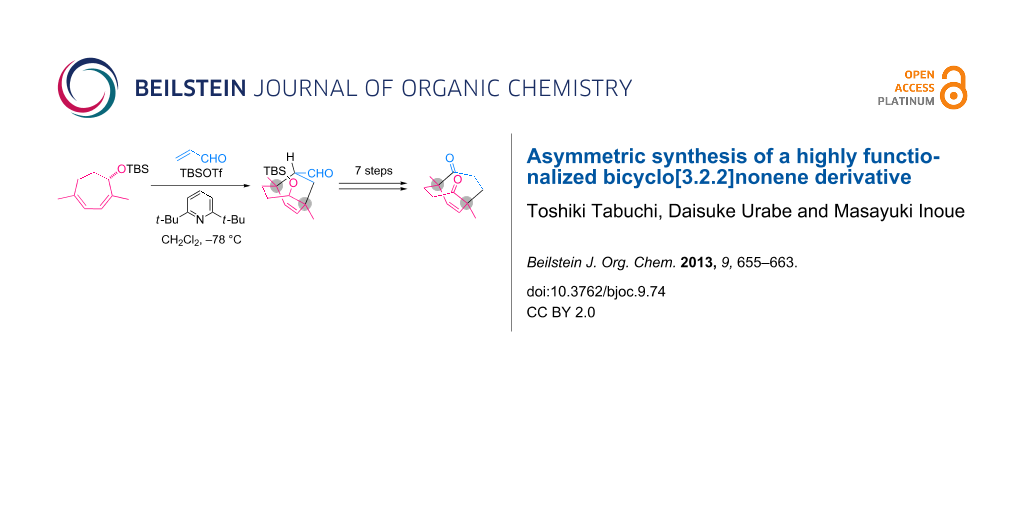
Abstract
The stereoselective Diels–Alder reaction between an optically active 1,4-dimethylcycloheptadiene and acrolein was effectively promoted by TBSOTf to produce a bicyclo[3.2.2]nonene derivative bearing two quaternary carbons. Seven additional transformations from the obtained bicycle delivered the C2-symmetric bicyclo[3.3.2]decene derivative, a key intermediate in our synthetic study of ryanodine.

Graphical Abstract
Introduction
Ryanodine (Scheme 1) [1-3] is a potent modulator of the intracellular calcium release channels, known as ryanodine receptors [4,5]. Its complex architecture, including eight contiguous tetrasubstituted carbons on the pentacyclic ABCDE-ring system, has posed a formidable synthetic challenge. To date, the only total synthesis of a compound in this class of natural products was reported by Deslongchamps, who constructed ryanodol in 1979 [6-9]. Most recently, we reported the synthesis of 9-demethyl-10,15-dideoxyryanodol [10] by taking advantage of the intrinsic C2-symmetry of the target molecule. In this synthesis, C2-symmetric compounds such as bicyclo[3.3.2]decene 1 were strategically designed, and application of pairwise functionalizations of these molecules minimized the total number of steps.
![[1860-5397-9-74-i1]](/bjoc/content/inline/1860-5397-9-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structure of ryanodine and the Diels–Alder reactions for construction of the potential intermediates of ryanodine.
Scheme 1: Structure of ryanodine and the Diels–Alder reactions for construction of the potential intermediate...
Bicyclo[3.3.2]decene 1 was prepared from C2-symmetric bicyclo[2.2.2]octene 2 through a ring-expansion reaction (Scheme 1) [11]. We reported the synthetic routes to racemic 2 and enantiomerically pure 2 from 3 and 5, respectively. Specifically, the dearomatizing Diels–Alder reaction between 2,5-dimethylbenzene-1,4-diol (3) and maleic anhydride lead to the construction of bicyclo[2.2.2]octene 4, which was then transformed into racemic 2 through electrolysis [11]. Alternatively, the Diels–Alder reaction between 3,6-dimethyl-o-quinone monoacetal 5 and 1,1-diethoxyethylene provided bicyclo[2.2.2]octene 6, which was then converted to enantiopure 2 via an enzymatic kinetic resolution [12]. The Diels–Alder reaction was effectively employed in both of these syntheses for construction of the two quaternary carbons at the C1 and C5 positions of ryanodine (highlighted in gray). However, the current route to (+)-2 from racemic 6 generated the unnecessary antipode. Therefore, development of an alternative asymmetric route to 1 was planned to further improve the overall practicality. Here we report an asymmetric Diels–Alder reaction for simultaneous installation of the C1- and C5-stereocenters using the optically active cycloheptadiene derivative 7, and its derivatization into bicyclo[3.3.2]decene 1.
We assumed that the C4-stereocenter of optically active seven-membered diene 7 would permit the requisite stereoselective Diels–Alder reaction (Scheme 1). Namely, the reaction between 7 and acrolein was expected to stereoselectively introduce the C1, C5 and C12 stereocenters to afford bicyclo[3.2.2]nonene 8. The C11-aldehyde of 8 was then to be utilized as a handle for the ring expansion to access 1. To the best of our knowledge, construction of the two quaternary carbons by the intermolecular Diels–Alder reaction of 1,4-disubstituted cycloheptadiene derivatives has not been reported [13,14].
Results and Discussion
The synthesis of optically active 7 began from crotyl chloride (Scheme 2). The carbon chain extension of crotyl chloride by treatment with acetylacetone and K2CO3 [15], followed by the addition of vinylmagnesium bromide [16], provided 9. The bromoetherification of tertiary alcohol 9 by using NBS led to tetrahydrofuran 10 as a diastereomeric mixture. Next, the base-induced elimination of HBr converted 10 to diene 11, which underwent the Claisen rearrangement at 170 °C to give rise to heptenone 12 [17-21]. The more thermodynamically stable silyl enol ether 13 was regioselectively formed from 12 under Holton’s conditions [22], and DDQ-mediated oxidation of 13 resulted in the formation of α,β-unsaturated ketone 14. Asymmetric reduction of ketone 14 was in turn realized by applying a stoichiometric amount of (R)-CBS and BH3·SMe2 to produce 15 (82% ee) [23]. The absolute configuration of C4 was determined as S by the modified Mosher method after acylation of 15 using (R)- and (S)-MTPACl [24]. Finally, the hydroxy group of 15 was protected as its TBS ether to afford 7.
