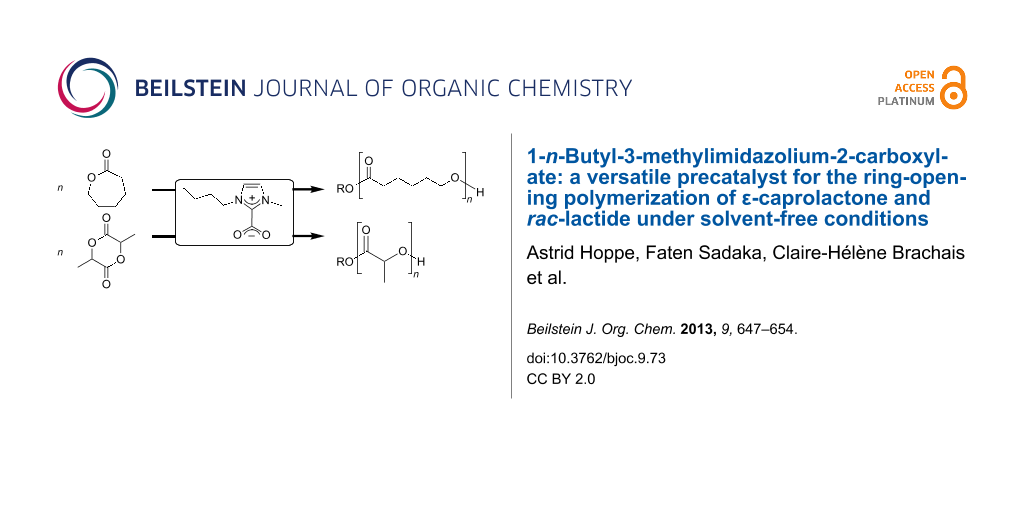
Abstract
The ring-opening polymerization of ε-caprolactone (ε-CL) and rac-lactide (rac-LA) under solvent-free conditions and using 1-n-butyl-3-methylimidazolium-2-carboxylate (BMIM-2-CO2) as precatalyst is described. Linear and star-branched polyesters were synthesized by successive use of benzyl alcohol, ethylene glycol, glycerol and pentaerythritol as initiator alcohols, and the products were fully characterized by 1H and 13C{1H} NMR spectroscopy, gel permeation chromatography (GPC), and differential scanning calorimetry (DSC). BMIM-2-CO2 acts as an N-heterocyclic carbene precursor, resulting from in situ decarboxylation, either by heating under vacuo (method A) or by addition of NaBPh4 (method B). Possible catalytic and deactivation mechanisms are proposed.

Graphical Abstract
Introduction
Poly(ε-caprolactone) (PCL) and polylactic acid (PLA) are biologically relevant aliphatic polyesters. Their applications vary, due to their compatibility with other polymers and their biodegradability, from packaging to pharmaceutics and medicine [1-6]. PCL serves for instance as a scaffold for tissue engineering [7,8] and as a carrier of stem cells [9], PLA as an implant material for stents and screws [10]. A general synthetic route for polyesters with controlled molecular weight and distribution requires the use of metal alkoxides and catalysts of transition, rare-earth and alkali metals, as initiators for the ring-opening polymerization (ROP) of cyclic esters. During the past decade, numerous articles and reviews have been published, which shows a growing interest for this research area [11-18]. However, in order to avoid the use of metal-based catalysts, which are unwanted in medical applications, great efforts have been made recently to develop metal-free organo-catalysts [19-27]. In this context, ROP promoted by N-heterocyclic carbenes (NHCs) is an alternative route. Indeed, previous studies have shown that NHCs provide versatile catalyst activities with high efficiencies and molecular-weight control [20,27-32]. Their selectivity and conversion rate are influenced by the nature of the monomer and the carbene used. Sterically encumbered carbenes, such as 1,3-dimesitylimidazol-2-ylidene (IMes, Figure 1), are known to be highly active for the ROP of rac-lactide [20,27,31,33], while less encumbered and electronically rich carbenes such as 1,3,4,5-tetramethylimidazol-2-ylidene (ImMe4, Figure 1) show a higher activity towards the ROP of ε-caprolactone [28,34]. Thus, the design of a versatile catalyst for both reactions still remains a challenge.
![[1860-5397-9-73-1]](/bjoc/content/figures/1860-5397-9-73-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular representations of N-heterocyclic carbenes and BMIM-2-CO2.
Figure 1: Molecular representations of N-heterocyclic carbenes and BMIM-2-CO2.
In this quest and encouraged by our previous results, we chose to explore the potential of zwitterionic 1-n-butyl-3-methylimidazolium-2-carboxylate (BMIM-2-CO2, Figure 1). Indeed, methylimidazolium-2-carboxylates can be easily synthesized with high yields by the one-pot reaction of dimethyl carbonate (DMC) with imidazole derivatives, making them very attractive and accessible compounds [35]. Therefore, they found applications as ligands in organometallic catalysis [36-41], as precursors of halide-free ionic liquids [42-45], and as organocatalysts in reactions involving carbon dioxide transfer in the formation of ketoacetates [46,47] and carboxylation of epoxides [48]. Some of us have recently reported a green application of BMIM-2-CO2, highlighting its efficiency for the conversion of raw glycerol to glycerol carbonate by transesterification [49]. More recently, this concept was extended to the synthesis of aliphatic polycarbonates, involving the transesterification of DMC with linear alkane diols under solvent-free conditions, and based on a two-step polymerization process [50]. The high reactivity of imidazolium-2-carboxylates can be explained by their facile decarboxylation, thus generating active carbene species, which occurs either by heating [51] or by the addition of Na+ or K+ (NaBPh4, KPF6) [47]. In this paper, we apply this dual approach to the solvent-free ROP of ε-caprolactone and rac-lactide using 1-n-butyl-3-methylimidazolium-2-carboxylate (BMIM-2-CO2) as a precatalyst.
