Abstract
Titanium dioxide (TiO2) nanofibers have been widely applied in various fields including photocatalysis, energy storage and solar cells due to the advantages of low cost, high abundance and nontoxicity. However, the low conductivity of ions and bulk electrons hinder its rapid development in lithium-ion batteries (LIB). In order to improve the electrochemical performances of TiO2 nanomaterials as anode for LIB, hierarchically porous TiO2 nanofibers with different tetrabutyl titanate (TBT)/paraffin oil ratios were prepared as anode for LIB via a versatile single-nozzle microemulsion electrospinning (ME-ES) method followed by calcining. The experimental results indicated that TiO2 nanofibers with the higher TBT/paraffin oil ratio demonstrated more axially aligned channels and a larger specific surface area. Furthermore, they presented superior lithium-ion storage properties in terms of specific capacity, rate capability and cycling performance compared with solid TiO2 nanofibers for LIB. The initial discharge and charge capacity of porous TiO2 nanofibers with a TBT/paraffin oil ratio of 2.25 reached up to 634.72 and 390.42 mAh·g−1, thus resulting in a coulombic efficiency of 61.51%; and the discharge capacity maintained 264.56 mAh·g−1 after 100 cycles, which was much higher than that of solid TiO2 nanofibers. TiO2 nanofibers with TBT/paraffin oil ratio of 2.25 still obtained a high reversible capacity of 204.53 mAh·g−1 when current density returned back to 40 mA·g−1 after 60 cycles at increasing stepwise current density from 40 mA·g−1 to 800 mA·g−1. Herein, hierarchically porous TiO2 nanofibers have the potential to be applied as anode for lithium-ion batteries in practical applications.
Introduction
The increasing demand for energy has heightened the need for the development of renewable resources because fossil fuels, which are currently the most common energy resources, are a finite resource [1-4]. Battery industry, which is considered to be a vital part in the field of renewable resources, has attracted much attention in recent years. Substantial effort has been made to improve the performance of electrode materials for lithium-ion batteries aiming at aspects including safety, energy density, lifetime and power density [5-8].
So far, among all the commercial lithium-ion batteries, graphite plays an extremely important role in anode materials; nevertheless, structural deformation, electrical disconnection and the initial loss of capacity hinder its further development [9,10]. Titanium dioxide (TiO2) is considered to be an alternative anode material to graphite, which can be attributed to the superior advantages of titanium dioxide such as low-cost, eco-friendliness, nontoxicity and high abundance [10]. Furthermore, safety and stability of titanium dioxide are higher than those of graphite, because since Li ions can be inserted into the TiO2 matrix at higher voltages (at least 1.5 V vs Li/Li+) [11-13]. TiO2 possesses a high theoretical specific capacity of 335 mAh·g−1 [14-16]. However, the poor rate performance and cycling performance has hindered the practical application of titanium dioxide in lithium-ion batteries. The low conductivities of ions and bulk electrons as well as the aggregation of nanoparticles are the major causes of poor rate performance and cycling performance.
One effective method to improve the electrochemical performance of TiO2 is to prepare nanostructures [17-20] that have advantages over normal structures including a large specific surface area, a high electrolyte–electrode contact area and excellent mass transport of products or reactants to active sites inside meso- or micropores. One-dimensional (1D) nanostructures such as nanofibers, nanotubes, nanowires and nanorods are considered to be promising electrode materials for excellent lithium-ion battery performance. This is partly because of their small size enabling fast electron transport and decreasing retardation at the interface [21,22]. Electrospinning, a simple and versatile method, has been widely used in fabricating various nanostructures. The structure of nanofibers can be conveniently adjusted by changing the experimental parameters, the composition of the precursor solution and the structure of the spinneret [23-25].
Core–shell nanofibers are normally prepared by coaxial electrospinning. However, coaxial electrospinning limits the further development of core–shell nanofibers because of its complicated spinneret, the limited number of suitable fluids and the unstable structure of the fibers. Recently the method of ME-ES has been developed to fabricate hierarchical nanofibers without the need of a complex spinneret. It is a simple and versatile way to prepare nanofibers with hollow or multichannel structures [26]. More importantly, various inorganic nanofibers can be fabricated easily by ME-ES including TiO2, SiO2, ZrO2, SnO2, V2O5, GeO2 and Al2O3. A microemulsion system was composed of a metal alkoxide and an oil phase as, respectively, continuous phase and dispersion phase in the system [27]. The microemulsion solution was electrospun to form nanofibers and, after calcination, the as-spun nanofibers partly decomposed to hierarchically porous inorganic nanofibers [26]. Chen et al. reported the fabrication of hierarchical TiO2 nanorods via ME-ES and the application as photoanode material for dye-sensitized solar cells [25]. According to Shi et al., highly porous SnO2/TiO2 composite nanofibers were prepared successfully by ME-ES and subsequent calcination [28].
There are various reports about porous TiO2 nanofibers as anode for LIB. Cho et al. prepared fiber-in-tube TiO2 nanofibers as anode material for LIB and the initial discharge and charge capacity were 231 and 170 mAh·g−1, respectively [29]. Furthermore multichannel hollow TiO2 nanofibers were fabricated by Tang et al. and demonstrated excellent rate capability and cycling performances when used as anode in LIB [30]. However, there is no report about hierarchically porous TiO2 nanofibers fabricated via ME-ES used as anode material for LIB. So in this work, the ME-ES solution was firstly electrospun to prepare nanofibers, then as-spun nanofibers were calcined at 500 °C to form porous TiO2 nanofibers. The electrochemical performances of hierarchically porous TiO2 nanofibers as anode material for LIB were investigated in detail.
Experimental
Materials
Tetrabutyl titanate (TBT), paraffin oil, absolute ethanol, acetic acid and cetyltrimethylammonium bromide (CTAB) were supplied by Shanghai Chemical Regents Co. (Shanghai, China). Additionally, polyvinyl pyrrolidone (PVP) (Mw = 1300000) was purchased from Tianjin Bodi Chemical Reagent Co., Ltd. (Tianjin, China). All chemicals were used as received without further purification.
