv) in dry THF (100 mL). The mixture was stirred at –78 °C for 90 min. Then, a saturated aqueous solution of NH4Cl (100 mL) was added slowly, and the resulting solution was allowed to warm to room temperature. Then, the solution was extracted twice with ethyl acetate. The combined organic extracts were dried over Na2SO4, filtered and evaporated. The crude product was added to a suspension of 10% Pd/C (5.00 g) in methanol (200 mL). The mixture was placed on a Parr apparatus and stirred under a hydrogen atmosphere (60 psi) for 4 days. The catalyst was removed by filtration on Celite®. The residue was concentrated in vacuum and dissolved in dichloromethane (200 mL) and water (200 mL). Then, sodium carbonate (42.4 g, 400 mmol, 4.0 equiv) and ethyl chloroformate (28.5 mL, 200 mmol, 2 equiv) were added dropwise. The resulting solution was stirred at room temperature for 3 h. The aqueous material was extracted with dichloromethane and the combined organic extracts were dried over Na2SO4, filtered and concentrated in vacuo. Purification by chromatography on silica gel (cyclohexane/EtOAc 9/1 to 5/5) afforded 10a as a yellow oil (21.4 g, 57% in three steps): [α]D25 −35.6 (c 0.99, CHCl3), lit.[32] [α]D25 −37.07 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.03 (br s, 1H, NH), 4.03 (m, 3H), 3.62 (s, 3H), 2.46 (d, J = 6.9 Hz, 2H), 1.16 (t, J = 6.9 Hz, 3H), 1.15 (d, J = 6.6 Hz, 3H). Spectral data are identical to those reported in [32].
General procedure for the synthesis of 13
(R)-Ethyl [5-(diethoxyphosphoryl)-4-oxopentan-2-yl]carbamate 13a: To a solution of diethyl methylphosphonate (5.8 mL, 39.7 mmol, 2.5 equiv) in anhydrous THF (15 mL) kept at −78 °C, was added dropwise n-butyl lithium (24.8 mL, 1.6 M in hexane, 39.7 mmol, 2.5 equiv). After 20 min at −78 °C, a solution of 10a (3 g, 15.9 mmol, 1 equiv) in anhydrous THF (15 mL) was added dropwise. After addition, the temperature of the reaction was kept at −78 °C for 30 min and then allowed to reach 0 °C over 1 h, and the reaction was quenched with a solution of ammonium chloride and extracted twice with ethyl acetate. After drying over MgSO4 and concentration under vacuum, the crude oil was first distilled at low pressure to remove excess diethyl methylphosphonate, and the residue was then purified by flash chromatography (eluent: cyclohexane/EtOAc 2/1 to EtOAc) afforded 13a as a yellow oil (3.3 g, 68% yield): [α]D25 +33.6 (c 1.17, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.03 (br s, 1H,), 4.16–3.94 (m, 7H), 3.08 (dd, J = 23.0, 14.0 Hz, 1H), 2.99 (dd, J = 22.6, 14.0 Hz, 1H), 2.84 (dd, J = 17.1, 6.0 Hz, 1H), 2.71 (dd, J = 17.1, 5.7 Hz, 1H), 1.33–1.21 (m, 6H), 1.15–1.20 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 200.6, 155.8, 62.6 (d, J = 6.6 Hz), 62.5 (d, J = 6.5 Hz), 60.5, 49.6, 43.5, 42.9 (d, J = 127.4 Hz), 20.7, 16.2, 16.1, 14.6; HRMS-ESI (M + Na), m/z calcd. for C12H24NO6PNa 332.1239, found 332.1239.
General procedure for the synthesis of 15
(R,E)-Ethyl [4-oxo-6-phenyl-hex-5-en-2-yl]carbamate 15a: To a solution of 13a (0.5 g, 1.6 mmol, 1 equiv) in THF (7 mL), was poured Ba(OH)2 (0.346 g, 2.0 mmol, 1.25 equiv) in one batch at room temperature. After 30 min, a solution of benzaldehyde (0.172 ml, 1.7 mmol, 1.05 equiv) in THF/H2O (40/1) (7 mL) was slowly added at room temperature. After 1 h, the reaction mixture was quenched with ammonium chloride and extracted three times with ethyl acetate. Then the organic layer was dried over MgSO4, concentrated under vacuum and purified by flash chromatography (eluent: cyclohexane to cyclohexane/EtOAc 8/2) to give 15a as a white solid (0.401 g, 95%): Mp 74 °C; [α]D25 +9.5 (c 1.21, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 16.7 Hz, 1H), 7.47 (dd, J = 7.8, 3.0 Hz, 1H), 7.33–7.30 (m, 3H), 6.65 (d, J = 16.7 Hz, 1H), 5.14 (s, 1H), 4.14–4.06 (m, 1H), 4.02 (q, J = 6.9 Hz, 2H), 2.95 (dd, J = 15.9, 4.2 Hz, 1H), 2.71 (dd, J = 15.9, 6.5 Hz, 1H), 1.19 (d, J = 6.8 Hz, 3H), 1.14 (t, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 198.8, 155.9, 143.4, 134.3, 130.6, 128.9, 128.4, 126.3, 60.6, 46.3, 44.1, 20.5, 14.6; HRMS-ESI (M + Na): calcd. for C15H19NO3Na 284.1263, found 284.1275.
