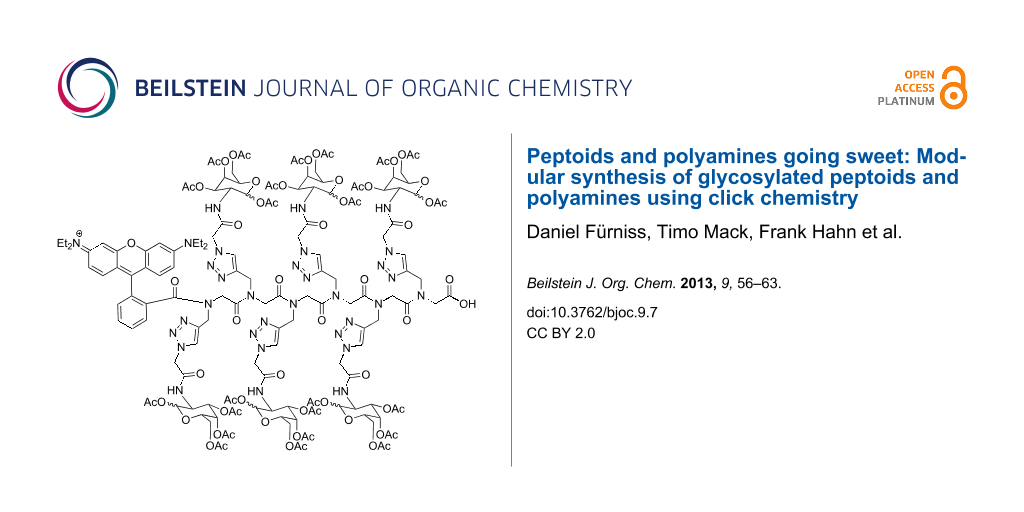
Abstract
Sugar moieties are present in a wide range of bioactive molecules. Thus, having versatile and fast methods for the decoration of biomimetic molecules with sugars is of fundamental importance. The glycosylation of peptoids and polyamines as examples of such biomimetic molecules is reported here. The method uses Cu-catalyzed azide alkyne cycloaddition to promote the reaction of azidosugars with either polyamines or peptoids. In addition, functionalized nucleic acids were attached to polyamines via the same route. Based on a modular solid-phase synthesis of peralkynylated peptoids with up to six alkyne groups, the latter were modified with azidosugar building blocks by using copper-catalyzed azide alkyne cycloadditions. In addition, the up-scaling of some particular azide-modified sugars is described.

Graphical Abstract
Introduction
To date, oligosaccharides have gained more and more interest as potential drugs in the treatment of a variety of diseases. However, the rendering of nucleic acids and oligosaccharides as therapeutically active substances often requires a derivatization or a chemical coupling reaction that permits the selective and simple formation of covalent adducts. Some modifications permit the attachment to other molecules through a variety of functional groups, such as amines (–NH2) and carboxylic acids (–COOH) resulting in peptide bonds, thiols (–SH) resulting in disulfides, thioethers or thioesters, aldehydes (–CHO) and hydroxy (–OH) groups. Nonetheless, coupling to these groups often requires a laborious protection of other reactive functional groups as they can compete in the coupling step. A matched pair of groups, which are selective in reacting with each other while being unreactive with other functional groups in the molecule would, therefore, be highly useful in the preparation of functional structures. Likewise, the coupling reaction should be permitted in hydrophilic solvents such as water or DMSO, since both unprotected nucleic acids and oligosaccharides, as well as many other biomacromolecules, prefer a hydrophilic reaction environment.
With the advent of mild and biocompatible conjugation methods such as the Staudinger ligation [1] or the copper-catalyzed alkyne azide cycloaddition (CuAAC) [2,3], a large number of versatile and functional bioconjugates are accessible for various applications in chemical biology [4].
To date, many therapeutically active molecules are synthetic derivatives of biomacromolecules that have to be soluble in hydrophilic environments to be taken up in vivo or in cell culture. Common solubilizers that enhance the cellular uptake are polyamines and other polycationic moieties such as particular peptoids.
Recently, polycationic polyamines have been shown to be efficacious in the cellular delivery of oligonucleotides such as DNA [5-7] and RNA [8-11]. Conjugates of polyamines with aliphatic lipids or cholesterol yielding, i.e., dioctadecylaminoglycylspermine (DOGS, transfectam) are well established reagents for the transfection of DNA and oligonucleotides [5-7,11-13] displaying only very little toxicity towards mammalian cells [11]. They have also been shown to function in the recognition of biomacromolecules. Likewise, other polycationic species, such as the N-alkylated glycine oligomers (peptoids) [14-25], have emerged as powerful tools in the context of drug delivery [26-29], peptidomimetics and other biologically relevant applications [28,30-32] as well as materials science [33,34].
During the last decade the synthesis of polyamines and peptoids has been well established on solid phases [26,27,29,35-39]. However, the on-bead addition of oligosaccharide or monosaccharide modifications are not known so far. The modification of polyamines or peptoids is usually achieved by alternation of the termini [36,40] or by direct use of different side-chain functionalities. For peptoids, CuAAC has already been used successfully to introduce diverse side-chain functionalities directly during solid-phase synthesis of peptoids starting from both, azido- and alkyne-functionalized side chains [41,42]. In addition CuAAC has also been used in order to constrain peptoid secondary structures [43].