Preparation of the spinning solution
0.5 g PVP and 0.3 g CTAB were firstly dissolved into 6.5 g ethanol and 0.4 g acetic acid followed by vigorously stirring to form a homogeneous solution. Subsequently, 1 g paraffin oil and stoichiometric amounts of TBT with different TBT/paraffin oil ratios (w/w) of 2.25, 1.9 and 1.55 were added slowly under stirring for 12 h to obtain a microemulsion.
Preparation of porous TiO2 nanofibers
The prepared microemulsion was filled in a 30 mL syringe with a blunt-end stainless steel needle, which had an inside diameter of 0.3 mm. Then the microemulsion was electrospun into nanofibers by a custom-made electrospinning setup consisting of a high voltage power supply, a syringe pump and a grounded nanofiber collector covered with aluminium foil. During electrospinning, a positive voltage of 20 kV was applied between the needle tip and grounded collector at a distance of 15 cm. The flow rate of the spinning solution was maintained at 2 mL·h−1. The as-spun nanofibers with TBT/paraffin oil ratios of 2.25, 1.9 and 1.55 were marked as sample A1, sample B1 and sample C1, respectively.
Subsequently, the as-spun nanofibrous mats were calcined in a quartz tube furnace in air atmosphere at 500 °C for 2 h to yield hierarchically porous TiO2 nanofibers. The porous TiO2 nanofibers obtained after calcination with TBT/paraffin oil ratios of 2.25, 1.9 and 1.55 were named as sample A2, sample B2 and sample C2, respectively. In other words, sample A2 corresponded to the calcined sample A1, sample B2 and sample C2 were the same naming method as sample A2.
Characterization
Thermogravimetric analysis (1100SF) was employed to characterize the thermal decomposition of as-spun TiO2 nanofibers in air atmosphere with a heating rate of 10 °C/min. X-ray diffraction patterns of porous TiO2 nanofibers were recorded on a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (wavelength λ = 1.54 Å) at a scanning speed of 4 °C/min. A Hitachi field emission scanning electron microscope (FE-SEM, SU4800) was employed to observe the surface morphology of porous TiO2 nanofibers. Prior to the FE-SEM examination, all the specimens were sputter-coated with gold to avoid charge accumulations. Transmission electron microscopy was conducted on a JEOL JEM-2100 transmission electron microscopy unit at an accelerating voltage of 120 kV. The specific surface area and pore structure of porous TiO2 nanofibers were characterized with a physisorption analyzer (ASAP 2020, Micromeritics).
Electrochemical measurements
The electrodes were prepared by mixing thoroughly porous TiO2 nanofibers (60 wt %), carbon black (20 wt %) and poly(tetrafluoroethylene) (20 wt %) to form a homogeneous paste. Subsequently, the paste was coated uniformly on the nickel net and then dried in a vacuum oven at 60 °C for 12 h. Prior to cell assembly, the electrodes were cut into circular disks with a diameter of 16 mm. The testing cells employed Celgard 2300 membrane as separator, lithium foil as both reference and counter electrode, and 1 M LiPF6 dissolved in ethylene carbonate (EC), dimethyl carbonate (DMC) and ethylene methyl carbonate (EMC) (1:1:1, v/v/v) as the electrolyte, respectively. Coin cells were assembled in an argon-filled glove box. The galvanostatic discharge–charge tests were conducted at the constant current density of 40 mA·g−1 in the voltage range of 0.01–3.0 V (vs Li+/Li). The cyclic voltammetry curves were tested at a scanning rate of 0.1 mV/s over the voltage range of 0.01–3.0 V (vs Li+/Li) with a CHI660e electrochemical workstation.
Results and Discussion
Thermal properties and structural morphology of porous TiO2 nanofibers
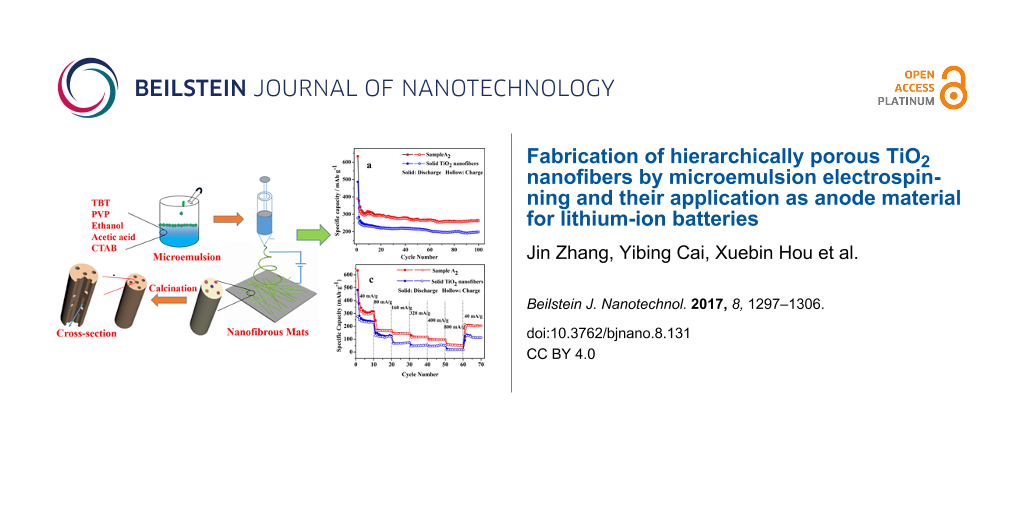
The fabrication process of hierarchically porous TiO2 nanofibers is presented in Figure 1. When paraffin oil was added dropwise into the solution, it floated on the surface of the solution. After stirring vigorously, the opaque precursor solution was prepared successfully. During the process of electrospinning, the viscous liquid was elongated quickly accompanied by the evaporation of solvent as well as the hydrolysis condensation of TBT. Herein, despite the fact that the oil droplets in the microemulsion were stable with a diameter of about 20 nm, several small oil droplets merged into larger ones and then were stretched strongly by electrostatic force along the direction of fluid jet, thus forming oriented and axially aligned oil droplets. After calcination, the axially aligned oil droplets would be removed, resulting in axially aligned pores with the diameters of tens of nanometers. It meant that the axially aligned pores were the vacancies of paraffin oil.
