Search results
Search for "dynamics" in Full Text gives 247 result(s) in Beilstein Journal of Organic Chemistry. Showing first 200.
A three-armed cryptand with triazine and pyridine units: synthesis, structure and complexation with polycyclic aromatic compounds
Beilstein J. Org. Chem. 2018, 14, 1370–1377, doi:10.3762/bjoc.14.115

- another major impediment to form full complexation, i.e., the guest molecule enters entirely inside the cryptand’s cavity, is the fitting dynamics. This process might have a strong entropic character and the energetic contribution can also give valuable information about the efficiency of the fitting
- . Accordingly, a further constrained geometry optimization was performed for the cryptand–anthracene case, where the distance between the 3,5-dicyanopyridine fragment of 2 and the carbon atom of the anthracene was kept constant for each optimization case. The inclusion dynamics of the anthracene in the cavity
- the anthracene depending on the constrained distance are: −6.00 kcal/mol (blue), −3.74 kcal/mol (green), −2.63 kcal/mol (red) and −0.76 kcal/mol (grey). This decrease of the binding energy tells us that even for the anthracene the fitting dynamics is not a straightforward process. Conclusion The 1,3,5









An overview of recent advances in duplex DNA recognition by small molecules
Beilstein J. Org. Chem. 2018, 14, 1051–1086, doi:10.3762/bjoc.14.93
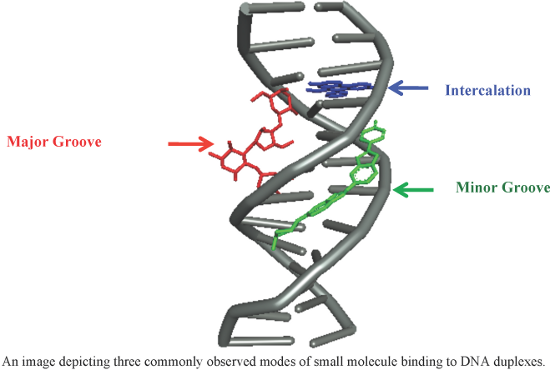
- binding affinity using sophisticated molecular dynamics approach with eight different nucleotides having variety of sequences. It has been observed that QUE appears to be a minor groove binder, whereas FLP involves in combined mode of interaction such as minor groove binding and intercalation. A set of






















Crystal structure of the inclusion complex of cholesterol in β-cyclodextrin and molecular dynamics studies
Beilstein J. Org. Chem. 2018, 14, 838–848, doi:10.3762/bjoc.14.69

- inclusion compound remains very stable in aqueous solution at both temperatures. Keywords: beta-cyclodextrin; cholesterol; crystal structure; molecular dynamics; Introduction Cholesterol ((3β)-cholest-5-en-3-ol, CHL, Figure 1a) is a polycyclic steroid that is synthesized in mammalian cells and has a
- supported by the above mentioned guest–host interactions. Therefore, cholesterol is always found in the cavity of the β-CD dimer, its size and shape prohibiting its diffusion in the crystal. Molecular dynamics The crystallographically determined atomic coordinates of the CHL/β-CD complex (host/guest
- stoichiometry 2:1) (Figure 2a) were subjected to equilibration and subsequent molecular dynamics simulations at both 300 and 340 K in explicit water solvent for almost 12 ns with the aim to monitor the dynamic behavior of CHL in β-CD in two different temperatures, study the host–guest interactions during the






Fluorogenic PNA probes
Beilstein J. Org. Chem. 2018, 14, 253–281, doi:10.3762/bjoc.14.17

- dT in the DNA strand with comparable affinity to that for A, and the base pairing resulted in fluorescence quenching. While the quenching of 2-AP can be useful for probing the structure and dynamics of the PNA and its DNA/RNA hybrids, the quenching effect is small and not highly specific as quenching
























Fluorescent nucleobase analogues for base–base FRET in nucleic acids: synthesis, photophysics and applications
Beilstein J. Org. Chem. 2018, 14, 114–129, doi:10.3762/bjoc.14.7

- acid bases inside the base-stack, base analogue donor and acceptor molecules complement external fluorophores like the Cy-, Alexa- and ATTO-dyes and enable detailed investigations of structure and dynamics of nucleic acid containing systems. The first base–base FRET pair, tCO–tCnitro, has recently been
- . Base–base FRET has been around for less than a decade, only in 2017 expanded beyond one FRET pair, and represents a highly promising structure and dynamics methodology for the field of nucleic acids. Here we bring up its advantages as well as disadvantages and touch upon potential future applications
- . Keywords: B-to-Z-DNA transition; fluorescent base analogues; FRET; netropsin; nucleic acid structure and dynamics; quadracyclic adenines; tricyclic cytosines; Z-DNA; Review Introduction The importance of nucleic acid structure and dynamics in the understanding of vital processes in living organisms has

















Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids – tutorial
Beilstein J. Org. Chem. 2018, 14, 84–105, doi:10.3762/bjoc.14.5
















Aminosugar-based immunomodulator lipid A: synthetic approaches
Beilstein J. Org. Chem. 2018, 14, 25–53, doi:10.3762/bjoc.14.3

- synthetic lipid A derivative had a bias towards MyD88- or TRIF-dependent immune responses [78]. 1.3. Synthesis of fluorescent-labeled lipid A analogues For studying the structural basis and the dynamics of TLR4-lipid A interplay, the application of labeled synthetic lipid A derivatives as versatile probes


















The use of 4,4,4-trifluorothreonine to stabilize extended peptide structures and mimic β-strands
Beilstein J. Org. Chem. 2017, 13, 2842–2853, doi:10.3762/bjoc.13.276

- dynamics simulations. CF3-threonine containing pentapeptides are more prone to mimic β-strands than their natural Ser and Thr pentapeptide analogues. The proof of concept that these fluorinated β-strand mimics are able to disrupt protein–protein interactions involving β-sheet structures is provided. The
- according to their central fluorinated or non-fluorinated residue, all-atom molecular dynamics (MD) simulations were performed using the GROMACS 4.5 package, with the OPLS-AA force field in combination with the SPC/E water model (for a complete description of the method, see Supporting Information File 1
- obtained through the aldol reaction of trifluoroacetaldehyde with the Ni(II) complex of the chiral Schiff base of glycine. The conformational analysis of these pentapeptides was conducted by the combined use of NMR spectroscopy and molecular dynamics simulations. NMR conformational studies showed that the








Binding abilities of polyaminocyclodextrins: polarimetric investigations and biological assays
Beilstein J. Org. Chem. 2017, 13, 2751–2763, doi:10.3762/bjoc.13.271

- occurrence of a significant interaction between the cationic polyamine pendant chains and the counter-anions of the buffer, probably by either ion pairing or formation of multiple hydrogen bonds. Consequently, the mobility of the chains and, in turn, the conformational dynamics of the entire CD scaffold, are









What contributes to an effective mannose recognition domain?
Beilstein J. Org. Chem. 2017, 13, 2584–2595, doi:10.3762/bjoc.13.255

- of F features the aromatic His189, that can engage in CH–π interactions, associated with contributions to the binding affinity in the range of 0–6.3 kJ/mol [55][56]. Analysis of the dynamics of mannose–lectin interactions (Figure 3). In a next step, the stability of H-bond and metal interactions, as
- well as the influence of highly mobile vs conserved waters were analyzed. For the assessment of the dynamic behavior of the ligand complexes of the seven calcium-dependent lectins, 20 ns molecular dynamics (MD) simulations were performed [57]. The most prominent interactions of O–C3 and O–C4 of the









The effect of milling frequency on a mechanochemical organic reaction monitored by in situ Raman spectroscopy
Beilstein J. Org. Chem. 2017, 13, 2160–2168, doi:10.3762/bjoc.13.216

- of reactive encounters between the particles. In contrast, ex situ gas chromatography studies of the Knoevenagel condensation between vanillin and barbituric acid in a planetary mill revealed a sigmoidal dependence of reaction yield with time [22]. Similarly, sigmoidal dynamics were detected by in









Remarkable functions of sn-3 hydroxy and phosphocholine groups in 1,2-diacyl-sn-glycerolipids to induce clockwise (+)-helicity around the 1,2-diacyl moiety: Evidence from conformation analysis by 1H NMR spectroscopy
Beilstein J. Org. Chem. 2017, 13, 1999–2009, doi:10.3762/bjoc.13.196

- the 1,3-isomer marginally affects the 1H NMR signals of 3. As shown in Table 1, entries 2 and 3, the solvents marginally affect the 1H NMR signals of 2. Clearly, sn-3 OH plays an essential role in the conformational dynamics, as shown above. The dynamic change is probably caused by solvation by
- hydration occurring around the carbonyl groups in the 1,2-diacyl moiety triggers the dynamics of the molecular alignments in liposomes. Probably, an analogous phenomenon related to the solvation around sn-3 OH was observed. Thus, solvation is thought to play a key role in the dynamic conformation change