CuAAC reactions for the attachment of sugar residues to peptoid backbones have been reported for some cases [44,45]; however, a fully glycosylated structure is unknown (for glycodendrons see [46]). In this study, we describe the first solid-phase synthesis of glycosylated polyamines and a fully glycosylated hexapeptoid.
Results and Discussion
Synthesis of azidosugars
Although the syntheses of the azidosugars 1–3 and 5 (Figure 1) were described before [47], we optimized and revised the procedure reported by Laughlin and Bertozzi [48] due to some difficulties in obtaining reproducible results.
![[1860-5397-9-7-1]](/bjoc/content/figures/1860-5397-9-7-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Azidosugars used in this study. The synthesis of the azidosugars 1–3 was modified from [47,48], compounds 4, 6 and 7 were commercially available.
Figure 1: Azidosugars used in this study. The synthesis of the azidosugars 1–3 was modified from [47,48], compounds 4...
Instead of 5.00 equiv of chloroacetic anhydride only 1.10 equiv were used. A change from LiN3 to NaN3, which is more stable and cheaper, in conjunction with lowering the amount of the azide salt from 5.00 equiv to less than 3.60 equiv gave similar results. The solvent was changed from DMF to MeOH. By applying revised conditions 55% yield (on a 4.64 mmol scale) was obtained with a comparable yield on a 1.16 mmol scale (61%). In contrast to the originally reported procedure, the purification and isolation of intermediates could be omitted. We tried to use fewer equivalents of sodium azide during the scale-up of the reaction but this caused a decrease of the overall reaction yields.
In all cases, the test reactions were performed starting from D-glucosamine hydrochloride as a model compound for the synthesis of 1,3,4,6-tetra-O-acetyl-N-azidoacetyl-D-glucosamine (Ac4GlcNAz = 1). Optimal variations from Bertozzi’s protocol for this model compound were the use of 1.10 equiv chloroacetic anhydride in the first step and 3.50 equiv sodium azide as well as methanol in the second step (for further details, see Table 1). Eventually, we applied these optimized reaction conditions to synthesize also 1,3,4,6-tetra-O-acetyl-N-azidoacetyl-D-galactosamine (Ac4GalNAz = 2) and 1,3,4,6-tetra-O-acetyl-N-azidoacetyl-D-mannosamine (Ac4ManNAz = 3) with similar yields under these optimized conditions (see Supporting Information File 1).
Table 1: Optimization of the synthesis of azide 1.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-7-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Reaction scale | Solvent (2nd step) | MN3 | Overall reaction yield (over 3 steps) |
|---|---|---|---|---|
| 1 | 1.16 mmol | DMF | 5.00 equiv (M = Li) | 61% |
| 2 | 4.64 mmol | DMF | 5.00 equiv (M = Na) | 23% |
| 3 | 4.64 mmol | MeOH | 2.10 equiv (M = Na) | 35% |
| 4 | 4.64 mmol | MeOH | 3.50 equiv (M = Na) | 55% |
| 5 | 13.4 mmol | MeOH | 2.10 equiv (M = Na) | 38% |
| 6 | 23.2 mmol | MeOH | 2.00 equiv (M = Na) | 42% |
Click reaction on alkynylated polyamines and peptoids
The covalent coupling of many biomacromolecules to solid-phase-bound polycationic moieties, such as polyamines or peptoids, often requires a hydrophilic reaction environment as well as very mild cleaving conditions of the final product from the solid support. To avoid a degradation of the coupled biomacromolecules at high concentrations of strong acids, a polystyrene resin was chosen that contains a tritylchloride linker. The resin was obtained by treatment of Merrifield resin with p-hydroxytriphenylmethyl alcohol and subsequent chlorination [49,50]. This tritylchloride linker allowed a mild cleavage of the acid labile products using less than 0.5% trifluoroacetic acid (TFA) in dichloromethane. The loading of the Merrifield resin occurred in pretty good yields as the ratio of the measured to the calculated loading value was 0.70 mmol/g to 0.78 mmol/g (Scheme 1).
![[1860-5397-9-7-i1]](/bjoc/content/inline/1860-5397-9-7-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction conditions and reagents: (a) Ns-chloride (6.00 equiv), 2,4,6-collidine (12.0 equiv), CH2Cl2, rt, 16 h; (b) 5-chloropent-1-yne (10.0 equiv), K2CO3 (15.0 equiv), DMF, 60 °C, 16 h; (c) azidosugars 6, 7 or AZT (4) (2.00 equiv/1.86 equiv), CuSO4·5H2O (0.500 equiv), sodium ascorbate (5.00 equiv), DMF/H2O (6:1), rt, 2 d; (d) DBU (20.0 equiv), β-mercaptoethanol (20.0 equiv), DMF, rt, 18 h; (e) 1% TFA in CH2Cl2, rt, 10 min.
Scheme 1: Reaction conditions and reagents: (a) Ns-chloride (6.00 equiv), 2,4,6-collidine (12.0 equiv), CH2Cl2...