![[2190-4286-8-131-1]](/bjnano/content/figures/2190-4286-8-131-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Fabrication procedure of porous TiO2 nanofibers via ME-ES.
Figure 1: Fabrication procedure of porous TiO2 nanofibers via ME-ES.
Thermogravimetry (TG) and differential thermogravimetry (DTG) curves of as-spun sample A1 are shown in Figure 2a. It can be seen that the total weight loss was approximately 35%, and whole decomposition process could be divided into three steps. The first step occurred between 25 to 150 °C and can be attributed to the evaporation of residual solvent and water. The second step from 160 to 370 °C corresponds to the major decomposition process of long molecular chains of PVP and paraffin oil as well as to the partial decomposition of TBT molecules, resulting in weight loss of 20%. The final decomposition step occurred in the temperature range from 380 to 500 °C and corresponds to the further decomposition of molecular chains of PVP and paraffin oil as well as the complete degradation of TBT, causing a weight loss of approximately 8%. It should be noted from Figure 2 that there was no obvious weight loss above 500 °C, suggesting the formation of stable TiO2 nanofibers. Figure 2b–d displays the surface SEM images of sample A2, sample B2 and sample C2. It can be observed that after calcination, the nanofibers had the length of a few micrometers and a diameter of hundreds of nanometers. Moreover, from Figure 2b–d, the nanofibers became increasingly brittle due to the decreased content of TBT in the microemulsion system.
![[2190-4286-8-131-2]](/bjnano/content/figures/2190-4286-8-131-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) TG and DTG curves of as-spun TiO2 nanofibers; surface SEM images after calcination of sample A2 (b), sample B2 (c) and sample C2 (d).
Figure 2: (a) TG and DTG curves of as-spun TiO2 nanofibers; surface SEM images after calcination of sample A2...
Figure 3 shows the representative XRD pattern of sample A2 calcined at 500 °C for 2 h in air atmosphere. All the peaks can be indexed to anatase TiO2 (JCPDS, No. 21-1272). The diffraction peaks at 2θ = 25.281, 37.800, 48.049, 53.890, 55.060, 62.688, 70.309 and 75.029° correspond to the (101), (004), (200), (105), (211), (204), (220) and (215) reflections, respectively. It is notable that there was no peak of any other phase in Figure 3, thus confirming the pure phase of prepared porous TiO2 nanofibers. Considering that all the porous TiO2 nanofibers were calcined at 500 °C, all of them should be expected to be anatase TiO2. More importantly, among the various polymorphs of TiO2, the anatase polymorph possesses an open crystal structure resulting from the stacking of zigzag units, which would benefit the intercalation/deintercalation of Li+ ions.
![[2190-4286-8-131-3]](/bjnano/content/figures/2190-4286-8-131-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Representative XRD pattern of porous TiO2 nanofibers.
Figure 3: Representative XRD pattern of porous TiO2 nanofibers.
Figure 4 displays the surface SEM images of sample A1 (a), sample B1 (c), and sample C1 (e) as well as cross-sectional SEM images of sample A2 (b), sample B2 (d), sample C2 (f) and solid TiO2 nanofibers (g), and representative TEM images of sample A2. In addition, the inset SEM images are the corresponding images with the higher magnification. The as-spun nanofibers have a smooth surface and relatively uniform distribution. Nevertheless, it can be observed that with decreasing relative content of TBT/paraffin oil, the surface morphology looks worse, and the distribution of the fiber diameter becomes increasingly non-uniform. This is because the butoxyl groups in TBT serve as additional surfactant, which is advantageous to form the stable interfaces between the two phases, thus forming a stable microemulsion. In other words, the decreased content of TBT caused increasing instability of the microemulsion, so that the morphology of the nanofibers became worse. In addition, the most significant phenomenon is that the number of channels in each nanofiber decreased as the content of paraffin oil increased. This is because the pores stemmed from the vacancies of oil droplets after calcination, and a larger oil droplets were formed when more paraffin oil was added into the microemulsion [31]. The SEM image of unmodified TiO2 nanofibers in Figure 4g reveals solid structures without pores in the cross-section. This sample was used as reference in the following investigation. As shown in Figure 4h, a representative TEM image of a single porous nanofiber confirms that the pores inside the fibers were oriented and axially aligned, which may be caused by both the strong stretching force from the strong electric field and the formation of larger oil droplets during electrospinning [25].
![[2190-4286-8-131-4]](/bjnano/content/figures/2190-4286-8-131-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Surface SEM images of sample A1 (a), sample B1 (c), and sample C1 (e); cross-sectional SEM images of sample A2 (b), sample B2 (d), sample C2 (f) and solid TiO2 nanofibers (g), inset SEM images were the corresponding images with higher magnification; and representative TEM images of sample A2 (h).
Figure 4: Surface SEM images of sample A1 (a), sample B1 (c), and sample C1 (e); cross-sectional SEM images o...
Nitrogen adsorption–desorption isotherms of sample A2, sample B2, sample C2 and solid TiO2 nanofibers are shown in Figure 5. All isotherms in Figure 5 belong to type IV accompanied with a hysteresis loop located at higher relative pressures [32,33]. The specific surface areas of sample A2, sample B2, sample C2 and solid TiO2 nanofibers were approximately 69.9, 48.2, 43.1 and 35.6 m2/g, respectively. It is noteworthy that the specific surface area of sample A2 was almost two times that of solid TiO2 nanofibers, which demonstrated the fact that ME-ES was indeed an effective method to enlarge the specific surface area. The specific surface area plays a very important role in the performance of electrodes for lithium-ion batteries. The merits of porous nanofibers with a higher specific surface area lie in the higher lithium-ion flux across the interfaces and the larger contact area between the electrode and electrolyte [2,34,35]. Herein, sample A2 should have the best performances as the electrode of lithium-ion battery in theory. In addition, Figure 5b displays the pore-size distribution of sample A2, sample B2 and sample C2. According to the analysis of pore-size distribution with the Barrett–Joyner–Halenda (BJH) method, the average pore size increased gradually, and the average pore diameter was 17.8, 18.5 and 20.1 nm for sample A2, sample B2, sample C2, respectively. Moreover, it can be observed clearly that the average diameter of macropores increased with the ratio of TBT/paraffin oil decreasing. The results confirmed the presence of mesopores and macropores in the porous TiO2 nanofibers.