![[1860-5397-9-74-i2]](/bjoc/content/inline/1860-5397-9-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Asymmetric synthesis of 7 and determination of the absolute configuration at C4 of 15 by the modified Mosher method. The numbers are differences in 1H chemical shifts between 16a and 16b (Δδ = δ16a − δ16b).
Scheme 2: Asymmetric synthesis of 7 and determination of the absolute configuration at C4 of 15 by the modifi...
We then explored the Diels–Alder reaction between 7 and acrolein to construct the bicyclo[3.2.2]nonene structure (Scheme 3). The Diels–Alder reaction under thermal conditions (100 °C) induced the decomposition of diene 7, and only the starting material was recovered after 10 h (26%). Because of the low reactivity of 7, we applied a Lewis acid to facilitate the reaction. However, the reaction of 7 and acrolein in the presence of BF3·OEt2 (50 mol %) led to formation of the unexpected bicyclo[2.2.2]octene skeletons 17a and 17b as a 2.9:1 mixture. Under these conditions, BF3·OEt2-promoted elimination of the allylic siloxy group in 7 generated triene 18, which then isomerized into 19 via a 6π-electrocyclic reaction. Diene 19, which appeared to be more reactive than the original diene 7, then underwent the Diels–Alder reaction to provide 17a and 17b.
![[1860-5397-9-74-i3]](/bjoc/content/inline/1860-5397-9-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Generation of 17 through the 6π-electrocyclic reaction and the Diels–Alder reaction.
Scheme 3: Generation of 17 through the 6π-electrocyclic reaction and the Diels–Alder reaction.
Formation of 17a and 17b in Scheme 3 confirmed that selective activation of acrolein in the presence of the Lewis-basic allylic oxygen was a prerequisite to the successful formation of 8. We anticipated that bulky trialkylsilyl triflates would function as such chemoselective Lewis acids, because the activation of the TBS-connected oxygen of 7 by the silyl triflate would cause highly unfavorable steric interactions. Indeed, the cycloaddition between 7 and acrolein proceeded even at −78 °C by the action of TMSOTf (50 mol %) in toluene to afford the bicyclo[3.2.2]nonene 8 and 20 along with a small amount of two other isomers 21ab (Table 1, entry 1). Among the various silyl triflates used (Table 1, entries 1–3), TBSOTf was found to be superior to TMSOTf or TIPSOTf in terms of the combined yield. Alteration of the amount of TBSOTf from 50 mol % (Table 1, entry 2) to 200 mol % (Table 1, entry 4) and change of the solvent from toluene (Table 1, entry 4) to CH2Cl2 (Table 1, entry 5) increased the yield of the adducts. It is particularly worthy of note that the use of 2,6-di-tert-butylpyridine in combination with 200 mol % of TBSOTf effectively inhibited the Lewis-acid-promoted elimination of the C4-oxy group, and that the ratio of 8 to 20 was improved from 1.6:1 to 3.4:1 by replacement of the solvent (Table 1, entries 4 and 5). Thus, we developed an effective method for synthesis of the requisite stereoisomer 8 by applying a TBSOTf-promoted Diels–Alder reaction [25,26]. Most importantly, the C4-stereocenter behaved as the control element to introduce the two quaternary carbons (C1 and 5) and the C12-stereocenter.
Table 1: Optimization of the Diels–Alder reaction.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-74-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | silyl triflate (R) | solvent | yieldb | ratioc (8/20/21ab) |
|---|---|---|---|---|
| 1 | TMS (50 mol %) | toluene | 31% | 1.9:1:0.6 |
| 2 | TBS (50 mol %) | toluene | 59% | 1.8:1:0.5 |
| 3 | TIPS (50 mol %) | toluene | 47% | 1.9:1:0.4 |
| 4d | TBS (200 mol %) | toluene | 69% | 1.6:1:0.4 |
| 5d | TBS (200 mol %) | CH2Cl2 | 88% | 3.4:1:0.5 |
aReaction was performed at 0.5 M. bCombined yield of 8, 20 and two other isomers 21a/21b. cRatio was determined by 1H NMR analysis. d2,6-Di-tert-butylpyridine (200 mol %) was added as a buffer.
The selective formation of 8 out of eight possible isomers is rationalized in Scheme 4. The endo-type transition states would be favored over their exo-type counterparts, and acrolein would approach from the bottom face of 7 to avoid steric interactions with the axially oriented C2- and C4-hydrogen atoms on the top face [27]. These considerations eliminate six out of the eight stereoisomeric transition states, and leave only TS-A and TS-B, which in fact correspond to the generated adducts 8 and 20, respectively. TS-A would be preferred over TS-B due to the unfavorable interaction of the two proximal TBS groups in TS-B, allowing formation of 8 as the major compound.
![[1860-5397-9-74-i4]](/bjoc/content/inline/1860-5397-9-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Rationale of the stereoselectivity of the Diels–Alder reaction.
Scheme 4: Rationale of the stereoselectivity of the Diels–Alder reaction.
Having synthesized the optically active 8, the next task was the preparation of C2-symmetric bicyclo[3.3.2]decene 1 from 8 (Scheme 5). The silyl enol ether formation of aldehyde 8 afforded 22 as a single stereoisomer, and the obtained 22 was oxidized with DMDO to provide α-hydroxy aldehyde 23 as a diastereomeric mixture (dr = 2.8:1). Compound 23 then reacted with benzyl hydroxylamine to produce oxime 24, LiAlH4-treatment of which led to 25. The regioselective ring expansion of seven-membered 25 was induced by treatment with NaNO2 in acetic acid [28,29], resulting in the formation of eight-membered 27 through the intermediary of 26. Finally, the desilylation of 27 with TBAF and the subsequent oxidation of the resultant hydroxy group delivered the symmetric diketone 1 in optically active form.