Supporting Information
| Supporting Information File 1: Experimental section, characterization data and spectra of all new compounds. | ||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Seebach, D.; Kimmerlin, T.; Šebesta, R.; Campo, M. A.; Beck, A. K. Tetrahedron 2004, 60, 7455–7506. doi:10.1016/j.tet.2004.06.043
Return to citation in text: [1] -
Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2
Return to citation in text: [1] -
Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957–5978. doi:10.1016/S0040-4020(02)00584-7
Return to citation in text: [1] -
Ishimaru, K.; Kojima, T. J. J. Org. Chem. 2000, 65, 8395–8398. doi:10.1021/jo0011888
Return to citation in text: [1] -
Davis, F. A.; Song, M.; Augustine, A. J. Org. Chem. 2006, 71, 2779–2786. doi:10.1021/jo052566h
Return to citation in text: [1] -
Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. Drug Discovery Today 2007, 12, 8–27. doi:10.1016/j.drudis.2006.11.004
Return to citation in text: [1] -
Verkade, J. M. M.; Van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutges, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29–41. doi:10.1039/b713885g
Return to citation in text: [1] -
Bhadury, P. S.; Song, B.-A. Curr. Org. Chem 2010, 14, 1989–2006. doi:10.2174/138527210792927564
Return to citation in text: [1] -
Probst, N.; Madarász, Á.; Valkonen, A.; Pápai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P. M. Angew. Chem., Int. Ed. 2012, 51, 8495–8499. doi:10.1002/anie.201203852
Return to citation in text: [1] [2] -
Jiang, C.; Zhong, F.; Lu, Y. Beilstein J. Org. Chem. 2012, 8, 1279–1283. doi:10.3762/bjoc.8.144
Return to citation in text: [1] [2] -
Brackenridge, I.; Davies, S. G.; Fenwick, D. R.; Ichihara, O.; Polywka, M. E. C. Tetrahedron 1999, 55, 533–540. doi:10.1016/S0040-4020(98)01051-5
Return to citation in text: [1] [2] -
Fleck, T. J.; McWhorter, W. W.; DeKam, R. N.; Pearlman, B. A. J. Org. Chem. 2003, 68, 9612–9617. doi:10.1021/jo0349633
Return to citation in text: [1] [2] -
Davis, F. A.; Theddu, N. J. Org. Chem. 2010, 75, 3814–3820. doi:10.1021/jo100680b
Return to citation in text: [1] -
Davis, F. A.; Xu, P. J. Org. Chem. 2011, 76, 3329–3337. doi:10.1021/jo2002352
Return to citation in text: [1] -
Abrunhosa-Thomas, I.; Roy, O.; Barra, M.; Besset, T.; Chalard, P.; Troin, Y. Synlett 2007, 1613–1615. doi:10.1055/s-2007-982547
Return to citation in text: [1] -
Poerwono, H.; Higashimaya, K.; Yamauchi, T.; Kubo, H.; Ohmiya, S.; Takahashi, H. Tetrahedron 1998, 54, 13955–13970. doi:10.1016/S0040-4020(98)00863-1
Return to citation in text: [1] -
Davis, F. A.; Xu, H.; Zhang, J. J. Org. Chem. 2007, 72, 2046–2052. doi:10.1021/jo062365t
Return to citation in text: [1] -
Sibi, M. P. Org. Prep. Proced. Int. 1993, 25, 15–40. doi:10.1080/00304949309457931
Return to citation in text: [1] [2] -
Wadsworth, W. S.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042
Return to citation in text: [1] -
Wadsworth, W. S.; Emmons, W. D. Org. Synth. 1973, Coll. Vol. 5, 547–563.