![[2190-4286-8-131-5]](/bjnano/content/figures/2190-4286-8-131-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: (a) Nitrogen adsorption–desorption curves of sample A2, sample B2, sample C2 and solid TiO2 nanofibers; (b) pore-size distribution of sample A2, sample B2, sample C2.
Figure 5: (a) Nitrogen adsorption–desorption curves of sample A2, sample B2, sample C2 and solid TiO2 nanofib...
Electrochemical performances
The electrochemical properties of TiO2 nanofiber electrodes were investigated by cyclic voltammetry (CV) test using lithium foil as counter electrode and reference electrode. The CV curves of sample A2 and solid TiO2 nanofibers examined in the range of 0.01–3 V (vs Li+/Li) at a scanning rate of 0.1 mV/s are shown in Figure S1 (Supporting Information File 1). Sample A2 has obvious reduction and oxidation peaks at about 1.49 and 2.33 V. The reduction peak corresponded to the intercalation of Li+ into interstitial octahedral sites of anatase TiO2 via a phase transition from tetragonal anatase to orthorhombic Li0.5TiO2 according to the scheme: xLi+ + TiO2+ xe− ↔ LixTiO2 [36,37]. There is no big difference between sample A2 and solid TiO2 nanofibers (Figure S1, Supporting Information File 1).
Figure 6 shows the representative galvanostatic charge–discharge curves of sample A2 (a), sample B2 (b) and sample C2 (c) at a current density of 40 mA·g−1. It is apparent that all the charge or discharge profiles were similar, thus implying that the processes of lithium insertion or extraction were the same. There were obvious voltage plateaus in all discharge curves at around 1.72 V and charge curves at around 1.93 V, which were due to the phase transition between tetragonal anatase and orthorhombic Li0.5TiO2 [38]. The initial discharge capacities of sample A2, sample B2 and sample C2 were 634.72, 583.44 and 522.65 mAh·g−1, and the corresponding charge capacities were 390.42, 367.85 and 279.82 mAh·g−1, resulting in coulombic efficiencies of 61.51%, 63.05% and 53.54%, respectively. It was worth noting that the specific capacities increased accordingly as the specific surface area of the samples increased, which further confirmed that specific surface area plays a vital role in the performance of the LIB anode material. Sample A2 with a specific surface area of 69.9 m2·g−1 exhibited the best performance among all the samples, and was chosen for further investigation.
![[2190-4286-8-131-6]](/bjnano/content/figures/2190-4286-8-131-6.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Galvanostatic charge–discharge curves of sample A2 (a), sample B2 (b) and sample C2 (c) for the first ten cycles at the current density of 40 mA·g−1.
Figure 6: Galvanostatic charge–discharge curves of sample A2 (a), sample B2 (b) and sample C2 (c) for the fir...
Figure 7 provides the cycling performance (a), coulombic efficiency (b) and rate capability (c) of sample A2 and solid TiO2 nanofibers. As can be seen from Figure 7a, there was relatively large capacity fading in the first several cycles for both samples, which may be ascribed to the partial irreversible decomposition of the electrolyte, the incomplete decomposition of the solid–electrolyte interface (SEI) or a partial irreversible intercalation of Li+ into TiO2 nanofibers [30]. The capacity remains relatively stable in the following cycles. The 100th discharge (charge) capacity of sample A2 and solid TiO2 nanofibers were 264.56 (261.61) and 198.96 (198.12) mAh·g−1, respectively. It is apparent that the cycling performance of sample A2 shows significant improvement compared with solid TiO2 nanofibers, which can be attributed to the larger specific surface area of sample A2. As mentioned in Figure 7, the coulombic efficiency of sample A2 was 61.51% for the first cycle, whereas the coulombic efficiency of solid TiO2 nanofibers was 57.21%. And the coulombic efficiency remained relatively constant in the following cycles due to the relatively stable cycling performance. The rate capability plays a crucial role in the high-power applications of LIB, therefore rate capability of both samples was evaluated by stepwise increasing the current density. The specific capacity decreased gradually with increasing current density. The electrode delivered reversible discharge capacities of 315.14, 166.47, 142.55, 117.56, 96.42, 50.195 mAh·g−1 at the current density of 40, 80, 160, 320, 400, 800 mA·g−1, respectively. Despite the fact that the capacity could not recovered fully, the corresponding specific capacity of sample A2 was as high as 204.53 mAh·g−1 when the current density returned back to 40 mA·g−1, which was much higher than the values of solid TiO2 nanofibers. Sample A2 as anode for LIB demonstrated excellent rate capability, which stemmed mostly from the hierarchically porous structure and the large specific surface area.
![[2190-4286-8-131-7]](/bjnano/content/figures/2190-4286-8-131-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Comparison of cycling performance (a), coulombic efficiency (b) and rate capability (c) of sample A2 and solid TiO2 nanofibers.
Figure 7: Comparison of cycling performance (a), coulombic efficiency (b) and rate capability (c) of sample A2...
Electrochemical impedance spectroscopy (EIS) was carried out on the electrode of sample A2 and solid TiO2 nanofibers. Nyquist impedance plots obtained for sample A2 and solid TiO2 nanofibers in a fresh cell are presented in Figure S2 (Supporting Information File 1). The plots for the two samples were similar, consisting of a compressed semicircle and a straight line. The semicircles in the high-frequency range represents the charge-transfer resistance (Rct) and the straight line in the low-frequency region represents the diffusion and accumulation process of lithium ions in the electrode [1,29,39]. As can be seen from Figure S2 (Supporting Information File 1), the charge–transfer resistance of sample A2 was about 235 Ω, whereas the charge–transfer resistance of solid TiO2 nanofibers was about 425 Ω. Herein, the lower charge–transfer resistance of sample A2 can be ascribed to the hierarchically porous structures of sample A2 facilitating charge transfer at the electrolyte–electrode interfaces [40-42] (Figure S2, Supporting Information File 1). Table 1 compares the electrochemical performances of sample A2 with other reference results.