The synthesis started with the assembly of the 2-nitrobenzenesulfonyl-(Nosyl, further abbreviated with Ns)-protected spermine backbone 8 on a solid phase via Fukuyama Ns strategy [51]. The next step was the Ns protection of the residual primary amine with 6.00 equiv of Ns-chloride and 12.0 equiv 2,4,6-collidine in CH2Cl2 followed by N-alkylation with 5-chloropent-1-yne to insert the terminal alkyne moiety. To accomplish that, we used 10.0 equiv of alkyne and 15.0 equiv of K2CO3 in DMF; the reaction led to resin 9 with virtually quantitative yield, as shown in Scheme 1. For the CuAAC with the azides moieties 4, 6 and 7, respectively, we used 0.500 equiv of CuSO4·5H2O and 5.00 equiv sodium ascorbate as the catalytic system, which is a slightly higher catalyst concentration than reported for the reaction in solution [52]. To ensure, that the reaction proceeds completely we chose 2 days of agitation at ambient temperature and obtained resin 10−12.
Finally, the Ns deprotection was achieved in 18 h with 20.0 equiv of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 20.0 equiv of β-mercaptoethanol. The cleavage from the resin was carried out with 1% TFA in dichloromethane at 10 min residence time. The products 13–15 were obtained with >90% yield, calculated on the initial loading of the resin (0.70 mmol/g). The 1H NMR data clearly showed the aromatic shift of the triazole proton at 8.02 ppm, 8.03 ppm and 7.94 ppm for 13–15, respectively. The 13C NMR spectra indicated the presence of the anomeric carbon atoms at 89.70 ppm for 13 as well as at 89.41 ppm and 105.47 ppm for 14.
In a different approach, we synthesized glycosylated spermine derivatives by our optimized procedure on a 2-chlorotrityl chloride resin [35,37-39]. The reaction of the resin 16 and 17 with the Ac4GlcNAz derivative 1 proceeded smoothly in the presence of copper ions. In some cases, changing the base from DIPEA to DBU was beneficial (Scheme 2).
![[1860-5397-9-7-i2]](/bjoc/content/inline/1860-5397-9-7-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of spermine conjugates 20,21 and 24,25. 2-chlorotrityl chloride resin was used as a solid support.
Scheme 2: Synthesis of spermine conjugates 20,21 and 24,25. 2-chlorotrityl chloride resin was used as a solid...
After these encouraging results, we turned our attention to peptoids. For the glycosylation of peptoids, we envisaged the generation of a fully glycosylated peptoid in order to investigate the compatibility of peptoid synthesis and decoration with sugars.
As a model compound we started with the synthesis of a hexaglycosylated peptoid hexamer. Therefore, we synthesized a hexaalkynated peptoid structure 26,27 on resin (Scheme 3).
![[1860-5397-9-7-i3]](/bjoc/content/inline/1860-5397-9-7-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of hexaalkynyl peptoids 26 and 27 on solid supports.
Scheme 3: Synthesis of hexaalkynyl peptoids 26 and 27 on solid supports.
The synthesis of a hexaalkynated peptoid backbone 26,27 was carried out on Rink or Barlos resin containing a 2-chlorotrityl chloride linker by standard methods using the submonomer strategy [14]. By using this method, the peptoid backbone is assembled in two subsequently repeated steps: In the first step (acylation), bromoacetic acid is reacted with the resin, and in the second step (amination) a primary amine is used to substitute the bromine to give the peptoid residue. This approach avoids the use of N-terminally protected monomers, which have to be synthesized in advance.
For the incorporation of the alkyne side chains we chose propargylamine as building block. A sixfold repetitive coupling sequence resulted in the peptoids, which were further modified with rhodamine B (Rho-CO2H) as an easily accessible and versatile fluorescent tag.
Rhodamine B was coupled to the N-terminus in order to provide a label for future biological applications, such as the study of the cellular uptake. For both resins, the peptoid synthesis was successful; the only differences are the functional groups on the C-terminus. After cleavage from Rink-amide resin with trifluoroacetic acid, an amide is obtained, whereas cleavage from 2-chlorotrityl chloride resin with hexafluoroisopropanol (HFIP) gives the carboxylic acid. The mild cleavage conditions of the 2-chlorotrityl linker did not harm the sugar moieties of the final glycosylated compound, and this linker was therefore favored over the Rink linker.
For the conjugation of Ac4GalNAz (2), the same conditions were used as described for the spermine conjugation (Scheme 2) with minor modifications. The CuAAC was carried out by using Cu(CH3CN)4PF6 in THF with 2,6-lutidine as base. Only 1.60 equiv of the azidosugar were necessary to achieve full conversion after 18 h, and no shorter oligomers were observed in the MALDI–TOF spectrum. Cleavage from the resin by treatment with 33% hexafluoroisopropanol (HFIP) in dichloromethane and subsequent HPLC purification resulted in the fully glycosylated hexameric product 28 (Scheme 4).
![[1860-5397-9-7-i4]](/bjoc/content/inline/1860-5397-9-7-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of a hexa-glycosylated peptoid 28.
Scheme 4: Synthesis of a hexa-glycosylated peptoid 28.
Conclusion
In conclusion, we were able to improve protocols for the synthesis of tetra-O-acetyl protected sugars. Applying this, better yields as well as upscaling was possible. With these sugar building blocks, functionalization of polyamine derivatives was possible directly on solid supports by using copper-catalyzed alkyne azide cycloaddition conditions. In addition to that, the functionalization of a peptoid-hexaalkyne was also possible. By using the more labile 2-chlorotrityl chloride resin, the cleavage of peptoid 28 could be achieved without degradation.