Results and Discussion
Two solvent-free polymerization procedures were developed in this study, named methods A and B (Scheme 1). Four initiator alcohols, with an increasing number of OH functions, were successively used: benzyl alcohol (1), ethylene glycol (2), glycerol (3) or pentaerythritol (4) (Scheme 1).
![[1860-5397-9-73-i1]](/bjoc/content/inline/1860-5397-9-73-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Ring-opening polymerization of ε-caprolactone and rac-lactide by using BMIM-2-CO2 as precatalyst (1 mol %), and in the presence of alcohol initiators ROH (1, 2, 3, 4). Method A: solvent-free conditions, in vacuo, 75 °C, 75 min; method B: solvent-free conditions, NaBPh4 (0.5 mol %) as decarboxylating agent, 75 °C, 120 min.
Scheme 1: Ring-opening polymerization of ε-caprolactone and rac-lactide by using BMIM-2-CO2 as precatalyst (1...
The ROPs of ε-CL and rac-LA were firstly investigated by heating in vacuo at 75 °C for 75 min (method A), studying the influence of the amount and the nature of the initiator alcohols. Linear and star-branched polyesters were synthesized and fully characterized by 1H and 13C{1H} NMR spectroscopy, gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). Polymerization data are summarized in Table 1 and Table 2.
Table 1: Ring-opening polymerization of ε-CL in the presence of alcohol initiator [benzyl alcohol (1), ethylene glycol (2), glycerol (3) or pentaerythritol (4)], initiated in vacuo at 75 °C for 75 min.
| entry |
1
mmol |
2
mmol |
3
mmol |
4
mmol |
M/Ia | I/Cb |
Mnc
(theor.) |
Mnd | Mne | PDIf |
conv.d
% |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 0.10 | — | — | — | 90 | 2 | 6927 | 2730 | 2950 | 1.40 | 100 |
| 1b | 0.48 | — | — | — | 19 | 8 | 2031 | 1130 | 2280 | 1.49 | 100 |
| 1c | 0.96 | — | — | — | 9 | 19 | 1120 | 680 | 780 | 1.53 | 100 |
| 1d | 1.92 | — | — | — | 5 | 38 | 629 | 450 | 340 | 1.93 | 100 |
| 2a | — | 0.18 | — | — | 50 | 3 | 4508 | 1090 | 1270 | 1.50 | 100 |
| 2b | — | 0.89 | — | — | 10 | 18 | 1158 | 600 | 490 | 1.86 | 100 |
| 2c | — | 1.79 | — | — | 5 | 36 | 621 | 400 | 360 | 2.22 | 100 |
| 2d | — | 3.58 | — | — | 3 | 72 | 346 | 290 | 310 | 1.95 | 100 |
| 3a | — | — | 0.14 | — | 64 | 3 | 5416 | 1580 | 2520 | 1.75 | 100 |
| 3b | — | — | 0.68 | — | 13 | 11 | 1477 | 1460 | 2390 | 1.91 | 100 |
| 3c | — | — | 1.37 | — | 7 | 23 | 817 | 770 | 830 | 1.98 | 100 |
| 3d | — | — | 2.74 | — | 3 | 46 | 411 | 660 | 750 | 2.06 | 100 |
| 4a | — | — | — | 0.15 | 60 | 3 | 5181 | 1960 | 11700 | 1.23 | 100 |
| 4b | — | — | — | 0.50 | 18 | 10 | 2009 | 1163 | 5840 | 1.50 | 100 |
| 4c | — | — | — | 0.96 | 10 | 19 | 1157 | 593 | 2340 | 1.66 | 100 |
| 4d | — | — | — | 1.99 | 5 | 40 | 639 | 479 | 1330 | 1.79 | 100 |
aMonomer to initiator ratio. bInitiator to catalyst ratio. cCalculated according to Equation 1 and Equation 2. dDetermined by 1H NMR spectroscopy. eDetermined by gel permeation chromatography and corrected with a coefficient of 0.45 [52]. fPolydispersity index determined by gel permeation chromatography.
Table 2: Ring-opening polymerization of rac-lactide in the presence of alcohol initiators [benzyl alcohol (1), ethylene glycol (2), glycerol (3)]a, initiated in vacuo at 75 °C for 75 min.
| entry |
1
mmol |
2
mmol |
3
mmol |
M/Ib | I/Cc |
Mnd
(theor.) |
Mne | Mnf | PDIg |
conv.e
% |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.96 | — | — | 7 | 20 | 885 | 973 | 320 | 1.62 | 67 |
| 2 | — | 1.79 | — | 4 | 36 | 549 | 494 | 350 | 1.21 | 48 |
| 3 | — | — | 1.37 | 5 | 27 | 762 | 524 | 390 | 1.16 | 50 |
aNo ROP was detected with pentaerythritol (4). bMonomer to initiator ratio. cInitiator to catalyst ratio. dCalculated according to Equation 1 and Equation 2. eDetermined by 1H NMR spectroscopy. fDetermined by gel permeation chromatography and corrected with a coefficient of 0.45 [52]. gPolydispersity index determined by gel permeation chromatography.
As shown in Table 1 and Table 2, full conversion is observed for all ROPs of ε-caprolactone initiated by the various alcohols while conversions in the range of 50–70% were obtained for the ROP of rac-lactide, which afforded oligomers. The incomplete conversion can be explained by the viscous nature of the reaction mixture, resulting in PLA formation and limiting the polymerization process. Besides, increasing the amount of alcohol and consequently the initiator/catalyst ratio causes a decrease of the molecular weight (Mn) and an increase of the polydispersity (PDI = Mw/Mn). This is closely linked to the degree of polymerization, designated as DPn and defined according to Equation 1 [53,54].