Table 1: Comparison of electrochemical performances of different TiO2 nanostructures.
| number | structures | reversible capacity (mAh·g−1) | charge–discharge rates (mA·g−1) | reference |
|---|---|---|---|---|
| 1 | TiO2 nanofibers with fiber-in-tube structure | 170 | 200 | [29] |
| 2 | mesoporous TiO2 nanotubes | 108 | 335 | [43] |
| 3 | multi-channel hollow TiO2 nanofibers | 212 | 168 | [30] |
| 4 | TiO2 nanoflakes | 261 | 33 | [44] |
| 5 | TiO2 hollow spheres | 148 | 850 | [45] |
| 6 | hierarchically porous TiO2 nanofibers | 205 | 40 | this work |
Conclusion
Hierarchically porous TiO2 nanofibers were fabricated successfully via single-nozzle microemulsion electrospinning. Moreover, multichannel TiO2 nanofibers displayed excellent lithium-ion storage properties compared with normal solid TiO2 nanofibers when used as anode for LIB. The initial discharge and charge capacities of porous TiO2 nanofibers with a TBT/paraffin oil ratio of 2.25 reached up to 634.72 and 390.42 mAh·g−1; and the discharge capacity was 264.56 mAh·g−1 after 100 cycles. In addition, TiO2 nanofibers with a TBT/paraffin oil ratio of 2.25 still obtained a high reversible capacity of 204.53 mAh·g−1 when the current density returned back to 40 mA·g-1. The results confirmed microemulsion electrospinning is indeed a simple and versatile method to prepare porous nanofibers with large specific surface area and the prepared nanofibers improve greatly their electrochemical performance in practical applications.
Supporting Information
The Supporting Information presents CV curves and Nyquist impedance plots.
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 138.3 KB | Download |
Acknowledgements
This research was financially supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions, 111 Project (B17021), Six Talent Peaks Project in Jiangsu Province (2014-XCL001), the Fundamental Research Funds for the Central Universities (JUSRP51621A), and the China Postdoctoral Science Foundation (No. 2015T80496).
References
-
Qiao, H.; Luo, L.; Chen, K.; Fei, Y.; Cui, R.; Wei, Q. Electrochim. Acta 2015, 160, 43–49. doi:10.1016/j.electacta.2015.02.012
Return to citation in text: [1] [2] -
Luo, L.; Qiao, H.; Chen, K.; Fei, Y.; Wei, Q. Electrochim. Acta 2015, 177, 283–289. doi:10.1016/j.electacta.2015.01.100
Return to citation in text: [1] [2] -
Zhang, X.; Ji, L.; Toprakci, O.; Liang, Y.; Alcoutlabi, M. Polym. Rev. 2011, 51, 239–264. doi:10.1080/15583724.2011.593390
Return to citation in text: [1] -
Tarascon, J.-M.; Armand, M. Nature 2001, 414, 359–367. doi:10.1038/35104644
Return to citation in text: [1] -
Nishi, Y. Chem. Rec. 2001, 1, 406–413. doi:10.1002/tcr.1024
Return to citation in text: [1] -
Li, H.; Wang, Z.; Chen, L.; Huang, X. Adv. Mater. 2009, 21, 4593–4607. doi:10.1002/adma.200901710
Return to citation in text: [1] -
Patil, A.; Patil, V.; Shin, D. W.; Choi, J.-W.; Paik, D.-S.; Yoon, S.-J. Mater. Res. Bull. 2008, 43, 1913–1942. doi:10.1016/j.materresbull.2007.08.031
Return to citation in text: [1] -
Prakash, R.; Fanselau, K.; Ren, S.; Mandal, T. K.; Kübel, C.; Hahn, H.; Fichtner, M. Beilstein J. Nanotechnol. 2013, 4, 699–704. doi:10.3762/bjnano.4.79
Return to citation in text: [1] -
Wang, F.; Chen, J.; Huang, K.; Liu, S. Sci. China, Ser. E: Technol. Sci. 2009, 52, 3219–3223. doi:10.1007/s11431-009-0310-2
Return to citation in text: [1] -
Xu, J.; Jia, C.; Cao, B.; Zhang, W. F. Electrochim. Acta 2007, 52, 8044–8047. doi:10.1016/j.electacta.2007.06.077
Return to citation in text: [1] [2] -
Wang, J.; Bai, Y.; Wu, M.; Jiang, Y.; Zhang, W. F. J. Power Sources 2009, 191, 614–618. doi:10.1016/j.jpowsour.2009.02.056
Return to citation in text: [1] -
Pfanzelt, M.; Kubiak, P.; Fleischhammer, M.; Wohlfahrt-Mehrens, M. J. Power Sources 2011, 196, 6815–6821. doi:10.1016/j.jpowsour.2010.09.109
Return to citation in text: [1] -