Supporting Information
| Supporting Information File 1: Methods and NMR spectra. | ||
| Format: PDF | Size: 3.8 MB | Download |
Acknowledgements
We acknowledge funding from the Landesgraduiertenprogramm Baden-Württemberg (fellowship to S.B.L.V), the Carl-Zeiss foundation (fellowship to D.F.), Theodor Laymann Foundation (fellowships to T.M. and F.H.), the Fonds der Chemischen Industrie, the SFB 645 (Project Z1), the Helmholtz Program “Biointerface” and the Center for Functionalized Nanostructures (CFN), Karlsruhe.
References
-
Schilling, C. I.; Jung, N.; Biskup, M.; Schepers, U.; Bräse, S. Chem. Soc. Rev. 2011, 40, 4840–4871. doi:10.1039/c0cs00123f
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Holub, J. M.; Kirshenbaum, K. Chem. Soc. Rev. 2010, 39, 1325–1337. doi:10.1039/b901977b
Return to citation in text: [1] -
Mack, K. D.; Walzem, R.; Zeldis, J. B. Am. J. Med. Sci. 1994, 307, 138–143. doi:10.1097/00000441-199402000-00013
Return to citation in text: [1] [2] -
Boletta, A.; Benigni, A.; Lutz, J.; Remuzzi, G.; Soria, M. R.; Monaco, L. Hum. Gene Ther. 1997, 8, 1243–1251. doi:10.1089/hum.1997.8.10-1243
Return to citation in text: [1] [2] -
Kichler, A.; Mechtler, K.; Behr, J.-P.; Wagner, E. Bioconjugate Chem. 1997, 8, 213–221. doi:10.1021/bc970009z
Return to citation in text: [1] [2] -
Lu, D.; Benjamin, R.; Kim, M.; Conry, R. M.; Curiel, D. T. Cancer Gene Ther. 1994, 1, 245–252.
Return to citation in text: [1] -
Aoki, Y.; Kawa, S.; Karasawa, Y.; Horiuchi, A.; Kiyosawa, K. Clin. Exp. Pharmacol. Physiol. 1998, 25, 702–705. doi:10.1111/j.1440-1681.1998.tb02279.x
Return to citation in text: [1] -
Wong, F. M. P.; MacAdam, S. A.; Kim, A.; Oja, C.; Ramsay, E. C.; Bally, M. B. J. Drug Targeting 2002, 10, 615–623. doi:10.1080/1061186021000066246
Return to citation in text: [1] -
Guy-Caffey, J. K.; Bodepudi, V.; Bishop, J. S.; Jayaraman, K.; Chaudhary, N. J. Biol. Chem. 1995, 270, 31391–31396. doi:10.1074/jbc.270.52.31391
Return to citation in text: [1] [2] [3] -
Lee, E. R.; Marshall, J.; Siegel, C. S.; Jiang, C.; Yew, N. S.; Nichols, M. R.; Nietupski, J. B.; Ziegler, R. J.; Lane, M. B.; Wang, K. X.; Wan, N. C.; Scheule, R. K.; Harris, D. J.; Smith, A. E.; Cheng, S. H. Hum. Gene Ther. 1996, 7, 1701–1717. doi:10.1089/hum.1996.7.14-1701
Return to citation in text: [1] -
Lee, J.-H.; Nam, S.-H.; Hwang, S.-B.; Hong, M.-G.; Kwon, J.-Y.; Joeng, K.-S.; Im, S.-H.; Shim, J.-W.; Park, M.-C. J. Biochem. Mol. Biol. 2004, 37, 107–113. doi:10.5483/BMBRep.2004.37.1.107
Return to citation in text: [1] -
Zuckermann, R. N.; Kerr, J. M.; Kent, S. B. H.; Moos, W. H. J. Am. Chem. Soc. 1992, 114, 10646–10647. doi:10.1021/ja00052a076
Return to citation in text: [1] [2] -
Wu, C. W.; Seurynck, S. L.; Lee, K. Y. C.; Barron, A. E. Chem. Biol. 2003, 10, 1057–1063. doi:10.1016/j.chembiol.2003.10.008
Return to citation in text: [1] -
Wender, P. A.; Mitchell, D. J.; Pattabiraman, K.; Pelkey, E. T.; Steinman, L.; Rothbard, J. B. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13003–13008. doi:10.1073/pnas.97.24.13003
Return to citation in text: [1] -
Sanborn, T. J.; Wu, C. W.; Zuckerman, R. N.; Barron, A. E. Biopolymers 2002, 63, 12–20. doi:10.1002/bip.1058
Return to citation in text: [1] -
Burkoth, T. S.; Fafarman, A. T.; Charych, D. H.; Connolly, M. D.; Zuckermann, R. N. J. Am. Chem. Soc. 2003, 125, 8841–8845. doi:10.1021/ja0352101
Return to citation in text: [1] -
Fafarman, A. T.; Borbat, P. P.; Freed, J. H.; Kirshenbaum, K. Chem. Commun. 2007, 377–379. doi:10.1039/b612198e
Return to citation in text: [1] -