Enzymatic synthesis of glycosides: from natural O- and N-glycosides to rare C- and S-glycosides
Beilstein J. Org. Chem. 2017, 13, 1857–1865, doi:10.3762/bjoc.13.180

- still not well understood and controlling this ratio remains a challenge that still need to be solved, even if the use of artificial donors [10], the bioengineering of these biocatalysts [61] and the study of internal water dynamics [62] for examples have permitted important progresses. Consequently









Correction: Dynamic behavior of rearranging carbocations – implications for terpene biosynthesis
Beilstein J. Org. Chem. 2017, 13, 1669–1669, doi:10.3762/bjoc.13.161

- Stephanie R. Hare Dean J. Tantillo Department of Chemistry, University of California–Davis, 1 Shields Avenue, Davis, CA 95616, USA 10.3762/bjoc.13.161 Keywords: carbocation; density functional theory; dynamics; mechanism; terpene; The originally published Figure 6 had several mis-drawn

2-Methyl-2,4-pentanediol (MPD) boosts as detergent-substitute the performance of ß-barrel hybrid catalyst for phenylacetylene polymerization
Beilstein J. Org. Chem. 2017, 13, 1498–1506, doi:10.3762/bjoc.13.148

- rhodium-based biohybrid catalyst. Unlike commonly used detergents such as sodium dodecyl sulfate or polyethylene polyethyleneglycol, MPD does not form micelles in solution. Molecular dynamics simulations revealed the effect and position of stabilizing MPD molecules. The advantage of the amphiphilic MPD
- carried out, reaching higher molecular weights and yields compared to catalysis with the micelle-forming refolding reagent PE–PEG. Minimum of MPD molecules was analyzed by molecular dynamics studies to enable refolding of SDS-denatured transmembrane protein FhuA ΔCVFtev [29]. This report aims to
- 1). Molecular dynamics (MD) simulations reveal an optimal minimum number of ≈200 MPD molecules for shielding the hydrophobic transmembrane region of FhuA ΔCVFtev MD simulations of FhuA ΔCVFtev were performed in a box with varying numbers of MPD molecules from 126 MPD, 189 MPD, 252 MPD to 378 MPD






Grip on complexity in chemical reaction networks
Beilstein J. Org. Chem. 2017, 13, 1486–1497, doi:10.3762/bjoc.13.147

- the predictability and perhaps the possibility to influence the dynamics of change, but science has yet to find an answer to this complexity [15]. One of the ultimate aims for systems like a living cell, is to understand how the interplay between molecular level events and network topology determines
- true when those changes induce systems beyond a critical value, where the resulting abrupt shifts or phase transitions become unpredictable [20][21][22]. The analysis of the structure and the dynamics of a complex web of intricate interactions is a risk in removing the link between molecular structure
- dissipative self-assembling structures, creating new forms of smart materials [31][32][33][34][35]. Yet, we are severely limited by too few examples of systems which are both extensive enough to exhibit dynamics, and at the same time, simple enough to be tunable [36]. In this perspective, we will attempt to









Framing major prebiotic transitions as stages of protocell development: three challenges for origins-of-life research
Beilstein J. Org. Chem. 2017, 13, 1388–1395, doi:10.3762/bjoc.13.135
- experimental challenges aimed at constructing protocell systems made of a diversity of functionally coupled components and, thereby, at characterizing more precisely the type of prebiotic evolutionary dynamics that such protocells could engage in. Keywords: functional integration; origins of life; prebiotic
- be grown and multiplied under lab conditions [31]; their rich inherent dynamics [32]; and the competition–selection experiments they make possible, if mixed with different liposome populations [33][34][35][36]. Thus, the interest of working with these model systems stems from the fact that they
- move the field of origins of life forward would be to couple simple chemistry to prebiotic vesicle dynamics: chemical reactions provide the power for endogenous synthesis and vesicles the adequate scaffolding for the functional integration of what is synthesized. We will proceed briefly with the issue

Synthesis of oligonucleotides on a soluble support
Beilstein J. Org. Chem. 2017, 13, 1368–1387, doi:10.3762/bjoc.13.134

- needed. Spectroscopic studies on structure, dynamics and recognition of ONs by other biopolymers, small molecules or metal complexes, for example, may consume ONs in amounts that cannot be conveniently reached by lab-scale solid-phase synthesizers. In addition to synthesis on a soluble support















Aqueous semisynthesis of C-glycoside glycamines from agarose
Beilstein J. Org. Chem. 2017, 13, 1222–1229, doi:10.3762/bjoc.13.121