Lee, S.; Ha, J.; Choi, J.; Song, T.; Lee, J. W.; Paik, U. ACS Appl. Mater. Interfaces 2013, 5, 11525–11529. doi:10.1021/am404082h
Return to citation in text: [1] -
Zhang, X.; Kumar, P. S.; Aravindan, V.; Liu, H. H.; Sundaramurthy, J.; Mhaisalkar, S. G.; Duong, M. H.; Ramakrishna, S.; Madhavi, S. J. Phys. Chem. C 2012, 116, 14780–14788. doi:10.1021/jp302574g
Return to citation in text: [1] -
Zhao, B.; Jiang, S.; Su, C.; Cai, R.; Ran, R.; Tadé, M. O.; Shao, Z. J. Mater. Chem. A 2013, 1, 12310–12320. doi:10.1039/C3TA12770B
Return to citation in text: [1] -
Pham-Cong, D.; Kim, J.-H.; Jeong, S.-Y.; Choi, J. H.; Kim, J.; Cho, C.-R. Electrochem. Commun. 2015, 60, 204–207. doi:10.1016/j.elecom.2015.09.018
Return to citation in text: [1] -
Fu, L. J.; Liu, H.; Zhang, H. P.; Li, C.; Zhang, T.; Wu, Y. P.; Holze, R.; Wu, H. Q. Electrochem. Commun. 2006, 8, 1–4. doi:10.1016/j.elecom.2005.10.006
Return to citation in text: [1] -
Yang, J.; Takeda, Y.; Imanishi, N.; Ichikawa, T.; Yamamoto, O. J. Power Sources 1999, 79, 220–224. doi:10.1016/S0378-7753(99)00180-9
Return to citation in text: [1] -
Wang, C.; Appleby, A. J.; Little, F. E. Solid State Ionics 2002, 147, 13–22. doi:10.1016/S0167-2738(02)00041-3
Return to citation in text: [1] -
Liu, M.; Xie, W.; Gu, L.; Qin, T.; Hou, X.; He, D. Beilstein J. Nanotechnol. 2016, 7, 1289–1295. doi:10.3762/bjnano.7.120
Return to citation in text: [1] -
Kim, G.; Jo, C.; Kim, W.; Chun, J.; Yoon, S.; Lee, J.; Choi, W. Energy Environ. Sci. 2013, 6, 2932–2938. doi:10.1039/c3ee41880d
Return to citation in text: [1] -
Wu, H. B.; Chen, J. S.; Hng, H. H.; Lou, X. W. Nanoscale 2012, 4, 2526–2542. doi:10.1039/c2nr11966h
Return to citation in text: [1] -
Bognitzki, M.; Czado, W.; Frese, T.; Schaper, A.; Hellwig, M.; Steinhart, M.; Greiner, A.; Wendorff, J. H. Adv. Mater. 2001, 13, 70–72. doi:10.1002/1521-4095(200101)13:1<70::AID-ADMA70>3.0.CO;2-H
Return to citation in text: [1] -
Xu, X.; Zhuang, X.; Chen, X.; Wang, X.; Yang, L.; Jing, X. Macromol. Rapid Commun. 2006, 27, 1637–1642. doi:10.1002/marc.200600384
Return to citation in text: [1] -
Chen, H.-Y.; Zhang, T.-L.; Fan, J.; Kuang, D.-B.; Su, C.-Y. ACS Appl. Mater. Interfaces 2013, 5, 9205–9211. doi:10.1021/am402853q
Return to citation in text: [1] [2] [3] -
Choi, K.-I.; Lee, S. H.; Park, J.-Y.; Choi, D.-Y.; Hwang, C.-H.; Lee, I.-H.; Chang, M. H. Mater. Lett. 2013, 112, 113–116. doi:10.1016/j.matlet.2013.08.101
Return to citation in text: [1] [2] -
Xu, X.; Chen, X.; Ma, P.; Wang, X.; Jing, X. Eur. J. Pharm. Biopharm. 2008, 70, 165–170. doi:10.1016/j.ejpb.2008.03.010
Return to citation in text: [1] -
Shi, H.; Zhou, M.; Song, D.; Pan, X.; Fu, J.; Zhou, J.; Ma, S.; Wang, T. Ceram. Int. 2014, 40, 10383–10393. doi:10.1016/j.ceramint.2014.02.124
Return to citation in text: [1] -
Cho, J. S.; Hong, Y. J.; Kang, Y. C. Chem. – Eur. J. 2015, 21, 11082–11087. doi:10.1002/chem.201500729
Return to citation in text: [1] [2] [3] -
Tang, K.; Yu, Y.; Mu, X.; van Aken, P. A.; Maier, J. Electrochem. Commun. 2013, 28, 54–57. doi:10.1016/j.elecom.2012.12.005
Return to citation in text: [1] [2] [3] -
Chen, H.; Di, J.; Wang, N.; Dong, H.; Wu, J.; Zhao, Y.; Yu, J.; Jiang, L. Small 2011, 7, 1779–1783. doi:10.1002/smll.201002376
Return to citation in text: [1] -
Luo, L.; Cui, R.; Liu, K.; Qiao, H.; Wei, Q. Ionics 2015, 21, 687–694. doi:10.1007/s11581-014-1213-1
Return to citation in text: [1] -
Sing, K. S. W. Pure Appl. Chem. 1982, 54, 2201–2218. doi:10.1351/pac198254112201
Return to citation in text: [1] -
Qiao, H.; Li, J.; Fu, J.; Kumar, D.; Wei, Q.; Cai, Y.; Huang, F. ACS Appl. Mater. Interfaces 2011, 3, 3704–3708. doi:10.1021/am200884k
Return to citation in text: [1] -
Cherian, C. T.; Sundaramurthy, J.; Reddy, M. V.; Kumar, P. S.; Mani, K.; Pliszka, D.; Sow, C. H.; Ramakrishna, S.; Chowdari, B. V. R. ACS Appl. Mater. Interfaces 2013, 5, 9957–9963. doi:10.1021/am401779p
Return to citation in text: [1] -
Morgan, B. J.; Madden, P. A. Phys. Rev. B 2012, 86, 035147. doi:10.1103/PhysRevB.86.035147