Gorske, B. C.; Jewell, S. A.; Guerard, E. J.; Blackwell, H. E. Org. Lett. 2005, 7, 1521–1524. doi:10.1021/ol0502984
Return to citation in text: [1] -
Kruijtzer, J. A. W.; Hofmeyer, L. J. F.; Heerma, W.; Versluis, C.; Liskamp, R. M. J. Chem.–Eur. J. 1998, 4, 1570–1580. doi:10.1002/(SICI)1521-3765(19980807)4:8<1570::AID-CHEM1570>3.0.CO;2-2
Return to citation in text: [1] -
Li, S.; Bowerman, D.; Marthandan, N.; Klyza, S.; Luebke, K. J.; Garner, H. R.; Kodadek, T. J. Am. Chem. Soc. 2004, 126, 4088–4089. doi:10.1021/ja039565w
Return to citation in text: [1] -
Olivos, H. J.; Alluri, P. G.; Reddy, M. M.; Salony, D.; Kodadek, T. Org. Lett. 2002, 4, 4057–4059. doi:10.1021/ol0267578
Return to citation in text: [1] -
Saha, U. K.; Roy, R. J. Chem. Soc., Chem. Commun. 1995, 2571–2573. doi:10.1039/C39950002571
Return to citation in text: [1] -
Saha, U. K.; Roy, R. Tetrahedron Lett. 1995, 36, 3635–3638. doi:10.1016/0040-4039(95)00620-R
Return to citation in text: [1] -
Schröder, T.; Schmitz, K.; Niemeier, N.; Balaban, T. S.; Krug, H. F.; Schepers, U.; Bräse, S. Bioconjugate Chem. 2007, 18, 342–354. doi:10.1021/bc0602073
Return to citation in text: [1] [2] -
Schröder, T.; Niemeier, N.; Afonin, S.; Ulrich, A. S.; Krug, H. F.; Bräse, S. J. Med. Chem. 2008, 51, 376–379. doi:10.1021/jm070603m
Return to citation in text: [1] [2] -
Seo, J.; Ren, G.; Liu, H.; Miao, Z.; Park, M.; Wang, Y.; Miller, T. M.; Barron, A. E.; Cheng, Z. Bioconjugate Chem. 2012, 23, 1069–1079. doi:10.1021/bc300091d
Return to citation in text: [1] [2] -
Eggenberger, K.; Birtalan, E.; Schröder, T.; Bräse, S.; Nick, P. ChemBioChem 2009, 10, 2504–2512. doi:10.1002/cbic.200900331
Return to citation in text: [1] [2] -
Aditya, A.; Kodadek, T. ACS Comb. Sci. 2012, 14, 164–169. doi:10.1021/co200195t
Return to citation in text: [1] -
Chen, X.; Wu, J.; Luo, Y.; Liang, X.; Supnet, C.; Kim, M. W.; Lotz, G. P.; Yang, G.; Muchowski, P. J.; Kodadek, T.; Bezprozvanny, I. Chem. Biol. 2011, 18, 1113–1125. doi:10.1016/j.chembiol.2011.06.010
Return to citation in text: [1] -
Levine, P. M.; Imberg, K.; Garabedian, M. J.; Kirshenbaum, K. J. Am. Chem. Soc. 2012, 134, 6912–6915. doi:10.1021/ja300170n
Return to citation in text: [1] -
Uejio, J. S.; Schwartz, C. P.; Duffin, A. M.; England, A.; Prendergast, D.; Saykally, R. J. J. Phys. Chem. B 2010, 114, 4702–4709. doi:10.1021/jp911007k
Return to citation in text: [1] -
Brown, N. J.; Johansson, J.; Barron, A. E. Acc. Chem. Res. 2008, 41, 1409–1417. doi:10.1021/ar800058t
Return to citation in text: [1] -
Hahn, F.; Müllen, K.; Schepers, U. Synlett 2008, 2785–2790. doi:10.1055/s-0028-1083566
Return to citation in text: [1] [2] -
Hahn, F.; Schepers, U. Solid Phase Chemistry for the Directed Synthesis of Biologically Active Polyamine Analogs, Derivatives, and Conjugates. In Combinatorial Chemistry on Solid Supports; Bräse, S., Ed.; Topics in Current Chemistry, Vol. 278; Springer: Berlin, 2007; pp 135–208. doi:10.1007/128_2007_135
Return to citation in text: [1] [2] -
Hahn, F.; Schepers, U. J. Comb. Chem. 2008, 10, 267–273. doi:10.1021/cc700119g
Return to citation in text: [1] [2] -
Hahn, F.; Schepers, U. Synlett 2009, 2755–2760. doi:10.1055/s-0029-1217987
Return to citation in text: [1] [2] -
Hahn, F.; Schmitz, K.; Balaban, T. S.; Bräse, S.; Schepers, U. ChemMedChem 2008, 3, 1185–1188. doi:10.1002/cmdc.200800013
Return to citation in text: [1] [2] -
Brown, N. J.; Dohm, M. T.; de la Serna, J. B.; Barron, A. E. Biophys. J. 2011, 101, 1076–1085. doi:10.1016/j.bpj.2011.04.055
Return to citation in text: [1] -
Fritz, D.; Bräse, S. Synlett 2010, 1544–1548. doi:10.1055/s-0029-1219925