- aminomonosaccharides 9 and 13 are interesting moieties, regarding their resemblance with the bioactive (+)-muscarine 14 (Figure 2). Indeed, ongoing docking and molecular dynamics experiments revealed the amino-AnGal moiety as a promising platform to launch the design of new mAChR modulators [11]. The differences in





Towards open-ended evolution in self-replicating molecular systems
Beilstein J. Org. Chem. 2017, 13, 1189–1203, doi:10.3762/bjoc.13.118

- to biology, it does not directly explain the emergence of the RNA and DNA dominated world as we know it, which would probably have required open-ended evolution. 3 Evolutionary dynamics of replicators 3.1 Enzyme mediated replication Iconic early experiments aiming to achieve Darwinian type evolution
- strands by creating heat gradients in pores that act as a thermal trap [38]. The thermal traps selectively retain longer DNA sequences, thereby effectively overcoming the inherent advantage of the replication of short sequences. 3.2 Dynamics of self-replicators Ashkenasy recently reported a peptide based














Glycoscience@Synchrotron: Synchrotron radiation applied to structural glycoscience
Beilstein J. Org. Chem. 2017, 13, 1145–1167, doi:10.3762/bjoc.13.114

- dynamics, plasticity of GTs and conformation changes that allow for substrate recognition and catalysis [37]. In plants, GTs are also involved in the biosynthesis of hemicellulose. Xyloglucan is one of the main hemicellulose components in the cell walls of dicots. Its biosynthesis involves different GTs
- manner is creating an opportunity to investigate the microstructure and non-equilibrium dynamics of soft matter on a length scale from a few angtroms to micrometers and on a timescale descending to the millisecond. Grazing incidence X-ray reflectometry Glycolipids: Despite their importance in the
- constitution and dynamics of plasma membranes, the structural and physicochemical features of gangliosides have been somehow neglected presumably because of the lack of appropriate experimental techniques. X-ray reflectometry is a surface-sensitive analytical technique based on the measure of the intensity of





















G-Protein coupled receptors: answers from simulations
Beilstein J. Org. Chem. 2017, 13, 1071–1078, doi:10.3762/bjoc.13.106

- Timothy Clark Computer-Chemie-Centrum, Department of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nuernberg, Naegelsbachstr. 25, 91052 Erlangen, Germany 10.3762/bjoc.13.106 Abstract Molecular-dynamics (MD) simulations are playing an increasingly important role in research into
- ; molecular dynamics; Introduction Evolution is a unique optimization mechanism. Firstly, it stops optimizing as soon as an acceptable solution is reached. There is no evolutionary pressure for elegance, simplicity or even effectiveness above the critical threshold. Secondly, because evolution always starts
- experimental findings [29] suggest that both an agonist ligand and a bound G-protein are necessary in order to activate GPCRs. It is therefore significant that the first molecular dynamics (MD) simulations of a ternary GPCR complex were reported only four years ago [30]. Such simulations are now commonplace





Glyco-gold nanoparticles: synthesis and applications
Beilstein J. Org. Chem. 2017, 13, 1008–1021, doi:10.3762/bjoc.13.100

- [69][70][71]. Moya and co-workers deeply studied the intracellular dynamics and aggregation of glucose-AuNPs by employing fluorescence correlation spectroscopy (FCS) [12]. They demonstrated that GAuNPs were ubiquitous distributed inside the cell as single NP or small aggregates, suggesting a strong








Continuous-flow processes for the catalytic partial hydrogenation reaction of alkynes
Beilstein J. Org. Chem. 2017, 13, 734–754, doi:10.3762/bjoc.13.73

- catalysis [103][104]. Monolithic reactors may surpass most drawbacks typical of packed-bed systems, including high pressure drops, low contacting efficiency, large distribution of residence times, formation of hot-spots or stagnation zones, which results in poorly controlled fluid dynamics, hence in low
- selectivity trend was explained in terms of both adsorption mode on and relative accessibility to Pd active sites, depending on surface potentials and hindrance of modifiers, on the basis of density functional theory and molecular dynamics calculations. The rationale was summarized in the so-called


















Membrane properties of hydroxycholesterols related to the brain cholesterol metabolism
Beilstein J. Org. Chem. 2017, 13, 720–727, doi:10.3762/bjoc.13.71

- fluorescence techniques [21]. Also, a decreased but still significant effect of the hydroxysterols on lipid condensation compared to native cholesterol was found in molecular dynamics simulations, which is probably caused by an increased tilt angle of the sterols to the membrane normal [8][21]. However, using