![[1860-5397-9-74-i5]](/bjoc/content/inline/1860-5397-9-74-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we developed a synthetic route to the optically active seven-membered 7 and established the TBSOTf-promoted stereoselective Diels–Alder reaction between 7 and acrolein to construct highly functionalized bicyclo[3.2.2]nonene 8 bearing two quaternary carbons. Seven additional transformations of 8, including the ring expansion of the seven-membered ring to an eight-membered ring, delivered C2-symmetric bicyclo[3.3.2]decene 1, which is the key intermediate in our synthetic studies of ryanodine.
Experimental
General: All reactions sensitive to air or moisture were carried out under argon or nitrogen atmosphere in dry, freshly distilled solvents under anhydrous conditions, unless otherwise noted. All other reagents were used as supplied unless otherwise stated. Analytical thin-layer chromatography (TLC) was performed by using E. Merck Silica gel 60 F254 precoated plates. Flash column chromatography was performed by using 40–50 µm Silica Gel 60N (Kanto Chemical Co., Inc.), 40–63 µm Silicagel 60 (Merck) or 32–53 µm Silica-gel BW-300 (Fuji Silysia Chemical Ltd.). Melting points were measured on a Yanaco MP-J3 micro melting-point apparatus and are uncorrected. Optical rotations were recorded on a JASCO DIP-1000 Digital Polarimeter. Infrared (IR) spectra were recorded on a JASCO FT/IR-4100 spectrometer. 1H and 13C NMR spectra were recorded on JEOL JNM-ECX-500, JNM-ECA-500, or JNM-ECS-400 spectrometers. Chemical shifts were reported in parts per million (ppm) on the δ scale relative to CHCl3 (δ 7.26 for 1H NMR), CDCl3 (δ 77.0 for 13C NMR), C6D5H (δ 7.16 for 1H NMR), C6D6 (δ 128.0 for 13C NMR), CO(CD3)(CD2H) (δ 2.05 for 1H NMR) as internal references. Signal patterns are indicated as s, singlet; d, doublet; t, triplet; q, quartet, m, multiplet. The numbering of the compounds corresponds to that of the natural products. High-resolution mass spectra were measured on Bruker microTOFII.
TMS-enol ether 13: Methylmagnesium bromide (3.0 M in Et2O, 8.5 mL, 26 mmol) was added to a solution of iPr2NH (3.9 mL, 28 mmol) in Et2O (170 mL) at 0 °C. The mixture was stirred at room temperature for 19 h, and cooled to −78 °C. Then, a solution of 12 [3.5 g as a 5.7:1 mixture of 12 (23 mmol) and Et2O] in Et2O (60 mL), TMSCl (9.1 mL, 72 mmol), Et3N (11.3 mL, 81 mmol), and HMPA (2.0 mL, 11 mmol) were successively added to the mixture. The reaction mixture was stirred at room temperature for 18 h and cooled to 0 °C. Phosphate buffer (pH 7, 100 mL) was added, and the resultant solution was extracted with Et2O (150 mL × 3). The combined organic layers were washed with H2O (30 mL) and brine (200 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (silica gel 200 g, pentane only) to afford silyl enol ether 13 (3.3 g, 16 mmol) and its regioisomer (344 mg, 1.64 mmol) in 70% and 7% yield, respectively. The synthesized silyl enol ether 13 was immediately subjected to the next reaction: colorless oil; 1H NMR (500 MHz, C6D6) δ 0.17 (9H, s, CH3 of TMS), 1.61 (3H, d, J = 1.2 Hz, H17), 1.73 (3H, s, H20), 2.05 (2H, t, J = 6.3 Hz, H2ab), 2.31–2.35 (2H, m, H3ab), 2.60 (2H, d, J = 5.7 Hz, H14ab), 5.53 (1H, tq, J = 5.7, 1.2 Hz, H15); 13C NMR (125 MHz, C6D6) δ 1.2, 19.3, 25.8, 30.9, 31.7, 32.8, 114.7, 123.3, 137.4, 146.4. The regioisomer of 13: colorless oil; 1H NMR (400 MHz, C6D6) δ 0.18 (9H, s, CH3 of TMS), 1.19 (3H, d, J = 6.9 Hz, H20), 1.67 (3H, br s, H17), 2.00 (1H, ddd, J = 14.6, 7.3, 7.3 Hz, H14a), 2.23–2.27 (1H, m, H14b), 2.32–2.40 (1H, m, H5), 2.46 (1H, dd, J = 17.9, 6.4 Hz, H2a), 2.62 (1H, dd, J = 17.9, 5.5 Hz, H2b), 4.95 (1H, dd, J = 6.4, 5.5 Hz, H3), 5.49 (1H, br ddq, J = 7.3, 7.3, 1.4 Hz, H15).
Diene 14: DDQ (7.1 g, 31 mmol) was added to a solution of 13 (3.3 g 16 mmol) and 2,6-lutidine (5.4 mL, 46 mmol) in benzene (30 mL) at 0 °C. The reaction mixture was stirred at room temperature for 20 min and filtered through a short column of Al2O3 with Et2O. The filtrate was concentrated. The residue was purified by flash column chromatography (silica gel 120 g, pentane/Et2O 10:1 to 5:1) to afford diene 14 (1.5 g, 11 mmol) in 69% yield: yellow oil; IR (neat) νmax: 2949, 1656, 1595, 1431, 1377 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.92 (3H, s, H20), 1.98 (3H, s, H17), 2.29 (2H, t, J = 6.3 Hz, H2ab), 2.64 (2H, t, J = 6.3 Hz, H3ab), 5.80 (1H, d, J = 8.0 Hz, H15), 6.51 (1H, d, J = 8.0 Hz, H14); 13C NMR (100 MHz, CDCl3) δ 20.4, 26.5, 28.1, 41.7, 122.4, 136.0, 137.0, 150.0, 201.3; HRMS–ESI (m/z): [M + Na]+ calcd for C9H12ONa, 159.0780; found, 159.0778.