Return to citation in text: [1] -
Della Monica, C.; Maulucci, N.; De Riccardis, F.; Izzo, I. Tetrahedron: Asymmetry 2003, 14, 3371–3378. doi:10.1016/S0957-4166(03)00622-0
Return to citation in text: [1] -
Rudisill, D. E.; Whitten, J. P. Synthesis 1994, 851–854. doi:10.1055/s-1994-25588
Return to citation in text: [1] -
Daly, M.; Cant, A. A.; Fowler, L. S.; Simpson, G. L.; Senn, H. M.; Sutherland, A. J. Org. Chem. 2012, 77, 10001–10009. doi:10.1021/jo3022583
Return to citation in text: [1] [2] -
Boeglin, D.; Heitz, A.; Martinez, J.; Fehrentz, J.-A. Eur. J. Org. Chem. 2003, 3139–3146. doi:10.1002/ejoc.200300148
Return to citation in text: [1] -
Davis, F. A.; Wu, Y. Org. Lett. 2004, 6, 1269–1272. doi:10.1021/ol049795v
Return to citation in text: [1] [2] -
Modica, E.; Compostella, F.; Colombo, D.; Franchini, L.; Cavallari, M.; Mori, L.; De Libero, G.; Panza, L.; Ronchetti, F. Org. Lett. 2006, 8, 3255–3258. doi:10.1021/ol061100y
Return to citation in text: [1] -
Wiemer, D. F. Tetrahedron 1997, 53, 16609–16644. doi:10.1016/S0040-4020(97)10305-2
A review on the preparation of nonracemic phosphonates.
Return to citation in text: [1] -
Rodriquez, M.; Bruno, I.; Cini, E.; Marchetti, M.; Taddei, M.; Gomez-Paloma, L. J. Org. Chem. 2006, 71, 103–107. doi:10.1021/jo0518250
Dimethyl methyl phosphonate (DMMP) is also used for this reaction.
Return to citation in text: [1] -
Paterson, I.; Lyothier, I. Org. Lett. 2004, 6, 4933–4936. doi:10.1021/ol0478842
Return to citation in text: [1] -
Anjum, A.; Hoegenauer, E. K.; Enev, V. S.; Hanbauer, M.; Kaehlig, H.; Öhler, E.; Mulzer, J. J. Org. Chem. 2003, 68, 3026–3042. doi:10.1021/jo026743f
Return to citation in text: [1] -
Kangani, C. O.; Brückner, A. M.; Curran, D. P. Org. Lett. 2005, 7, 379–382. doi:10.1021/ol0478279
Return to citation in text: [1] -
Cooper, J.; Knight, D. W.; Gallagher, P. T. J. Chem. Soc., Perkin Trans. 1 1991, 705–713. doi:10.1039/P19910000705
Return to citation in text: [1] [2]
| 29. | Paterson, I.; Lyothier, I. Org. Lett. 2004, 6, 4933–4936. doi:10.1021/ol0478842 |
| 23. | Daly, M.; Cant, A. A.; Fowler, L. S.; Simpson, G. L.; Senn, H. M.; Sutherland, A. J. Org. Chem. 2012, 77, 10001–10009. doi:10.1021/jo3022583 |
| 1. | Seebach, D.; Kimmerlin, T.; Šebesta, R.; Campo, M. A.; Beck, A. K. Tetrahedron 2004, 60, 7455–7506. doi:10.1016/j.tet.2004.06.043 |
| 6. | Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. Drug Discovery Today 2007, 12, 8–27. doi:10.1016/j.drudis.2006.11.004 |
| 7. | Verkade, J. M. M.; Van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutges, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29–41. doi:10.1039/b713885g |
| 8. | Bhadury, P. S.; Song, B.-A. Curr. Org. Chem 2010, 14, 1989–2006. doi:10.2174/138527210792927564 |
| 9. | Probst, N.; Madarász, Á.; Valkonen, A.; Pápai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P. M. Angew. Chem., Int. Ed. 2012, 51, 8495–8499. doi:10.1002/anie.201203852 |
| 10. | Jiang, C.; Zhong, F.; Lu, Y. Beilstein J. Org. Chem. 2012, 8, 1279–1283. doi:10.3762/bjoc.8.144 |
| 21. | Della Monica, C.; Maulucci, N.; De Riccardis, F.; Izzo, I. Tetrahedron: Asymmetry 2003, 14, 3371–3378. doi:10.1016/S0957-4166(03)00622-0 |
| 4. | Ishimaru, K.; Kojima, T. J. J. Org. Chem. 2000, 65, 8395–8398. doi:10.1021/jo0011888 |
| 5. | Davis, F. A.; Song, M.; Augustine, A. J. Org. Chem. 2006, 71, 2779–2786. doi:10.1021/jo052566h |
| 22. | Rudisill, D. E.; Whitten, J. P. Synthesis 1994, 851–854. doi:10.1055/s-1994-25588 |