Return to citation in text: [1] -
Roy, O.; Faure, S.; Thery, V.; Didierjean, C.; Taillefumier, C. Org. Lett. 2008, 10, 921–924. doi:10.1021/ol7030763
Return to citation in text: [1] -
Holub, J. M.; Jang, H.; Kirshenbaum, K. Org. Lett. 2007, 9, 3275–3278. doi:10.1021/ol071169l
Return to citation in text: [1] -
Norgren, A. S.; Budke, C.; Majer, Z.; Heggemann, C.; Koop, T.; Sewald, N. Synthesis 2009, 488–494. doi:10.1055/s-0028-1083302
Return to citation in text: [1] -
Disney, M. D.; Lee, M. M.; Pushechnikov, A.; Childs-Disney, J. L. ChemBioChem 2010, 11, 375–382. doi:10.1002/cbic.200900716
Return to citation in text: [1] -
Papadopoulos, A.; Shiao, T. C.; Roy, R. Mol. Pharmaceutics 2012, 9, 394–403. doi:10.1021/mp200490b
Return to citation in text: [1] -
Saxon, E.; Bertozzi, C. R. Science 2000, 287, 2007–2010. doi:10.1126/science.287.5460.2007
Return to citation in text: [1] [2] -
Laughlin, S. T.; Bertozzi, C. R. Nat. Protoc. 2007, 2, 2930–2944. doi:10.1038/nprot.2007.422
Return to citation in text: [1] [2] -
Hidai, Y.; Kan, T.; Fukuyama, T. Tetrahedron Lett. 1999, 40, 4711–4714. doi:10.1016/S0040-4039(99)00851-5
Return to citation in text: [1] -
Hidai, Y.; Kan, T.; Fukuyama, T. Chem. Pharm. Bull. 2000, 48, 1570–1576. doi:10.1248/cpb.48.1570
Return to citation in text: [1] -
Kan, T.; Fukuyama, T. Chem. Commun. 2004, 4, 353–359. doi:10.1039/b311203a
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.3.CO;2-X
Return to citation in text: [1]
| 1. | Schilling, C. I.; Jung, N.; Biskup, M.; Schepers, U.; Bräse, S. Chem. Soc. Rev. 2011, 40, 4840–4871. doi:10.1039/c0cs00123f |
| 8. | Lu, D.; Benjamin, R.; Kim, M.; Conry, R. M.; Curiel, D. T. Cancer Gene Ther. 1994, 1, 245–252. |
| 9. | Aoki, Y.; Kawa, S.; Karasawa, Y.; Horiuchi, A.; Kiyosawa, K. Clin. Exp. Pharmacol. Physiol. 1998, 25, 702–705. doi:10.1111/j.1440-1681.1998.tb02279.x |
| 10. | Wong, F. M. P.; MacAdam, S. A.; Kim, A.; Oja, C.; Ramsay, E. C.; Bally, M. B. J. Drug Targeting 2002, 10, 615–623. doi:10.1080/1061186021000066246 |
| 11. | Guy-Caffey, J. K.; Bodepudi, V.; Bishop, J. S.; Jayaraman, K.; Chaudhary, N. J. Biol. Chem. 1995, 270, 31391–31396. doi:10.1074/jbc.270.52.31391 |
| 43. | Holub, J. M.; Jang, H.; Kirshenbaum, K. Org. Lett. 2007, 9, 3275–3278. doi:10.1021/ol071169l |
| 5. | Mack, K. D.; Walzem, R.; Zeldis, J. B. Am. J. Med. Sci. 1994, 307, 138–143. doi:10.1097/00000441-199402000-00013 |
| 6. | Boletta, A.; Benigni, A.; Lutz, J.; Remuzzi, G.; Soria, M. R.; Monaco, L. Hum. Gene Ther. 1997, 8, 1243–1251. doi:10.1089/hum.1997.8.10-1243 |
| 7. | Kichler, A.; Mechtler, K.; Behr, J.-P.; Wagner, E. Bioconjugate Chem. 1997, 8, 213–221. doi:10.1021/bc970009z |
| 44. | Norgren, A. S.; Budke, C.; Majer, Z.; Heggemann, C.; Koop, T.; Sewald, N. Synthesis 2009, 488–494. doi:10.1055/s-0028-1083302 |
| 45. | Disney, M. D.; Lee, M. M.; Pushechnikov, A.; Childs-Disney, J. L. ChemBioChem 2010, 11, 375–382. doi:10.1002/cbic.200900716 |
| 4. | Holub, J. M.; Kirshenbaum, K. Chem. Soc. Rev. 2010, 39, 1325–1337. doi:10.1039/b901977b |
| 36. | Hahn, F.; Schepers, U. Solid Phase Chemistry for the Directed Synthesis of Biologically Active Polyamine Analogs, Derivatives, and Conjugates. In Combinatorial Chemistry on Solid Supports; Bräse, S., Ed.; Topics in Current Chemistry, Vol. 278; Springer: Berlin, 2007; pp 135–208. doi:10.1007/128_2007_135 |
| 40. | Brown, N. J.; Dohm, M. T.; de la Serna, J. B.; Barron, A. E. Biophys. J. 2011, 101, 1076–1085. doi:10.1016/j.bpj.2011.04.055 |
| 2. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 3. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 41. | Fritz, D.; Bräse, S. Synlett 2010, 1544–1548. doi:10.1055/s-0029-1219925 |