Diene 15: BH3·SMe2 (220 μL, 2.3 mmol) was added to a solution of (R)-2-Me-CBS-oxazaborolidine (1.0 M in toluene, 2.3 mL, 2.3 mmol) in THF (6 mL) at 0 °C. The solution was stirred for 30 min at 0 °C and cooled to −78 °C. Then a solution of diene 14 (265 mg, 1.94 mmol) in THF (3 mL) was added at −78 °C. The reaction mixture was warmed to 0 °C over 15 min, and H2O (10 mL) was added. The resultant solution was extracted with Et2O (6 mL × 3). The combined organic layers were washed with brine (5 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (silica gel 5 g, pentane/Et2O 20:1 to 5:1) to afford 15 (261 mg, 1.89 mmol) in 97% yield. The enantiopurity of 15 was determined as 82% ee by comparison of the integrations of the H14 peak at 5.78 and 5.72 ppm on 1H NMR after the esterification with (S)-MTPACl; yellow oil; [α]D20 −402 (c 0.995, CHCl3); IR (neat) νmax: 3336, 2965, 2920, 2880, 1434, 1056, 1013 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.54 (1H, br s, OH), 1.75 (1H, dddd, J = 13.7, 11.9, 2.8, 2.8 Hz, H3a), 1.85 (3H, s, H17 or 20), 1.95 (3H, s, H17 or 20), 2.00–2.15 (2H, m, H2a and 3b), 2.32–2.39 (1H, br dd, J = 16.5, 11.9 Hz, H2b), 4.20 (1H, m, H4), 5.52 (1H, br d, J = 7.8 Hz, H14 or 15), 5.61 (1H, d, J = 7.8 Hz, H14 or 15); 13C NMR (125 MHz, C6D6) δ 24.0, 26.6, 29.2, 33.2, 72.4, 120.7, 122.6, 140.2, 142.9; HRMS–ESI (m/z): [M + Na]+ calcd for C9H14ONa, 161.0937; found, 161.0934.
Diene 7: The mixture of 15 (259 mg, 1.88 mmol), imidazole (300 mg, 4.41 mmol) and TBSCl (330 mg, 2.19 mmol) in DMF (6.0 mL) was stirred for 8.5 h at room temperature and cooled to 0 °C. H2O (10 mL) was added to the reaction mixture, and the resultant solution was stirred for 30 min. The mixture was extracted with EtOAc (6 mL × 3). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (silica gel 5 g, hexane/EtOAc 1:0 to 100:1) to afford 7 (404 mg, 1.60 mmol) in 85% yield; colorless oil; [α]D20 −247 (c 1.04, CHCl3); IR (neat) νmax: 2956, 2928, 2856, 1472, 1436, 1253 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.085 (3H, s, CH3 of TBS), 0.093 (3H, s, CH3 of TBS), 0.91 (9H, s, t-Bu of TBS), 1.75 (1H, dddd, J = 13.3, 10.5, 2.3, 2.3 Hz, H3a), 1.83 (3H, s, H17), 1.85 (3H, s, H20), 1.92–2.00 (1H, dddd, J = 13.3, 8.7, 6.4, 2.3 Hz, H3b), 2.04 (1H, ddd, J = 16.5, 8.7, 2.3 Hz, H2a), 2.40 (1H, br dd, J = 16.5, 10.5 Hz, H2b), 4.22 (1H, br d, J = 6.4 Hz, H4), 5.50 (1H, d, J = 7.8 Hz, H15), 5.54, (1H, d, J = 7.8 Hz, H14); 13C NMR (100 MHz, CDCl3) δ −4.3, −4.1, 18.5, 24.0, 26.2, 26.9, 30.0, 34.9, 73.4, 120.5, 121.5, 141.4, 143.2; HRMS–ESI (m/z): [M + Na]+ calcd for C15H28OSiNa, 275.1802; found, 275.1801.
Cycloadduct 8 and 20: TBSOTf (530 μL, 2.3 mmol) was added to a solution of 7 (297 mg, 1.18 mmol), acrolein (390 μL, 5.8 mmol) and 2,6-di-tert-butylpyridine (530 μL, 2.3 mmol) in CH2Cl2 (2.4 mL) at −78 °C. The reaction mixture was stirred for 15 min at −78 °C, and then saturated aqueous NaHCO3 (3 mL) was added. The resultant mixture was extracted with EtOAc (5 mL × 3). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (silica gel 12 g, hexane/CH2Cl2 4:1 to 3:1) to afford pure 20 (23 mg, 75 μmol), a mixture of 20, 21a, 21b and 8 (215 mg, 0.67 mmol), and pure 8 (87 mg, 0.29 mmol) in 88% combined yield. 8: colorless oil; [α]D20 +25.6 (c 1.13, CHCl3); IR (neat) νmax: 2954, 2930, 1724, 1253, 1070 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.02 (6H, s, CH3 of TBS × 2), 0.88 (9H, s, t-Bu of TBS), 1.02 (3H, s, H17 or 20), 1.03 (3H, s, H17 or 20), 1.21 (1H, dd, J = 14.6, 4.1 Hz, H6a), 1.42 (1H, ddd, J = 13.7, 5.5, 5.0 Hz, H2a), 1.56 (1H, ddd, J = 13.7, 11.5, 5.0 Hz, H2b), 1.72 (1H, dddd, J = 14.6, 11.5, 9.2, 5.5 Hz, H3a), 1.84 (1H, dddd, J = 14.6, 5.0, 5.0, 5.0 Hz, H3b), 2.34 (1H, dd, J = 14.6, 9.6 Hz, H6b), 2.57 (1H, ddd, J = 9.6, 5.5, 4.1 Hz, H12), 3.38 (1H, dd, J = 9.2, 5.0 Hz, H4), 5.76 (1H, d, J = 9.2 Hz, H14), 5.81 (1H, d, J = 9.2 Hz, H15), 9.29 (1H, d, J = 5.5 Hz, CHO); 13C NMR (100 MHz, CDCl3) δ −4.8, −4.2, 18.1, 25.9, 26.7, 27.0, 29.2, 34.5, 35.3, 40.2, 40.4, 56.5, 71.7, 138.1, 138.3, 204.4; HRMS–ESI (m/z): [M + Na]+ calcd for C18H32O2SiNa, 331.2064; found, 331.2063. 20: IR (neat) νmax: 2954, 2930, 2856, 1724, 1253, 1070 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.02 (6H, s, CH3 of TBS × 2), 0.88 (9H, s, t-Bu of TBS), 1.02 (3H, s, CH3CCHCHO), 1.06 (3H, s, CH3CCH2), 1.34 (1H, ddd, J = 13.7, 6.0, 3.7 Hz, CCHAHBCH2), 1.52–1.59 (2H, m, CCHAHBCH2 and CCHAHBCHCHO), 1.70–1.87 (2H, m, CCH2CH2CH(OTBS)), 1.91 (1H, dd, J = 14.7, 10.1 Hz, CCHAHBCHCHO), 2.97 (1H, ddd, J = 10.1, 5.0, 5.0 Hz, CHCHO), 3.32 (1H, dd, J = 9.2, 5.5 Hz, CH(OTBS)), 5.55 (1H, d, J = 9.2 Hz, CCH=CHCCH2), 5.97 (1H, d, J = 9.2 Hz, CCH=CHCCH2), 9.37 (1H, d, J = 5.0 Hz, CHO); 13C NMR (125 MHz, CDCl3) δ −4.8, −4.2, 18.1, 24.1, 25.9, 29.7, 33.8, 34.1, 34.6, 38.0, 41.8, 50.6, 73.5, 134.1, 142.4, 204.3; HRMS–ESI (m/z): [M + Na]+ calcd for C18H32O2SiNa, 331.2064; found, 331.2057.