![[1860-5397-9-73-i4]](/bjoc/content/inline/1860-5397-9-73-i4.svg?max-width=590&scale=1.18182)
![[1860-5397-9-73-i5]](/bjoc/content/inline/1860-5397-9-73-i5.svg?max-width=590&scale=1.18182)
For the ROP of ε-CL, Table 1 shows discrepancies between theoretical and experimental values of Mn, despite a complete conversion. These deviations, which are significant in some cases, can be explained by a backbiting process and the involvement of redistribution reactions (inter- and intra-transesterification reactions) [55]. In all cases, the values of both experimental and calculated Mn are low, and the PDIs measured by GPC indicate a poor control of molecular weight distribution.
An alternate approach for the solvent-free ROP (method B, Scheme 1), based on the use of sodium cations as decarboxylation agent and circumventing vacuum conditions, was also tested [47]. Polymerization data are summarized in Table 3. A conversion of 67% for ε-caprolactone and 83% for rac-lactide could be achieved by heating to 75 °C for 75 min in the presence of NaBPh4. Raising the heating time did not lead to a higher conversion.
Table 3: ROP of ε-CL and rac-lactide in the presence of NaBPh4 and benzyl alcohol (1), or ethylene glycol (2), at 75 °C for 75 min.
| entry | cyclic ester |
1
mmol |
2
mmol |
M/Ia | I/Cb |
Mnc
(theor) |
Mnd | conv.d [%] | PDId |
|---|---|---|---|---|---|---|---|---|---|
| 1 | ε-CL | 0.96 | — | 9 | 19 | 1119 | 565 | 67 | 1.38 |
| 2 | rac-LA | 0.96 | — | 7 | 19 | 1388 | 540 | 83 | 1.59 |
| 3 | rac-LA | — | 1.79 | 3 | 36 | 767 | 813 | 90 | 1.33 |
aMonomer to initiator ratio. bInitiator to catalyst ratio. cCalculated according to Equation 1. dDetermined by 1H NMR spectroscopy. dPolydispersity index determined by gel permeation chromatography.
Both polymerization pathways (A and B) lead to the desired polymers. Heating in vacuo is more efficient for the ROP of ε-caprolactone for which a conversion of 100% was always observed. However the highest conversion for rac-lactide of 83% was achieved by using NaBPh4 as decarboxylating agent. Alcohols 1–4 initiate the ring-opening polymerization and form linear (benzyl alcohol, ethylene glycol) and star-branched (glycerol, pentaerythritol) polyesters. In comparison to their linear analogues, star-branched polymers feature lower crystallinity, lower melt viscosities and smaller hydrodynamic volume [56-58]. The crystallinity (defined according to Equation 3 with ΔHc being the crystallization enthalpy, and ΔH' the enthalpy of fusion of PCL, ΔH' = 161.1 J·g−1) affects directly the melting temperature (Tm). Thus, a decrease of molecular weight causes a decrease of the Tm. Analysis of the crystallinity of the various polycaprolactones by differential scanning calorimetry is summarized in Table 4. DSC profiles are depicted in Supporting Information File 2 (Figure S1).
![[1860-5397-9-73-i6]](/bjoc/content/inline/1860-5397-9-73-i6.svg?max-width=590&scale=1.18182)
Table 4: DSC data recorded for several polycaprolactones synthesized in this study.
| entrya | alcohol |
Mn
(g·mol−1)b |
Tc (°C) |
ΔHc
(J·g−1) |
Tm (°C) |
ΔHm
(J·g−1) |
Hc/H’
(%)c,d |
|---|---|---|---|---|---|---|---|
| 1b | 1 | 1130 | 20.36 | 80.01 | 46.71 | 88.56 | 50 |
| 3b | 3 | 1460 | 22.17 | 71.40 | 47.27 | 71.06 | 44 |
| 4b | 4 | 1163 | 24.39 | 56.72 | 47.08 | 56.71 | 35 |
aCorresponds to the same entries of Table 1. bDetermined by 1H NMR spectroscopy. cPercentage of crystallinity. dH’(PCL) = 161.1 J·g−1.
Samples with molecular weight in the 1100–1500 Da range (Table 4), show similar Tm and Tc, although the endings of the chains are different. However, values for the crystallinity decrease from 50% (benzyl alcohol, 1) through 44% (glycerol, 2) to 35% (pentaerythritol, 4).
From a mechanistic point of view, the formation of the target polymers can be postulated by the in situ decarboxylation of BMIM-2-CO2, which generates free N-heterocyclic carbene species. In the absence of BMIM-2-CO2, the mixture of ε-caprolactone or rac-lactide and initiator alcohols remained unchanged under otherwise identical conditions, even for an extended period of time, underlining the decisive role of the imidazolium-2-carboxylate species in the polymerization process. Moreover and under atmospheric pressure, we have observed from thermogravimetric measurements that BMIM-2-CO2 is stable up to 90 °C and then undergoes a substantial weight-loss, corroborating a previous report published by Louie and co-workers [51]. Under the temperature conditions of ROP reactions, i.e., 75 °C, the decarboxylation is induced either in vacuo (method A) or by the addition of NaBPh4 (method B). On the basis of previous studies reported by Waymouth and Hedrick [34], we suggest that the in situ generated N-heterocyclic carbene acts as a nucleophilic species and leads to the opening of cyclic esters (initiation step, Scheme 2). The protonation of the formed zwitterionic acylimidazole intermediate by the alcohol initiator followed by the attack of the thereby obtained alkoxide regenerates the carbene species. The chain propagation is by the attack of the newly formed hydroxy-terminated monomer (I) on another zwitterionic intermediate (propagation step, Scheme 2). As a consequence, if the ratio of monomer to alcohol initiator is too low, the latter can compete with the formed hydroxy-terminated monomers/polymers to initiate the formation of a new polymer chain, and the propagation is stopped. In this case oligomers rather than polymers are detected. Further experimental attempts to trap carbene species and the proposed intermediates are currently in progress in our laboratory.
![[1860-5397-9-73-i2]](/bjoc/content/inline/1860-5397-9-73-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Possible mechanism for the synthesis of PCL and PLA by ROP using BMIM-2-CO2 as precatalyst.