| 42. | Roy, O.; Faure, S.; Thery, V.; Didierjean, C.; Taillefumier, C. Org. Lett. 2008, 10, 921–924. doi:10.1021/ol7030763 |
| 26. | Schröder, T.; Schmitz, K.; Niemeier, N.; Balaban, T. S.; Krug, H. F.; Schepers, U.; Bräse, S. Bioconjugate Chem. 2007, 18, 342–354. doi:10.1021/bc0602073 |
| 27. | Schröder, T.; Niemeier, N.; Afonin, S.; Ulrich, A. S.; Krug, H. F.; Bräse, S. J. Med. Chem. 2008, 51, 376–379. doi:10.1021/jm070603m |
| 28. | Seo, J.; Ren, G.; Liu, H.; Miao, Z.; Park, M.; Wang, Y.; Miller, T. M.; Barron, A. E.; Cheng, Z. Bioconjugate Chem. 2012, 23, 1069–1079. doi:10.1021/bc300091d |
| 29. | Eggenberger, K.; Birtalan, E.; Schröder, T.; Bräse, S.; Nick, P. ChemBioChem 2009, 10, 2504–2512. doi:10.1002/cbic.200900331 |
| 33. | Uejio, J. S.; Schwartz, C. P.; Duffin, A. M.; England, A.; Prendergast, D.; Saykally, R. J. J. Phys. Chem. B 2010, 114, 4702–4709. doi:10.1021/jp911007k |
| 34. | Brown, N. J.; Johansson, J.; Barron, A. E. Acc. Chem. Res. 2008, 41, 1409–1417. doi:10.1021/ar800058t |
| 14. | Zuckermann, R. N.; Kerr, J. M.; Kent, S. B. H.; Moos, W. H. J. Am. Chem. Soc. 1992, 114, 10646–10647. doi:10.1021/ja00052a076 |
| 15. | Wu, C. W.; Seurynck, S. L.; Lee, K. Y. C.; Barron, A. E. Chem. Biol. 2003, 10, 1057–1063. doi:10.1016/j.chembiol.2003.10.008 |
| 16. | Wender, P. A.; Mitchell, D. J.; Pattabiraman, K.; Pelkey, E. T.; Steinman, L.; Rothbard, J. B. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13003–13008. doi:10.1073/pnas.97.24.13003 |
| 17. | Sanborn, T. J.; Wu, C. W.; Zuckerman, R. N.; Barron, A. E. Biopolymers 2002, 63, 12–20. doi:10.1002/bip.1058 |
| 18. | Burkoth, T. S.; Fafarman, A. T.; Charych, D. H.; Connolly, M. D.; Zuckermann, R. N. J. Am. Chem. Soc. 2003, 125, 8841–8845. doi:10.1021/ja0352101 |
| 19. | Fafarman, A. T.; Borbat, P. P.; Freed, J. H.; Kirshenbaum, K. Chem. Commun. 2007, 377–379. doi:10.1039/b612198e |
| 20. | Gorske, B. C.; Jewell, S. A.; Guerard, E. J.; Blackwell, H. E. Org. Lett. 2005, 7, 1521–1524. doi:10.1021/ol0502984 |
| 21. | Kruijtzer, J. A. W.; Hofmeyer, L. J. F.; Heerma, W.; Versluis, C.; Liskamp, R. M. J. Chem.–Eur. J. 1998, 4, 1570–1580. doi:10.1002/(SICI)1521-3765(19980807)4:8<1570::AID-CHEM1570>3.0.CO;2-2 |
| 22. | Li, S.; Bowerman, D.; Marthandan, N.; Klyza, S.; Luebke, K. J.; Garner, H. R.; Kodadek, T. J. Am. Chem. Soc. 2004, 126, 4088–4089. doi:10.1021/ja039565w |
| 23. | Olivos, H. J.; Alluri, P. G.; Reddy, M. M.; Salony, D.; Kodadek, T. Org. Lett. 2002, 4, 4057–4059. doi:10.1021/ol0267578 |
| 24. | Saha, U. K.; Roy, R. J. Chem. Soc., Chem. Commun. 1995, 2571–2573. doi:10.1039/C39950002571 |
| 25. | Saha, U. K.; Roy, R. Tetrahedron Lett. 1995, 36, 3635–3638. doi:10.1016/0040-4039(95)00620-R |
| 26. | Schröder, T.; Schmitz, K.; Niemeier, N.; Balaban, T. S.; Krug, H. F.; Schepers, U.; Bräse, S. Bioconjugate Chem. 2007, 18, 342–354. doi:10.1021/bc0602073 |
| 27. | Schröder, T.; Niemeier, N.; Afonin, S.; Ulrich, A. S.; Krug, H. F.; Bräse, S. J. Med. Chem. 2008, 51, 376–379. doi:10.1021/jm070603m |
| 29. | Eggenberger, K.; Birtalan, E.; Schröder, T.; Bräse, S.; Nick, P. ChemBioChem 2009, 10, 2504–2512. doi:10.1002/cbic.200900331 |
| 35. | Hahn, F.; Müllen, K.; Schepers, U. Synlett 2008, 2785–2790. doi:10.1055/s-0028-1083566 |
| 36. | Hahn, F.; Schepers, U. Solid Phase Chemistry for the Directed Synthesis of Biologically Active Polyamine Analogs, Derivatives, and Conjugates. In Combinatorial Chemistry on Solid Supports; Bräse, S., Ed.; Topics in Current Chemistry, Vol. 278; Springer: Berlin, 2007; pp 135–208. doi:10.1007/128_2007_135 |