Oxime 24: TMSOTf (220 μL, 1.2 mmol) was added to a solution of 8 (122 mg, 0.395 mmol) and Et3N (330 μL, 2.4 mmol) in CH2Cl2 (2.0 mL) at 0 °C. The reaction mixture was stirred at room temperature for 15 h and cooled to 0 °C. Phosphate buffer (pH 7, 5 mL) was added, and the resultant mixture was extracted with EtOAc (6 mL × 3). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was passed through a short column (silica gel 100 mg, hexane/EtOAc 2:1) to afford the crude TMS enol ether 22, which was used in the next reaction without further purification. DMDO (58 mM in acetone, 6.8 mL, 0.39 mmol) was added to a solution of the above crude TMS enol ether 22 in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 10 min at 0 °C, and then isoprene (39 μL, 0.39 mmol) was added. The resultant solution was concentrated. The residue was roughly purified by flash column chromatography (silica gel 24 g, hexane/EtOAc 1:0 to 10:1) to afford hydroxy aldehyde 23 (93 mg) as a diastereomixture (2.8:1), which was used in the next reaction without further purification. A mixture of the above crude 23, BnONH2·HCl (135 mg, 0.846 mmol), and MS4Å (93 mg) in THF (3.0 mL) was stirred at room temperature for 22 h. The reaction mixture was filtered through a pad of Celite with EtOAc (15 mL), and the filtrate was concentrated. The residue was purified by flash column chromatography (silica gel 15g, hexane/toluene 2:1 to 0:1 then hexane/CH2Cl2 1:1 to 1:3) to afford oxime 24a (81 mg, 0.19 mmol) and 24b (29 mg, 67 μmol) in 48% and 17% yield, respectively, over three steps. 24a: colorless oil: [α]D20 +60 (c 0.73, CHCl3); IR (neat) νmax: 3522, 2954, 2929, 2856, 1455, 1363, 1254, 1067 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.006 (3H, s, CH3 of TBS), 0.012 (3H, s, CH3 of TBS), 0.88 (9H, s, t-Bu of TBS), 0.98 (3H, s, H17 or 20), 1.02 (3H, s, H17 or 20), 1.33 (1H, d, J = 16.1 Hz, H6a), 1.36–1.57 (3H, m, H2ab and 3a), 1.85 (1H, dddd, J = 14.9, 5.8, 5.8, 5.8 Hz, H3b), 2.81 (1H, d, J = 16.1 Hz, H6b), 2.94 (1H, s, OH), 3.40 (1H, dd, J = 6.3, 5.8 Hz, H4), 5.13 (2H, s, OCH2Ph), 5.73 (1H, d, J = 9.2 Hz, H14 or 15), 5.79 (1H, d, J = 9.2 Hz, H14 or 15), 7.30–7.39 (5H, m, aromatic), 7.71 (1H, s, CHNOBn); 13C NMR (125 MHz, CDCl3) δ −4.8, −4.3, 18.1, 23.3, 25.9, 27.0, 32.6, 33.9, 39.7, 42.6, 42.9, 71.0, 76.3, 78.0, 128.0, 128.42, 128.44, 137.1, 137.3, 138.4, 152.8; HRMS–ESI (m/z): [M + Na]+ calcd for C25H39NO3SiNa, 452.2591; found, 452.2589. 24b: IR (neat) νmax: 3525, 3025, 2955, 2928, 2856 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.01 (3H, s, CH3 of TBS), 0.02 (3H, s, CH3 of TBS), 0.83 (3H, s, H17), 0.88 (9H, s, t-Bu of TBS), 0.95 (3H, s, H20), 1.33–1.42 (1H, m, H2a), 1.50 (1H, d, J = 15.1 Hz, H6a), 1.80–1.88 (2H, m, H2b and 3a), 1.96–2.07 (1H, m, H3b), 2.44 (1H, d, J = 15.1 Hz, H6b), 3.42 (1H, dd, J = 8.2, 6.0 Hz, H4), 3.48 (1H, s, OH), 5.06 (2H, s, OCH2Ph), 5.62 (1H, d, J = 9.6 Hz, H14), 5.65 (1H, d, J = 9.6 Hz, H15), 7.27–7.35 (5H, m, aromatic), 7.39 (1H, s, CHNOBn); 13C NMR (100 MHz, CDCl3) δ −4.8, −4.1, 18.1, 23.5, 25.9, 26.7, 34.3, 34.9, 41.4, 41.5, 42.3, 71.6, 76.2, 78.1, 127.9, 128.3, 128.4, 137.2, 137.6, 138.3, 156.2; HRMS–ESI (m/z): [M + Na]+ calcd for C25H39NO3SiNa, 452.2591; found, 452.2580.