Scheme 2: Possible mechanism for the synthesis of PCL and PLA by ROP using BMIM-2-CO2 as precatalyst.
Regarding method B, which uses NaBPh4 as the decarboxylating agent, the reaction does not occur in vacuo. Thus, and on the basis of a previous study reported by Tommasi et al. [47], we suggest that the liberated carbon dioxide reacts with the alcohol initiator ROH, leading to the formation of a ROC(O)O− anion (monobenzylcarbonate if benzyl alcohol is used as initiator, Scheme 3). The expelled proton reacts with the carbene species and deactivates the catalyst. The decrease of the amount of the initiating alcohol and the bit by bit deactivation of BMIM-2-CO2 can explain the incomplete conversion observed by comparison with method A.
![[1860-5397-9-73-i3]](/bjoc/content/inline/1860-5397-9-73-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: In situ formation of monobenzylcarbonate and deactivation of N-heterocyclic carbene.
Scheme 3: In situ formation of monobenzylcarbonate and deactivation of N-heterocyclic carbene.
Furthermore, this dependence between the amount of alcohol and the length of the chain confirms the proposed monomer-activated mechanism for the ring-opening polymerization of ε-caprolactone and rac-lactide (Scheme 2) [20,27,28,33,49].
Conclusion
In this contribution, we have reported that BMIM-2-CO2 can be an active precatalyst for the solvent-free ring-opening polymerization of ε-caprolactone and rac-lactide. A conversion of 100% could be achieved for ε-CL by using different alcohols in vacuo, while the addition of NaBPh4 appears to be more efficient for rac-LA, reaching a conversion of 83%. Thus, in view of these results, BMIM-2-CO2 can be considered as a potential versatile precatalyst. In addition, its facile preparation, in only one-step with a satisfactory yield (66%) and from commercial n-butylimidazole and dimethyl carbonate, increases its attractiveness and its green character compared to the direct use of stable and active N-heterocarbenes, which require several synthetic steps [59]. Therefore, we support the use of BMIM-2-CO2 as a well-adapted alternative with respect to green polymerization approaches, (solvent-free conditions, no metal-based catalyst) as reported in this manuscript. However, the formation of higher molecular weight polymers with low polydispersities still remains to be improved. Further work in this direction is underway.
References
-
Albertsson, A.-C.; Varma, I. K. Biomacromolecules 2003, 4, 1466–1486. doi:10.1021/bm034247a
Return to citation in text: [1] -
Sune Negre, J. M.; Manich Bou, A.; Tico Grau, J. R.; Bel Prieto, E. Cienc. Pharm. 1997, 7, 79–86.
Return to citation in text: [1] -
Molpeceres, J.; Guzman, M.; Aberturas, M. R.; Chacon, M.; Berges, L. J. Pharm. Sci. 1996, 85, 206–213. doi:10.1021/js950164r
Return to citation in text: [1] -
Ohya, Y. Bio Ind. 2007, 24, 16–27.
Return to citation in text: [1] -
Viinikainen, A.-K.; Göransson, H.; Huovinen, K.; Kellomäki, M.; Törmälä, P.; Rokkanen, P. J. Mater. Sci.: Mater. Med. 2009, 20, 1963–1969. doi:10.1007/s10856-009-3747-8
Return to citation in text: [1] -
Jérôme, C.; Lecomte, P. Adv. Drug Delivery Rev. 2008, 60, 1056–1076. doi:10.1016/j.addr.2008.02.008
Return to citation in text: [1] -
Hess, R.; Douglas, T.; Myers, K. A.; Rentsch, B.; Rentsch, C.; Worch, H.; Shrive, N. G.; Hart, D. A.; Scharnweber, D. J. Biomech. Eng. 2010, 132, 021001. doi:10.1115/1.4000194
Return to citation in text: [1] -
Oyana, A.; Uchida, M.; Choong, C.; Triffitt, J.; Jones, J.; Ito, A. Biomaterials 2005, 26, 2407–2413. doi:10.1016/j.biomaterials.2004.07.048
Return to citation in text: [1] -
Chang, K.-Y.; Hung, L.-H.; Chu, I.-M.; Ko, C.-S.; Lee, Y.-D. J. Biomed. Mater. Res., Part A 2010, 92A, 712–723. doi:10.1002/jbm.a.32198
Return to citation in text: [1] -
Hietala, E.-M.; Maasilta, P.; Juuti, H.; Nuutinen, J.-P.; Harjula, A. L. J.; Salminen, U.-S.; Lassila, R. Thromb. Haemostasis 2004, 92, 1394–1401. doi:10.1160/TH04-02-0124
Return to citation in text: [1] -
Kricheldorf, H. R.; Berl, M.; Scharnagel, N. Macromolecules 1988, 21, 286–293. doi:10.1021/ma00180a002
Return to citation in text: [1] -
O’Keefe, B. J.; Hillmyer, M. A.; Tolman, W. B. J. Chem. Soc., Dalton Trans. 2001, 2215–2224. doi:10.1039/B104197P
Return to citation in text: [1] -
Gibson, V. C.; Marshall, E. L. Compr. Coord. Chem. II 2004, 9, 1–74. doi:10.1016/B0-08-043748-6/09010-1