| 37. | Hahn, F.; Schepers, U. J. Comb. Chem. 2008, 10, 267–273. doi:10.1021/cc700119g |
| 38. | Hahn, F.; Schepers, U. Synlett 2009, 2755–2760. doi:10.1055/s-0029-1217987 |
| 39. | Hahn, F.; Schmitz, K.; Balaban, T. S.; Bräse, S.; Schepers, U. ChemMedChem 2008, 3, 1185–1188. doi:10.1002/cmdc.200800013 |
| 11. | Guy-Caffey, J. K.; Bodepudi, V.; Bishop, J. S.; Jayaraman, K.; Chaudhary, N. J. Biol. Chem. 1995, 270, 31391–31396. doi:10.1074/jbc.270.52.31391 |
| 5. | Mack, K. D.; Walzem, R.; Zeldis, J. B. Am. J. Med. Sci. 1994, 307, 138–143. doi:10.1097/00000441-199402000-00013 |
| 6. | Boletta, A.; Benigni, A.; Lutz, J.; Remuzzi, G.; Soria, M. R.; Monaco, L. Hum. Gene Ther. 1997, 8, 1243–1251. doi:10.1089/hum.1997.8.10-1243 |
| 7. | Kichler, A.; Mechtler, K.; Behr, J.-P.; Wagner, E. Bioconjugate Chem. 1997, 8, 213–221. doi:10.1021/bc970009z |
| 11. | Guy-Caffey, J. K.; Bodepudi, V.; Bishop, J. S.; Jayaraman, K.; Chaudhary, N. J. Biol. Chem. 1995, 270, 31391–31396. doi:10.1074/jbc.270.52.31391 |
| 12. | Lee, E. R.; Marshall, J.; Siegel, C. S.; Jiang, C.; Yew, N. S.; Nichols, M. R.; Nietupski, J. B.; Ziegler, R. J.; Lane, M. B.; Wang, K. X.; Wan, N. C.; Scheule, R. K.; Harris, D. J.; Smith, A. E.; Cheng, S. H. Hum. Gene Ther. 1996, 7, 1701–1717. doi:10.1089/hum.1996.7.14-1701 |
| 13. | Lee, J.-H.; Nam, S.-H.; Hwang, S.-B.; Hong, M.-G.; Kwon, J.-Y.; Joeng, K.-S.; Im, S.-H.; Shim, J.-W.; Park, M.-C. J. Biochem. Mol. Biol. 2004, 37, 107–113. doi:10.5483/BMBRep.2004.37.1.107 |
| 28. | Seo, J.; Ren, G.; Liu, H.; Miao, Z.; Park, M.; Wang, Y.; Miller, T. M.; Barron, A. E.; Cheng, Z. Bioconjugate Chem. 2012, 23, 1069–1079. doi:10.1021/bc300091d |
| 30. | Aditya, A.; Kodadek, T. ACS Comb. Sci. 2012, 14, 164–169. doi:10.1021/co200195t |
| 31. | Chen, X.; Wu, J.; Luo, Y.; Liang, X.; Supnet, C.; Kim, M. W.; Lotz, G. P.; Yang, G.; Muchowski, P. J.; Kodadek, T.; Bezprozvanny, I. Chem. Biol. 2011, 18, 1113–1125. doi:10.1016/j.chembiol.2011.06.010 |
| 32. | Levine, P. M.; Imberg, K.; Garabedian, M. J.; Kirshenbaum, K. J. Am. Chem. Soc. 2012, 134, 6912–6915. doi:10.1021/ja300170n |
| 48. | Laughlin, S. T.; Bertozzi, C. R. Nat. Protoc. 2007, 2, 2930–2944. doi:10.1038/nprot.2007.422 |
| 46. | Papadopoulos, A.; Shiao, T. C.; Roy, R. Mol. Pharmaceutics 2012, 9, 394–403. doi:10.1021/mp200490b |
| 47. | Saxon, E.; Bertozzi, C. R. Science 2000, 287, 2007–2010. doi:10.1126/science.287.5460.2007 |
| 35. | Hahn, F.; Müllen, K.; Schepers, U. Synlett 2008, 2785–2790. doi:10.1055/s-0028-1083566 |
| 37. | Hahn, F.; Schepers, U. J. Comb. Chem. 2008, 10, 267–273. doi:10.1021/cc700119g |
| 38. | Hahn, F.; Schepers, U. Synlett 2009, 2755–2760. doi:10.1055/s-0029-1217987 |
| 39. | Hahn, F.; Schmitz, K.; Balaban, T. S.; Bräse, S.; Schepers, U. ChemMedChem 2008, 3, 1185–1188. doi:10.1002/cmdc.200800013 |
| 14. | Zuckermann, R. N.; Kerr, J. M.; Kent, S. B. H.; Moos, W. H. J. Am. Chem. Soc. 1992, 114, 10646–10647. doi:10.1021/ja00052a076 |
| 52. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.3.CO;2-X |
| 47. | Saxon, E.; Bertozzi, C. R. Science 2000, 287, 2007–2010. doi:10.1126/science.287.5460.2007 |
| 48. | Laughlin, S. T.; Bertozzi, C. R. Nat. Protoc. 2007, 2, 2930–2944. doi:10.1038/nprot.2007.422 |
| 49. | Hidai, Y.; Kan, T.; Fukuyama, T. Tetrahedron Lett. 1999, 40, 4711–4714. doi:10.1016/S0040-4039(99)00851-5 |
| 50. | Hidai, Y.; Kan, T.; Fukuyama, T. Chem. Pharm. Bull. 2000, 48, 1570–1576. doi:10.1248/cpb.48.1570 |
© 2013 Fürniss et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)