Ketone 27: LiAlH4 (2.0 M in THF, 380 μL, 0.76 mmol) was added to a solution of oxime 24 (110 mg, 0.256 mmol, a 2.8:1 diastereomixture of 24a and 24b) in THF at 0 °C. The reaction mixture was stirred at 0 °C for 10 min, at room temperature for 2.5 h, and at 40 °C for 2 h. After additional LiAlH4 (2.0 M in THF, 380 μL, 0.76 mmol) was added, the reaction mixture was stirred at 50 °C for a further 3.5 h. LiAlH4 (2.0 M in THF, 380 μL, 0.76 mmol) was added again, and the reaction mixture was stirred for a further 1 h. After the reaction mixture was cooled to 0 °C, H2O (87 μL) was added. The resultant solution was stirred for 1 h at room temperature, and then 15% aqueous NaOH (87 μL) and H2O (260 μL) were added. The solution was stirred for 9 h. The resultant mixture was filtered through a pad of Celite with THF (30 mL), and the filtrate was concentrated to afford amino alcohol 25, which was used in the next reaction without further purification. A solution of NaNO2 (141 mg, 2.04 mmol) in H2O (1.3 mL) was added to a solution of the above crude amino alcohol 25 in H2O (3.8 mL) and AcOH (1.0 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 h, and then saturated aqueous NaHCO3 (30 mL) was added. The resultant mixture was extracted with EtOAc (8 mL × 4). The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (silica gel 18 g, hexane/CH2Cl2 3:1 to 0:1) to afford ketone 27 (20 mg, 65 μmol) in 25% yield over two steps: colorless oil; [α]D20 +134 (c 0.165, CHCl3); IR (neat) νmax: 2954, 2929, 2856, 1703, 1462, 1063 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.01 (6H, s, CH3 of TBS × 2), 0.86 (9H, s, t-Bu of TBS), 1.11 (3H, s, H17 or 20), 1.12 (3H, s, H17 or 20), 1.41 (1H, ddd, J = 14.9, 8.1, 2.3 Hz, H6a), 1.52–1.72 (3H, m, H2ab and 3a), 1.75–1.81 (1H, m, H3b), 2.25 (1H, ddd, J = 12.6, 5.2, 2.3 Hz, H11a), 2.34 (1H, ddd, J = 14.9, 12.6, 5.2 Hz, H6b), 2.93 (1H, ddd, J = 12.6, 12.6, 8.1 Hz, H11b), 3.40 (1H, dd, J = 10.3, 4.6 Hz, H4), 5.56 (1H, d, J = 10.9 Hz, H14), 5.73 (1H, d, J = 10.9 Hz, H15); 13C NMR (125 MHz, CDCl3) δ −4.7, −4.2, 18.0, 25.9, 26.4, 29.0, 31.2, 34.6, 40.0, 42.2, 42.4, 50.3, 73.7, 137.6, 139.4, 218.5; HRMS–ESI (m/z): [M + Na]+ calcd for C18H32O2SiNa, 331.2064; found, 331.2078.
C2-symmetric diketone 1: TBAF (1.0 M in THF, 130 μL, 0.13 mmol) was added to a solution of 27 (13 mg, 42 μmol) in THF (0.9 mL) at room temperature. The reaction mixture was stirred for 1.5 h at room temperature and at 40 °C for 1.5 h. TBAF (1.0 M in THF, 83 μL, 83 μmol) was added again to the reaction mixture, and the reaction mixture was stirred at 40 °C for a further 16.5 h. The reaction mixture was cooled to room temperature, and passed through a short column of silica gel with Et2O. The filtrate was concentrated to afford crude alcohol, which was used in the next reaction without further purification. A mixture of the above crude alcohol, NaHCO3 (33 mg, 0.39 mmol), and Dess–Martin periodinane (53 mg, 0.13 mmol) in CH2Cl2 (0.8 mL) was stirred at room temperature for 2.5 h, and then saturated aqueous Na2S2O3 (3 mL) was added. The resultant mixture was extracted with Et2O (4 mL × 3). The combined organic layers were washed with brine (5 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (silica gel 1 g, pentane/Et2O 5:1 to 2:1) to afford C2-symmetric diketone 1 (7.5 mg, 39 μmol) in 93% yield over two steps: crystal; m.p. 55 °C; [α]D20 +260 (c 0.37, CHCl3). Other analytical data of 1 were identical to those reported by our group previously [11].