Return to citation in text: [1] -
Dove, A. P.; Gibson, V. C.; Marshall, E. L.; White, A. J. P.; Williams, D. J. Chem. Commun. 2001, 283–284. doi:10.1039/b008770j
Return to citation in text: [1] -
Chisholm, M. H.; Delbridge, E. E. New J. Chem. 2003, 27, 1167–1176. doi:10.1039/b300101f
Return to citation in text: [1] -
Duda, A.; Penczek, S. Macromolecules 1995, 28, 5981–5992. doi:10.1021/ma00122a001
Return to citation in text: [1] -
Biela, T.; Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Macromol. Symp. 2006, 240, 47–55. doi:10.1002/masy.200650807
Return to citation in text: [1] -
Palard, I.; Schappacher, M.; Soum, A.; Guillaume, S. M. Polym. Int. 2006, 55, 1132–1137. doi:10.1002/pi.1984
Return to citation in text: [1] -
Kiesewetter, M. K.; Shin, E. J.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2010, 43, 2093–2107. doi:10.1021/ma9025948
Return to citation in text: [1] -
Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, 5813–5840. doi:10.1021/cr068415b
Return to citation in text: [1] [2] [3] [4] -
Jeong, W.; Hedrick, J. L.; Waymouth, R. M. J. Am. Chem. Soc. 2007, 129, 8414–8415. doi:10.1021/ja072037q
Return to citation in text: [1] -
Matsumura, S. Adv. Polym. Sci. 2006, 194, 95–132. doi:10.1007/12_030
Return to citation in text: [1] -
Gross, R. A.; Kumar, A.; Kalra, B. Chem. Rev. 2001, 101, 2097–2124. doi:10.1021/cr0002590
Return to citation in text: [1] -
Liu, J.; Liu, L. Macromolecules 2004, 37, 2674–2676. doi:10.1021/ma0348066
Return to citation in text: [1] -
Casas, J.; Persson, P. V.; Iversen, T.; Córdova, A. Adv. Synth. Catal. 2004, 346, 1087–1089. doi:10.1002/adsc.200404082
Return to citation in text: [1] -
Dove, A. P.; Pratt, R. C.; Lohmeijer, B. G. G.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 13798–13799. doi:10.1021/ja0543346
Return to citation in text: [1] -
Connor, E. F.; Nyce, G. W.; Myers, M.; Möck, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, 914–915. doi:10.1021/ja0173324
Return to citation in text: [1] [2] [3] [4] -
Kamber, N. E.; Jeong, W.; Gonzalez, S.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2009, 42, 1634–1639. doi:10.1021/ma802618h
Return to citation in text: [1] [2] [3] -
Dove, A. P.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Hagberg, E. C.; Nyce, G. W.; Waymouth, R. M.; Hedrick, J. L. Polymer 2006, 47, 4018–4025. doi:10.1016/j.polymer.2006.02.037
Return to citation in text: [1] -
Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. J. Am. Chem. Soc. 2009, 131, 3201–3209. doi:10.1021/ja809246f
Return to citation in text: [1] -
Dove, A. P.; Li, H.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Waymouth, R. M.; Hedrick, J. L. Chem. Commun. 2006, 2881–2883. doi:10.1039/b601393g
Return to citation in text: [1] [2] -
Culkin, D. A.; Jeong, W.; Csihony, S.; Gomez, E. D.; Balsara, N. P.; Hedrick, J. L.; Waymouth, R. M. Angew. Chem., Int. Ed. 2007, 46, 2627–2630. doi:10.1002/anie.200604740
Return to citation in text: [1] -
Csihony, S.; Culkin, D. A.; Sentman, A. C.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 9079–9084. doi:10.1021/ja050909n
Return to citation in text: [1] [2] -
Kamber, N. E.; Kiesewetter, M. K.; Hedrick, J. L.; Waymouth, R. M. Polym. Prepr. 2006, 47, 570–571.
Return to citation in text: [1] [2] -
Holbrey, J. D.; Reichert, W. M.; Tkatchenko, I.; Bouajila, E.; Walter, O.; Tommasi, I.; Rogers, R. D. Chem. Commun. 2003, 28–29. doi:10.1039/b211519k
Return to citation in text: [1] -
Azouri, M.; Andrieu, J.; Picquet, M.; Cattey, H. Inorg. Chem. 2009, 48, 1236–1242. doi:10.1021/ic801870w
Return to citation in text: [1] -
Azouri, M.; Andrieu, J.; Picquet, M.; Richard, P.; Hanquet, B.; Tkatchenko, I. Eur. J. Inorg. Chem. 2007, 4877–4883. doi:10.1002/ejic.200700590
Return to citation in text: [1] -
Voutchkova, A. M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R. H. J. Am. Chem. Soc. 2007, 129, 12834–12846. doi:10.1021/ja0742885
Return to citation in text: [1] -
Tudose, A.; Delaude, L.; André, B.; Demonceau, A. Tetrahedron Lett. 2006, 47, 8529–8533. doi:10.1016/j.tetlet.2006.09.139
Return to citation in text: [1] -
Tudose, A.; Demonceau, A.; Delaude, L. J. Organomet. Chem. 2006, 691, 5356–5365. doi:10.1016/j.jorganchem.2006.07.035
Return to citation in text: [1] -
Voutchkova, A. M.; Appelhans, L. N.; Chianese, A. R.; Crabtree, R. H. J. Am. Chem. Soc. 2005, 127, 17624–17625. doi:10.1021/ja056625k
Return to citation in text: [1] -