Return to citation in text: [1] -
Moriguchi, I.; Hidaka, R.; Yamada, H.; Kudo, T.; Murakami, H.; Nakashima, N. Adv. Mater. 2006, 18, 69–73. doi:10.1002/adma.200501366
Return to citation in text: [1] -
Wang, J.; Polleux, J.; Lim, J.; Dunn, B. J. Phys. Chem. C 2007, 111, 14925–14931. doi:10.1021/jp074464w
Return to citation in text: [1] -
Guo, B.; Wang, X.; Fulvio, P. F.; Chi, M.; Mahurin, S. M.; Sun, X.-G.; Dai, S. Adv. Mater. 2011, 23, 4661–4666. doi:10.1002/adma.201102032
Return to citation in text: [1] -
Aricò, A. S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; van Schalkwijk, W. Nat. Mater. 2005, 4, 366–377. doi:10.1038/nmat1368
Return to citation in text: [1] -
Bruce, P. G.; Scrosati, B.; Tarascon, J.-M. Angew. Chem., Int. Ed. 2008, 47, 2930–2946. doi:10.1002/anie.200702505
Return to citation in text: [1] -
Ma, H.; Zhang, S.; Ji, W.; Tao, Z.; Chen, J. J. Am. Chem. Soc. 2008, 130, 5361–5367. doi:10.1021/ja800109u
Return to citation in text: [1] -
Xing, Z.; Asiri, A. M.; Obaid, A. Y.; Sun, X.; Ge, X. RSC Adv. 2014, 4, 9061–9063. doi:10.1039/c3ra47239f
Return to citation in text: [1] -
Nguyen-Phan, T.-D.; Pham, V. H.; Kweon, H.; Chung, J. S.; Kim, E. J.; Hur, S. H.; Shin, E. W. J. Colloid Interface Sci. 2012, 367, 139–147. doi:10.1016/j.jcis.2011.10.021
Return to citation in text: [1] -
Ding, S.; Chen, J. S.; Wang, Z.; Yan, L. C.; Madhavi, S.; Hu, X.; Lou, X. W. J. Mater. Chem. 2011, 21, 1677–1680. doi:10.1039/C0JM03650A
Return to citation in text: [1]
| 30. | Tang, K.; Yu, Y.; Mu, X.; van Aken, P. A.; Maier, J. Electrochem. Commun. 2013, 28, 54–57. doi:10.1016/j.elecom.2012.12.005 |
| 44. | Nguyen-Phan, T.-D.; Pham, V. H.; Kweon, H.; Chung, J. S.; Kim, E. J.; Hur, S. H.; Shin, E. W. J. Colloid Interface Sci. 2012, 367, 139–147. doi:10.1016/j.jcis.2011.10.021 |
| 45. | Ding, S.; Chen, J. S.; Wang, Z.; Yan, L. C.; Madhavi, S.; Hu, X.; Lou, X. W. J. Mater. Chem. 2011, 21, 1677–1680. doi:10.1039/C0JM03650A |
| 1. | Qiao, H.; Luo, L.; Chen, K.; Fei, Y.; Cui, R.; Wei, Q. Electrochim. Acta 2015, 160, 43–49. doi:10.1016/j.electacta.2015.02.012 |
| 2. | Luo, L.; Qiao, H.; Chen, K.; Fei, Y.; Wei, Q. Electrochim. Acta 2015, 177, 283–289. doi:10.1016/j.electacta.2015.01.100 |
| 3. | Zhang, X.; Ji, L.; Toprakci, O.; Liang, Y.; Alcoutlabi, M. Polym. Rev. 2011, 51, 239–264. doi:10.1080/15583724.2011.593390 |
| 4. | Tarascon, J.-M.; Armand, M. Nature 2001, 414, 359–367. doi:10.1038/35104644 |
| 11. | Wang, J.; Bai, Y.; Wu, M.; Jiang, Y.; Zhang, W. F. J. Power Sources 2009, 191, 614–618. doi:10.1016/j.jpowsour.2009.02.056 |
| 12. | Pfanzelt, M.; Kubiak, P.; Fleischhammer, M.; Wohlfahrt-Mehrens, M. J. Power Sources 2011, 196, 6815–6821. doi:10.1016/j.jpowsour.2010.09.109 |
| 13. | Lee, S.; Ha, J.; Choi, J.; Song, T.; Lee, J. W.; Paik, U. ACS Appl. Mater. Interfaces 2013, 5, 11525–11529. doi:10.1021/am404082h |
| 29. | Cho, J. S.; Hong, Y. J.; Kang, Y. C. Chem. – Eur. J. 2015, 21, 11082–11087. doi:10.1002/chem.201500729 |
| 10. | Xu, J.; Jia, C.; Cao, B.; Zhang, W. F. Electrochim. Acta 2007, 52, 8044–8047. doi:10.1016/j.electacta.2007.06.077 |
| 30. | Tang, K.; Yu, Y.; Mu, X.; van Aken, P. A.; Maier, J. Electrochem. Commun. 2013, 28, 54–57. doi:10.1016/j.elecom.2012.12.005 |
| 9. | Wang, F.; Chen, J.; Huang, K.; Liu, S. Sci. China, Ser. E: Technol. Sci. 2009, 52, 3219–3223. doi:10.1007/s11431-009-0310-2 |
| 10. | Xu, J.; Jia, C.; Cao, B.; Zhang, W. F. Electrochim. Acta 2007, 52, 8044–8047. doi:10.1016/j.electacta.2007.06.077 |
| 25. | Chen, H.-Y.; Zhang, T.-L.; Fan, J.; Kuang, D.-B.; Su, C.-Y. ACS Appl. Mater. Interfaces 2013, 5, 9205–9211. doi:10.1021/am402853q |
| 5. | Nishi, Y. Chem. Rec. 2001, 1, 406–413. doi:10.1002/tcr.1024 |
| 6. | Li, H.; Wang, Z.; Chen, L.; Huang, X. Adv. Mater. 2009, 21, 4593–4607. doi:10.1002/adma.200901710 |