Supporting Information
| Supporting Information File 1: Experimental procedures and NMR spectra of all newly synthesized compounds. | ||
| Format: PDF | Size: 1.5 MB | Download |
References
-
Rogers, E. F.; Koniuszy, F. R.; Shavel, J., Jr.; Folkers, K. J. Am. Chem. Soc. 1948, 70, 3086–3088. doi:10.1021/ja01189a074
Return to citation in text: [1] -
Wiesner, K.; Valenta, Z.; Findlay, J. A. Tetrahedron Lett. 1967, 8, 221–223. doi:10.1016/S0040-4039(00)90521-5
Return to citation in text: [1] -
Srivastava, S. N.; Przybylska, M. Can. J. Chem. 1968, 46, 795–797. doi:10.1139/v68-133
Return to citation in text: [1] -
Betzenhauser, M. J.; Marks, A. R. Pfluegers Arch. 2010, 460, 467–480. doi:10.1007/s00424-010-0794-4
Return to citation in text: [1] -
Mackrill, J. J. Biochem. Pharmacol. 2010, 79, 1535–1543. doi:10.1016/j.bcp.2010.01.014
Return to citation in text: [1] -
Bélanger, A.; Berney, D. J. F.; Borschberg, H.-J.; Brousseau, R.; Doutheau, A.; Durand, R.; Katayama, H.; Lapalme, R.; Leturc, D. M.; Liao, C.-C.; MacLachlan, F. N.; Maffrand, J.-P.; Marazza, F.; Martino, R.; Moreau, C.; Saint-Laurent, L.; Saintonge, R.; Soucy, P.; Ruest, L.; Deslongchamps, P. Can. J. Chem. 1979, 57, 3348–3354. doi:10.1139/v79-547
Return to citation in text: [1] -
Deslongchamps, P.; Bélanger, A.; Berney, D. J. F.; Borschberg, H.-J.; Brousseau, R.; Doutheau, A.; Durand, R.; Katayama, H.; Lapalme, R.; Leturc, D. M.; Liao, C.-C.; MacLachlan, F. N.; Maffrand, J.-P.; Marazza, F.; Martino, R.; Moreau, C.; Ruest, L.; Saint-Laurent, L.; Saintonge, R.; Soucy, P. Can. J. Chem. 1990, 68, 186–192. doi:10.1139/v90-024
Return to citation in text: [1] -
Sieburth, S. M.; Santos, E. D. Tetrahedron Lett. 1994, 35, 8127–8130. doi:10.1016/0040-4039(94)88261-4
Return to citation in text: [1] -
Wood, J. L.; Graeber, J. K.; Njardarson, J. T. Tetrahedron 2003, 59, 8855–8858. doi:10.1016/j.tet.2003.05.001
Return to citation in text: [1] -
Urabe, D.; Nagatomo, M.; Hagiwara, K.; Masuda, K.; Inoue, M. Chem. Sci. 2013, 4, 1615–1619. doi:10.1039/c3sc00023k
Return to citation in text: [1] -
Hagiwara, K.; Himuro, M.; Hirama, M.; Inoue, M. Tetrahedron Lett. 2009, 50, 1035–1037. doi:10.1016/j.tetlet.2008.12.054
Return to citation in text: [1] [2] [3] -
Iwatsu, M.; Urabe, D.; Inoue, M. Heterocycles 2010, 82, 491–504. doi:10.3987/COM-10-S(E)22
Return to citation in text: [1] -
Spahić, B.; Thu, T. T. M.; Schlosser, M. Helv. Chim. Acta 1980, 63, 1236–1241. doi:10.1002/hlca.19800630515
Return to citation in text: [1] -
Guo, M.; Minuti, L.; Taticchi, A.; Wenkert, E. J. Org. Chem. 1990, 55, 1366–1368. doi:10.1021/jo00291a054
Return to citation in text: [1] -
Barbot, F.; Mesnard, D.; Miginiac, L. Org. Prep. Proced. Int. 1978, 10, 261–266. doi:10.1080/00304947809355047
Return to citation in text: [1] -
Cane, D. E.; Thomas, P. J. J. Am. Chem. Soc. 1984, 106, 5295–5303. doi:10.1021/ja00330a044
Return to citation in text: [1] -
Demole, E.; Enggist, P.; Borer, M. C. Helv. Chim. Acta 1971, 54, 1845–1864. doi:10.1002/hlca.19710540712
Return to citation in text: [1] -
Marcos, I. S.; Oliva, I. M.; Moro, R. F.; Díez, D.; Urones, J. G. Tetrahedron 1994, 50, 12655–12672. doi:10.1016/S0040-4020(01)89399-6
Return to citation in text: [1] -
González, A. G.; Darias, J.; Martin, J. D.; Melián, M. A. Tetrahedron Lett. 1978, 19, 481–482. doi:10.1016/S0040-4039(01)91461-3
Return to citation in text: [1] -
Brown, H. L.; Buchanan, G. L.; O’Donnell, J. J. Chem. Soc., Perkin Trans. 1 1979, 1740–1742. doi:10.1039/P19790001740
Return to citation in text: [1] -
Nakashima, K.; Inoue, K.; Sono, M.; Tori, M. J. Org. Chem. 2002, 67, 6034–6040. doi:10.1021/jo020287d
Return to citation in text: [1] -
Krafft, M. E.; Holton, R. A. Tetrahedron Lett. 1983, 24, 1345–1348. doi:10.1016/S0040-4039(00)81652-4
Return to citation in text: [1] -
Corey, E. J.; Shibata, S.; Bakshi, R. K. J. Org. Chem. 1988, 53, 2861–2863. doi:10.1021/jo00247a044
Return to citation in text: [1] -
Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006
Return to citation in text: [1] -
Lamy-Schelkens, H.; Giomi, D.; Ghosez, L. Tetrahedron Lett. 1989, 30, 5887–5890. doi:10.1016/S0040-4039(01)93497-5
Return to citation in text: [1] -
Lamy-Schelkens, D.; Ghosez, L. Tetrahedron Lett. 1989, 30, 5891–5894. doi:10.1016/S0040-4039(01)93498-7
Return to citation in text: [1] -
Hua, Z.; Chen, L.; Mei, Y.; Jin, Z. Tetrahedron Lett. 2009, 50, 6621–6623. doi:10.1016/j.tetlet.2009.09.055
Return to citation in text: [1] -