Rijksen, C.; Rogers, R. D. J. Org. Chem. 2008, 73, 5582–5584. doi:10.1021/jo800578b
Return to citation in text: [1] -
Bridges, N. J.; Hines, C. C.; Smiglak, M.; Rogers, R. D. Chem.–Eur. J. 2007, 13, 5207–5212. doi:10.1002/chem.200700055
Return to citation in text: [1] -
Smiglak, M.; Holbrey, J. D.; Griffin, S. T.; Reichert, W. M.; Swatloski, R. P.; Katritzky, A. R.; Yang, H.; Zhang, D.; Kirichenko, K.; Rogers, R. D. Green Chem. 2007, 9, 90–98. doi:10.1039/b610421e
Return to citation in text: [1] -
Picquet, M.; Poinsot, D.; Stutzmann, S.; Tkatchenko, I.; Tommasi, I.; Wasserscheid, P.; Zimmermann, J. Top. Catal. 2004, 29, 139–143. doi:10.1023/B:TOCA.0000029796.11969.ec
Return to citation in text: [1] -
Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2006, 47, 6453–6456. doi:10.1016/j.tetlet.2006.06.106
Return to citation in text: [1] -
Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2005, 46, 2141–2145. doi:10.1016/j.tetlet.2005.01.106
Return to citation in text: [1] [2] [3] [4] -
Zhou, H.; Zhang, W.-Z.; Liu, C.-H.; Qu, J.-P.; Lu, X.-B. J. Org. Chem. 2008, 73, 8039–8044. doi:10.1021/jo801457r
Return to citation in text: [1] -
Naik, P. U.; Petitjean, L.; Refes, K.; Picquet, M.; Plasseraud, L. Adv. Synth. Catal. 2009, 351, 1753–1756. doi:10.1002/adsc.200900280
Return to citation in text: [1] [2] -
Naik, P. U.; Refes, K.; Sadaka, F.; Brachais, C.-H.; Boni, G.; Couvercelle, J.-P.; Picquet, M.; Plasseraud, L. Polym. Chem. 2012, 3, 1475–1480. doi:10.1039/c2py20056b
Return to citation in text: [1] -
Van Ausdall, B. R.; Glass, J. L.; Wiggins, K. M.; Aarif, A. M.; Louie, J. J. Org. Chem. 2009, 74, 7935–7942. doi:10.1021/jo901791k
Return to citation in text: [1] [2] -
Biela, T.; Duda, A.; Penczek, S. Macromol. Symp. 2002, 183, 1–10. doi:10.1002/1521-3900(200207)183:1<1::AID-MASY1>3.0.CO;2-Q
Return to citation in text: [1] [2] -
Cowie, J. M. G. Polymers: Chemistry and Physics of Modern Materials, 2nd ed.; Blackie Academic & Professional: London, 1991.
Return to citation in text: [1] -
Gilbert, R. G.; Hess, M.; Jenkins, A. D.; Jones, R. G.; Kratochvil, P.; Stepto, R. F. T. Pure Appl. Chem. 2009, 81, 351–353. doi:10.1351/PAC-REC-08-05-02
Return to citation in text: [1] -
Labet, M.; Thielemans, W. Chem. Soc. Rev. 2009, 38, 3484–3504. doi:10.1039/b820162p
Return to citation in text: [1] -
Sanda, F.; Sanada, H.; Shibasaki, Y.; Endo, T. Macromolecules 2002, 35, 680–683. doi:10.1021/ma011341f
Return to citation in text: [1] -
Peppas, N. A.; Argade, A.; Bhargava, S. J. Appl. Polym. Sci. 2003, 87, 322–327. doi:10.1002/app.11444
Return to citation in text: [1] -
Lemmouchi, Y.; Perry, M. C.; Amass, A. J.; Chakraborty, K.; Schacht, E. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 3975–3985. doi:10.1002/pola.22151
Return to citation in text: [1] -
Arduengo, A. J., III; Krafczyk, R.; Schmutzler, R.; Craig, H. A.; Goerlich, J. R.; Marshall, W. J.; Unverzagt, M. Tetrahedron 1999, 55, 14523–14534. doi:10.1016/S0040-4020(99)00927-8
Return to citation in text: [1]
| 20. | Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, 5813–5840. doi:10.1021/cr068415b |
| 27. | Connor, E. F.; Nyce, G. W.; Myers, M.; Möck, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, 914–915. doi:10.1021/ja0173324 |
| 28. | Kamber, N. E.; Jeong, W.; Gonzalez, S.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2009, 42, 1634–1639. doi:10.1021/ma802618h |
| 33. | Csihony, S.; Culkin, D. A.; Sentman, A. C.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 9079–9084. doi:10.1021/ja050909n |
| 49. | Naik, P. U.; Petitjean, L.; Refes, K.; Picquet, M.; Plasseraud, L. Adv. Synth. Catal. 2009, 351, 1753–1756. doi:10.1002/adsc.200900280 |
| 59. | Arduengo, A. J., III; Krafczyk, R.; Schmutzler, R.; Craig, H. A.; Goerlich, J. R.; Marshall, W. J.; Unverzagt, M. Tetrahedron 1999, 55, 14523–14534. doi:10.1016/S0040-4020(99)00927-8 |
| 1. | Albertsson, A.-C.; Varma, I. K. Biomacromolecules 2003, 4, 1466–1486. doi:10.1021/bm034247a |
| 2. | Sune Negre, J. M.; Manich Bou, A.; Tico Grau, J. R.; Bel Prieto, E. Cienc. Pharm. 1997, 7, 79–86. |
| 3. | Molpeceres, J.; Guzman, M.; Aberturas, M. R.; Chacon, M.; Berges, L. J. Pharm. Sci. 1996, 85, 206–213. doi:10.1021/js950164r |
| 4. | Ohya, Y. Bio Ind. 2007, 24, 16–27. |
| 5. | Viinikainen, A.-K.; Göransson, H.; Huovinen, K.; Kellomäki, M.; Törmälä, P.; Rokkanen, P. J. Mater. Sci.: Mater. Med. 2009, 20, 1963–1969. doi:10.1007/s10856-009-3747-8 |