| 23. | Daly, M.; Cant, A. A.; Fowler, L. S.; Simpson, G. L.; Senn, H. M.; Sutherland, A. J. Org. Chem. 2012, 77, 10001–10009. doi:10.1021/jo3022583 |
| 24. | Boeglin, D.; Heitz, A.; Martinez, J.; Fehrentz, J.-A. Eur. J. Org. Chem. 2003, 3139–3146. doi:10.1002/ejoc.200300148 |
| 25. | Davis, F. A.; Wu, Y. Org. Lett. 2004, 6, 1269–1272. doi:10.1021/ol049795v |
| 26. | Modica, E.; Compostella, F.; Colombo, D.; Franchini, L.; Cavallari, M.; Mori, L.; De Libero, G.; Panza, L.; Ronchetti, F. Org. Lett. 2006, 8, 3255–3258. doi:10.1021/ol061100y |
| 27. |
Wiemer, D. F. Tetrahedron 1997, 53, 16609–16644. doi:10.1016/S0040-4020(97)10305-2
A review on the preparation of nonracemic phosphonates. |
| 28. |
Rodriquez, M.; Bruno, I.; Cini, E.; Marchetti, M.; Taddei, M.; Gomez-Paloma, L. J. Org. Chem. 2006, 71, 103–107. doi:10.1021/jo0518250
Dimethyl methyl phosphonate (DMMP) is also used for this reaction. |
| 3. | Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957–5978. doi:10.1016/S0040-4020(02)00584-7 |
| 18. | Sibi, M. P. Org. Prep. Proced. Int. 1993, 25, 15–40. doi:10.1080/00304949309457931 |
| 2. | Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2 |
| 19. | Wadsworth, W. S.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042 |
| 20. | Wadsworth, W. S.; Emmons, W. D. Org. Synth. 1973, Coll. Vol. 5, 547–563. |
| 15. | Abrunhosa-Thomas, I.; Roy, O.; Barra, M.; Besset, T.; Chalard, P.; Troin, Y. Synlett 2007, 1613–1615. doi:10.1055/s-2007-982547 |
| 18. | Sibi, M. P. Org. Prep. Proced. Int. 1993, 25, 15–40. doi:10.1080/00304949309457931 |
| 32. | Cooper, J.; Knight, D. W.; Gallagher, P. T. J. Chem. Soc., Perkin Trans. 1 1991, 705–713. doi:10.1039/P19910000705 |
| 13. | Davis, F. A.; Theddu, N. J. Org. Chem. 2010, 75, 3814–3820. doi:10.1021/jo100680b |
| 14. | Davis, F. A.; Xu, P. J. Org. Chem. 2011, 76, 3329–3337. doi:10.1021/jo2002352 |
| 11. | Brackenridge, I.; Davies, S. G.; Fenwick, D. R.; Ichihara, O.; Polywka, M. E. C. Tetrahedron 1999, 55, 533–540. doi:10.1016/S0040-4020(98)01051-5 |
| 12. | Fleck, T. J.; McWhorter, W. W.; DeKam, R. N.; Pearlman, B. A. J. Org. Chem. 2003, 68, 9612–9617. doi:10.1021/jo0349633 |
| 32. | Cooper, J.; Knight, D. W.; Gallagher, P. T. J. Chem. Soc., Perkin Trans. 1 1991, 705–713. doi:10.1039/P19910000705 |
| 11. | Brackenridge, I.; Davies, S. G.; Fenwick, D. R.; Ichihara, O.; Polywka, M. E. C. Tetrahedron 1999, 55, 533–540. doi:10.1016/S0040-4020(98)01051-5 |
| 12. | Fleck, T. J.; McWhorter, W. W.; DeKam, R. N.; Pearlman, B. A. J. Org. Chem. 2003, 68, 9612–9617. doi:10.1021/jo0349633 |
| 30. | Anjum, A.; Hoegenauer, E. K.; Enev, V. S.; Hanbauer, M.; Kaehlig, H.; Öhler, E.; Mulzer, J. J. Org. Chem. 2003, 68, 3026–3042. doi:10.1021/jo026743f |
| 9. | Probst, N.; Madarász, Á.; Valkonen, A.; Pápai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P. M. Angew. Chem., Int. Ed. 2012, 51, 8495–8499. doi:10.1002/anie.201203852 |
| 10. | Jiang, C.; Zhong, F.; Lu, Y. Beilstein J. Org. Chem. 2012, 8, 1279–1283. doi:10.3762/bjoc.8.144 |
| 16. | Poerwono, H.; Higashimaya, K.; Yamauchi, T.; Kubo, H.; Ohmiya, S.; Takahashi, H. Tetrahedron 1998, 54, 13955–13970. doi:10.1016/S0040-4020(98)00863-1 |
| 17. | Davis, F. A.; Xu, H.; Zhang, J. J. Org. Chem. 2007, 72, 2046–2052. doi:10.1021/jo062365t |
| 31. | Kangani, C. O.; Brückner, A. M.; Curran, D. P. Org. Lett. 2005, 7, 379–382. doi:10.1021/ol0478279 |
© 2013 Abrunhosa-Thomas et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)