Greenhouse, R.; Borden, W. T.; Ravindranathan, T.; Hirotsu, K.; Clardy, J. J. Am. Chem. Soc. 1977, 99, 6955–6961. doi:10.1021/ja00463a030
Return to citation in text: [1] -
Krow, G. R. Tetrahedron 1987, 43, 3–38. doi:10.1016/S0040-4020(01)89927-0
Return to citation in text: [1]
| 28. | Greenhouse, R.; Borden, W. T.; Ravindranathan, T.; Hirotsu, K.; Clardy, J. J. Am. Chem. Soc. 1977, 99, 6955–6961. doi:10.1021/ja00463a030 |
| 29. | Krow, G. R. Tetrahedron 1987, 43, 3–38. doi:10.1016/S0040-4020(01)89927-0 |
| 11. | Hagiwara, K.; Himuro, M.; Hirama, M.; Inoue, M. Tetrahedron Lett. 2009, 50, 1035–1037. doi:10.1016/j.tetlet.2008.12.054 |
| 1. | Rogers, E. F.; Koniuszy, F. R.; Shavel, J., Jr.; Folkers, K. J. Am. Chem. Soc. 1948, 70, 3086–3088. doi:10.1021/ja01189a074 |
| 2. | Wiesner, K.; Valenta, Z.; Findlay, J. A. Tetrahedron Lett. 1967, 8, 221–223. doi:10.1016/S0040-4039(00)90521-5 |
| 3. | Srivastava, S. N.; Przybylska, M. Can. J. Chem. 1968, 46, 795–797. doi:10.1139/v68-133 |
| 11. | Hagiwara, K.; Himuro, M.; Hirama, M.; Inoue, M. Tetrahedron Lett. 2009, 50, 1035–1037. doi:10.1016/j.tetlet.2008.12.054 |
| 25. | Lamy-Schelkens, H.; Giomi, D.; Ghosez, L. Tetrahedron Lett. 1989, 30, 5887–5890. doi:10.1016/S0040-4039(01)93497-5 |
| 26. | Lamy-Schelkens, D.; Ghosez, L. Tetrahedron Lett. 1989, 30, 5891–5894. doi:10.1016/S0040-4039(01)93498-7 |
| 10. | Urabe, D.; Nagatomo, M.; Hagiwara, K.; Masuda, K.; Inoue, M. Chem. Sci. 2013, 4, 1615–1619. doi:10.1039/c3sc00023k |
| 27. | Hua, Z.; Chen, L.; Mei, Y.; Jin, Z. Tetrahedron Lett. 2009, 50, 6621–6623. doi:10.1016/j.tetlet.2009.09.055 |
| 6. | Bélanger, A.; Berney, D. J. F.; Borschberg, H.-J.; Brousseau, R.; Doutheau, A.; Durand, R.; Katayama, H.; Lapalme, R.; Leturc, D. M.; Liao, C.-C.; MacLachlan, F. N.; Maffrand, J.-P.; Marazza, F.; Martino, R.; Moreau, C.; Saint-Laurent, L.; Saintonge, R.; Soucy, P.; Ruest, L.; Deslongchamps, P. Can. J. Chem. 1979, 57, 3348–3354. doi:10.1139/v79-547 |
| 7. | Deslongchamps, P.; Bélanger, A.; Berney, D. J. F.; Borschberg, H.-J.; Brousseau, R.; Doutheau, A.; Durand, R.; Katayama, H.; Lapalme, R.; Leturc, D. M.; Liao, C.-C.; MacLachlan, F. N.; Maffrand, J.-P.; Marazza, F.; Martino, R.; Moreau, C.; Ruest, L.; Saint-Laurent, L.; Saintonge, R.; Soucy, P. Can. J. Chem. 1990, 68, 186–192. doi:10.1139/v90-024 |
| 8. | Sieburth, S. M.; Santos, E. D. Tetrahedron Lett. 1994, 35, 8127–8130. doi:10.1016/0040-4039(94)88261-4 |
| 9. | Wood, J. L.; Graeber, J. K.; Njardarson, J. T. Tetrahedron 2003, 59, 8855–8858. doi:10.1016/j.tet.2003.05.001 |
| 23. | Corey, E. J.; Shibata, S.; Bakshi, R. K. J. Org. Chem. 1988, 53, 2861–2863. doi:10.1021/jo00247a044 |
| 4. | Betzenhauser, M. J.; Marks, A. R. Pfluegers Arch. 2010, 460, 467–480. doi:10.1007/s00424-010-0794-4 |
| 5. | Mackrill, J. J. Biochem. Pharmacol. 2010, 79, 1535–1543. doi:10.1016/j.bcp.2010.01.014 |
| 24. | Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006 |
| 15. | Barbot, F.; Mesnard, D.; Miginiac, L. Org. Prep. Proced. Int. 1978, 10, 261–266. doi:10.1080/00304947809355047 |
| 17. | Demole, E.; Enggist, P.; Borer, M. C. Helv. Chim. Acta 1971, 54, 1845–1864. doi:10.1002/hlca.19710540712 |
| 18. | Marcos, I. S.; Oliva, I. M.; Moro, R. F.; Díez, D.; Urones, J. G. Tetrahedron 1994, 50, 12655–12672. doi:10.1016/S0040-4020(01)89399-6 |
| 19. | González, A. G.; Darias, J.; Martin, J. D.; Melián, M. A. Tetrahedron Lett. 1978, 19, 481–482. doi:10.1016/S0040-4039(01)91461-3 |
| 20. | Brown, H. L.; Buchanan, G. L.; O’Donnell, J. J. Chem. Soc., Perkin Trans. 1 1979, 1740–1742. doi:10.1039/P19790001740 |
| 21. | Nakashima, K.; Inoue, K.; Sono, M.; Tori, M. J. Org. Chem. 2002, 67, 6034–6040. doi:10.1021/jo020287d |
| 13. | Spahić, B.; Thu, T. T. M.; Schlosser, M. Helv. Chim. Acta 1980, 63, 1236–1241. doi:10.1002/hlca.19800630515 |
| 14. | Guo, M.; Minuti, L.; Taticchi, A.; Wenkert, E. J. Org. Chem. 1990, 55, 1366–1368. doi:10.1021/jo00291a054 |
| 22. | Krafft, M. E.; Holton, R. A. Tetrahedron Lett. 1983, 24, 1345–1348. doi:10.1016/S0040-4039(00)81652-4 |
| 12. | Iwatsu, M.; Urabe, D.; Inoue, M. Heterocycles 2010, 82, 491–504. doi:10.3987/COM-10-S(E)22 |
| 11. | Hagiwara, K.; Himuro, M.; Hirama, M.; Inoue, M. Tetrahedron Lett. 2009, 50, 1035–1037. doi:10.1016/j.tetlet.2008.12.054 |
| 16. | Cane, D. E.; Thomas, P. J. J. Am. Chem. Soc. 1984, 106, 5295–5303. doi:10.1021/ja00330a044 |
© 2013 Tabuchi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)