| 6. | Jérôme, C.; Lecomte, P. Adv. Drug Delivery Rev. 2008, 60, 1056–1076. doi:10.1016/j.addr.2008.02.008 |
| 11. | Kricheldorf, H. R.; Berl, M.; Scharnagel, N. Macromolecules 1988, 21, 286–293. doi:10.1021/ma00180a002 |
| 12. | O’Keefe, B. J.; Hillmyer, M. A.; Tolman, W. B. J. Chem. Soc., Dalton Trans. 2001, 2215–2224. doi:10.1039/B104197P |
| 13. | Gibson, V. C.; Marshall, E. L. Compr. Coord. Chem. II 2004, 9, 1–74. doi:10.1016/B0-08-043748-6/09010-1 |
| 14. | Dove, A. P.; Gibson, V. C.; Marshall, E. L.; White, A. J. P.; Williams, D. J. Chem. Commun. 2001, 283–284. doi:10.1039/b008770j |
| 15. | Chisholm, M. H.; Delbridge, E. E. New J. Chem. 2003, 27, 1167–1176. doi:10.1039/b300101f |
| 16. | Duda, A.; Penczek, S. Macromolecules 1995, 28, 5981–5992. doi:10.1021/ma00122a001 |
| 17. | Biela, T.; Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Macromol. Symp. 2006, 240, 47–55. doi:10.1002/masy.200650807 |
| 18. | Palard, I.; Schappacher, M.; Soum, A.; Guillaume, S. M. Polym. Int. 2006, 55, 1132–1137. doi:10.1002/pi.1984 |
| 49. | Naik, P. U.; Petitjean, L.; Refes, K.; Picquet, M.; Plasseraud, L. Adv. Synth. Catal. 2009, 351, 1753–1756. doi:10.1002/adsc.200900280 |
| 10. | Hietala, E.-M.; Maasilta, P.; Juuti, H.; Nuutinen, J.-P.; Harjula, A. L. J.; Salminen, U.-S.; Lassila, R. Thromb. Haemostasis 2004, 92, 1394–1401. doi:10.1160/TH04-02-0124 |
| 50. | Naik, P. U.; Refes, K.; Sadaka, F.; Brachais, C.-H.; Boni, G.; Couvercelle, J.-P.; Picquet, M.; Plasseraud, L. Polym. Chem. 2012, 3, 1475–1480. doi:10.1039/c2py20056b |
| 9. | Chang, K.-Y.; Hung, L.-H.; Chu, I.-M.; Ko, C.-S.; Lee, Y.-D. J. Biomed. Mater. Res., Part A 2010, 92A, 712–723. doi:10.1002/jbm.a.32198 |
| 46. | Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2006, 47, 6453–6456. doi:10.1016/j.tetlet.2006.06.106 |
| 47. | Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2005, 46, 2141–2145. doi:10.1016/j.tetlet.2005.01.106 |
| 7. | Hess, R.; Douglas, T.; Myers, K. A.; Rentsch, B.; Rentsch, C.; Worch, H.; Shrive, N. G.; Hart, D. A.; Scharnweber, D. J. Biomech. Eng. 2010, 132, 021001. doi:10.1115/1.4000194 |
| 8. | Oyana, A.; Uchida, M.; Choong, C.; Triffitt, J.; Jones, J.; Ito, A. Biomaterials 2005, 26, 2407–2413. doi:10.1016/j.biomaterials.2004.07.048 |
| 48. | Zhou, H.; Zhang, W.-Z.; Liu, C.-H.; Qu, J.-P.; Lu, X.-B. J. Org. Chem. 2008, 73, 8039–8044. doi:10.1021/jo801457r |
| 28. | Kamber, N. E.; Jeong, W.; Gonzalez, S.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2009, 42, 1634–1639. doi:10.1021/ma802618h |
| 34. | Kamber, N. E.; Kiesewetter, M. K.; Hedrick, J. L.; Waymouth, R. M. Polym. Prepr. 2006, 47, 570–571. |
| 36. | Azouri, M.; Andrieu, J.; Picquet, M.; Cattey, H. Inorg. Chem. 2009, 48, 1236–1242. doi:10.1021/ic801870w |
| 37. | Azouri, M.; Andrieu, J.; Picquet, M.; Richard, P.; Hanquet, B.; Tkatchenko, I. Eur. J. Inorg. Chem. 2007, 4877–4883. doi:10.1002/ejic.200700590 |
| 38. | Voutchkova, A. M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R. H. J. Am. Chem. Soc. 2007, 129, 12834–12846. doi:10.1021/ja0742885 |
| 39. | Tudose, A.; Delaude, L.; André, B.; Demonceau, A. Tetrahedron Lett. 2006, 47, 8529–8533. doi:10.1016/j.tetlet.2006.09.139 |
| 40. | Tudose, A.; Demonceau, A.; Delaude, L. J. Organomet. Chem. 2006, 691, 5356–5365. doi:10.1016/j.jorganchem.2006.07.035 |
| 41. | Voutchkova, A. M.; Appelhans, L. N.; Chianese, A. R.; Crabtree, R. H. J. Am. Chem. Soc. 2005, 127, 17624–17625. doi:10.1021/ja056625k |
| 20. | Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, 5813–5840. doi:10.1021/cr068415b |
| 27. | Connor, E. F.; Nyce, G. W.; Myers, M.; Möck, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, 914–915. doi:10.1021/ja0173324 |
| 31. | Dove, A. P.; Li, H.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Waymouth, R. M.; Hedrick, J. L. Chem. Commun. 2006, 2881–2883. doi:10.1039/b601393g |
| 33. | Csihony, S.; Culkin, D. A.; Sentman, A. C.; Dove, A. P.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 9079–9084. doi:10.1021/ja050909n |
| 42. | Rijksen, C.; Rogers, R. D. J. Org. Chem. 2008, 73, 5582–5584. doi:10.1021/jo800578b |
| 43. | Bridges, N. J.; Hines, C. C.; Smiglak, M.; Rogers, R. D. Chem.–Eur. J. 2007, 13, 5207–5212. doi:10.1002/chem.200700055 |
| 44. | Smiglak, M.; Holbrey, J. D.; Griffin, S. T.; Reichert, W. M.; Swatloski, R. P.; Katritzky, A. R.; Yang, H.; Zhang, D.; Kirichenko, K.; Rogers, R. D. Green Chem. 2007, 9, 90–98. doi:10.1039/b610421e |
| 45. | Picquet, M.; Poinsot, D.; Stutzmann, S.; Tkatchenko, I.; Tommasi, I.; Wasserscheid, P.; Zimmermann, J. Top. Catal. 2004, 29, 139–143. doi:10.1023/B:TOCA.0000029796.11969.ec |
| 20. | Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, 5813–5840. doi:10.1021/cr068415b |
| 27. | Connor, E. F.; Nyce, G. W.; Myers, M.; Möck, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, 914–915. doi:10.1021/ja0173324 |
| 28. | Kamber, N. E.; Jeong, W.; Gonzalez, S.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2009, 42, 1634–1639. doi:10.1021/ma802618h |
| 29. | Dove, A. P.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Hagberg, E. C.; Nyce, G. W.; Waymouth, R. M.; Hedrick, J. L. Polymer 2006, 47, 4018–4025. doi:10.1016/j.polymer.2006.02.037 |
| 30. | Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. J. Am. Chem. Soc. 2009, 131, 3201–3209. doi:10.1021/ja809246f |
| 31. | Dove, A. P.; Li, H.; Pratt, R. C.; Lohmeijer, B. G. G.; Culkin, D. A.; Waymouth, R. M.; Hedrick, J. L. Chem. Commun. 2006, 2881–2883. doi:10.1039/b601393g |
| 32. | Culkin, D. A.; Jeong, W.; Csihony, S.; Gomez, E. D.; Balsara, N. P.; Hedrick, J. L.; Waymouth, R. M. Angew. Chem., Int. Ed. 2007, 46, 2627–2630. doi:10.1002/anie.200604740 |
| 19. | Kiesewetter, M. K.; Shin, E. J.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2010, 43, 2093–2107. doi:10.1021/ma9025948 |
| 20. | Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C.; Lohmeijer, B. G. G.; Hedrick, J. L. Chem. Rev. 2007, 107, 5813–5840. doi:10.1021/cr068415b |
| 21. | Jeong, W.; Hedrick, J. L.; Waymouth, R. M. J. Am. Chem. Soc. 2007, 129, 8414–8415. doi:10.1021/ja072037q |
| 22. | Matsumura, S. Adv. Polym. Sci. 2006, 194, 95–132. doi:10.1007/12_030 |
| 23. | Gross, R. A.; Kumar, A.; Kalra, B. Chem. Rev. 2001, 101, 2097–2124. doi:10.1021/cr0002590 |
| 24. | Liu, J.; Liu, L. Macromolecules 2004, 37, 2674–2676. doi:10.1021/ma0348066 |
| 25. | Casas, J.; Persson, P. V.; Iversen, T.; Córdova, A. Adv. Synth. Catal. 2004, 346, 1087–1089. doi:10.1002/adsc.200404082 |
| 26. | Dove, A. P.; Pratt, R. C.; Lohmeijer, B. G. G.; Waymouth, R. M.; Hedrick, J. L. J. Am. Chem. Soc. 2005, 127, 13798–13799. doi:10.1021/ja0543346 |
| 27. | Connor, E. F.; Nyce, G. W.; Myers, M.; Möck, A.; Hedrick, J. L. J. Am. Chem. Soc. 2002, 124, 914–915. doi:10.1021/ja0173324 |
| 35. | Holbrey, J. D.; Reichert, W. M.; Tkatchenko, I.; Bouajila, E.; Walter, O.; Tommasi, I.; Rogers, R. D. Chem. Commun. 2003, 28–29. doi:10.1039/b211519k |
| 52. | Biela, T.; Duda, A.; Penczek, S. Macromol. Symp. 2002, 183, 1–10. doi:10.1002/1521-3900(200207)183:1<1::AID-MASY1>3.0.CO;2-Q |
| 51. | Van Ausdall, B. R.; Glass, J. L.; Wiggins, K. M.; Aarif, A. M.; Louie, J. J. Org. Chem. 2009, 74, 7935–7942. doi:10.1021/jo901791k |
| 47. | Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2005, 46, 2141–2145. doi:10.1016/j.tetlet.2005.01.106 |
| 34. | Kamber, N. E.; Kiesewetter, M. K.; Hedrick, J. L.; Waymouth, R. M. Polym. Prepr. 2006, 47, 570–571. |
| 47. | Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2005, 46, 2141–2145. doi:10.1016/j.tetlet.2005.01.106 |
| 56. | Sanda, F.; Sanada, H.; Shibasaki, Y.; Endo, T. Macromolecules 2002, 35, 680–683. doi:10.1021/ma011341f |
| 57. | Peppas, N. A.; Argade, A.; Bhargava, S. J. Appl. Polym. Sci. 2003, 87, 322–327. doi:10.1002/app.11444 |
| 58. | Lemmouchi, Y.; Perry, M. C.; Amass, A. J.; Chakraborty, K.; Schacht, E. J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 3975–3985. doi:10.1002/pola.22151 |
| 51. | Van Ausdall, B. R.; Glass, J. L.; Wiggins, K. M.; Aarif, A. M.; Louie, J. J. Org. Chem. 2009, 74, 7935–7942. doi:10.1021/jo901791k |
| 55. | Labet, M.; Thielemans, W. Chem. Soc. Rev. 2009, 38, 3484–3504. doi:10.1039/b820162p |
| 47. | Tommasi, I.; Sorentino, F. Tetrahedron Lett. 2005, 46, 2141–2145. doi:10.1016/j.tetlet.2005.01.106 |
| 52. | Biela, T.; Duda, A.; Penczek, S. Macromol. Symp. 2002, 183, 1–10. doi:10.1002/1521-3900(200207)183:1<1::AID-MASY1>3.0.CO;2-Q |
| 53. | Cowie, J. M. G. Polymers: Chemistry and Physics of Modern Materials, 2nd ed.; Blackie Academic & Professional: London, 1991. |
| 54. | Gilbert, R. G.; Hess, M.; Jenkins, A. D.; Jones, R. G.; Kratochvil, P.; Stepto, R. F. T. Pure Appl. Chem. 2009, 81, 351–353. doi:10.1351/PAC-REC-08-05-02 |
© 2013 Hoppe et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)