| 7. | Patil, A.; Patil, V.; Shin, D. W.; Choi, J.-W.; Paik, D.-S.; Yoon, S.-J. Mater. Res. Bull. 2008, 43, 1913–1942. doi:10.1016/j.materresbull.2007.08.031 |
| 8. | Prakash, R.; Fanselau, K.; Ren, S.; Mandal, T. K.; Kübel, C.; Hahn, H.; Fichtner, M. Beilstein J. Nanotechnol. 2013, 4, 699–704. doi:10.3762/bjnano.4.79 |
| 28. | Shi, H.; Zhou, M.; Song, D.; Pan, X.; Fu, J.; Zhou, J.; Ma, S.; Wang, T. Ceram. Int. 2014, 40, 10383–10393. doi:10.1016/j.ceramint.2014.02.124 |
| 23. | Bognitzki, M.; Czado, W.; Frese, T.; Schaper, A.; Hellwig, M.; Steinhart, M.; Greiner, A.; Wendorff, J. H. Adv. Mater. 2001, 13, 70–72. doi:10.1002/1521-4095(200101)13:1<70::AID-ADMA70>3.0.CO;2-H |
| 24. | Xu, X.; Zhuang, X.; Chen, X.; Wang, X.; Yang, L.; Jing, X. Macromol. Rapid Commun. 2006, 27, 1637–1642. doi:10.1002/marc.200600384 |
| 25. | Chen, H.-Y.; Zhang, T.-L.; Fan, J.; Kuang, D.-B.; Su, C.-Y. ACS Appl. Mater. Interfaces 2013, 5, 9205–9211. doi:10.1021/am402853q |
| 27. | Xu, X.; Chen, X.; Ma, P.; Wang, X.; Jing, X. Eur. J. Pharm. Biopharm. 2008, 70, 165–170. doi:10.1016/j.ejpb.2008.03.010 |
| 21. | Kim, G.; Jo, C.; Kim, W.; Chun, J.; Yoon, S.; Lee, J.; Choi, W. Energy Environ. Sci. 2013, 6, 2932–2938. doi:10.1039/c3ee41880d |
| 22. | Wu, H. B.; Chen, J. S.; Hng, H. H.; Lou, X. W. Nanoscale 2012, 4, 2526–2542. doi:10.1039/c2nr11966h |
| 26. | Choi, K.-I.; Lee, S. H.; Park, J.-Y.; Choi, D.-Y.; Hwang, C.-H.; Lee, I.-H.; Chang, M. H. Mater. Lett. 2013, 112, 113–116. doi:10.1016/j.matlet.2013.08.101 |
| 17. | Fu, L. J.; Liu, H.; Zhang, H. P.; Li, C.; Zhang, T.; Wu, Y. P.; Holze, R.; Wu, H. Q. Electrochem. Commun. 2006, 8, 1–4. doi:10.1016/j.elecom.2005.10.006 |
| 18. | Yang, J.; Takeda, Y.; Imanishi, N.; Ichikawa, T.; Yamamoto, O. J. Power Sources 1999, 79, 220–224. doi:10.1016/S0378-7753(99)00180-9 |
| 19. | Wang, C.; Appleby, A. J.; Little, F. E. Solid State Ionics 2002, 147, 13–22. doi:10.1016/S0167-2738(02)00041-3 |
| 20. | Liu, M.; Xie, W.; Gu, L.; Qin, T.; Hou, X.; He, D. Beilstein J. Nanotechnol. 2016, 7, 1289–1295. doi:10.3762/bjnano.7.120 |
| 14. | Zhang, X.; Kumar, P. S.; Aravindan, V.; Liu, H. H.; Sundaramurthy, J.; Mhaisalkar, S. G.; Duong, M. H.; Ramakrishna, S.; Madhavi, S. J. Phys. Chem. C 2012, 116, 14780–14788. doi:10.1021/jp302574g |
| 15. | Zhao, B.; Jiang, S.; Su, C.; Cai, R.; Ran, R.; Tadé, M. O.; Shao, Z. J. Mater. Chem. A 2013, 1, 12310–12320. doi:10.1039/C3TA12770B |
| 16. | Pham-Cong, D.; Kim, J.-H.; Jeong, S.-Y.; Choi, J. H.; Kim, J.; Cho, C.-R. Electrochem. Commun. 2015, 60, 204–207. doi:10.1016/j.elecom.2015.09.018 |
| 26. | Choi, K.-I.; Lee, S. H.; Park, J.-Y.; Choi, D.-Y.; Hwang, C.-H.; Lee, I.-H.; Chang, M. H. Mater. Lett. 2013, 112, 113–116. doi:10.1016/j.matlet.2013.08.101 |
| 32. | Luo, L.; Cui, R.; Liu, K.; Qiao, H.; Wei, Q. Ionics 2015, 21, 687–694. doi:10.1007/s11581-014-1213-1 |
| 33. | Sing, K. S. W. Pure Appl. Chem. 1982, 54, 2201–2218. doi:10.1351/pac198254112201 |
| 31. | Chen, H.; Di, J.; Wang, N.; Dong, H.; Wu, J.; Zhao, Y.; Yu, J.; Jiang, L. Small 2011, 7, 1779–1783. doi:10.1002/smll.201002376 |
| 25. | Chen, H.-Y.; Zhang, T.-L.; Fan, J.; Kuang, D.-B.; Su, C.-Y. ACS Appl. Mater. Interfaces 2013, 5, 9205–9211. doi:10.1021/am402853q |
| 29. | Cho, J. S.; Hong, Y. J.; Kang, Y. C. Chem. – Eur. J. 2015, 21, 11082–11087. doi:10.1002/chem.201500729 |
| 43. | Xing, Z.; Asiri, A. M.; Obaid, A. Y.; Sun, X.; Ge, X. RSC Adv. 2014, 4, 9061–9063. doi:10.1039/c3ra47239f |
| 1. | Qiao, H.; Luo, L.; Chen, K.; Fei, Y.; Cui, R.; Wei, Q. Electrochim. Acta 2015, 160, 43–49. doi:10.1016/j.electacta.2015.02.012 |
| 29. | Cho, J. S.; Hong, Y. J.; Kang, Y. C. Chem. – Eur. J. 2015, 21, 11082–11087. doi:10.1002/chem.201500729 |
| 39. | Guo, B.; Wang, X.; Fulvio, P. F.; Chi, M.; Mahurin, S. M.; Sun, X.-G.; Dai, S. Adv. Mater. 2011, 23, 4661–4666. doi:10.1002/adma.201102032 |
| 40. | Aricò, A. S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; van Schalkwijk, W. Nat. Mater. 2005, 4, 366–377. doi:10.1038/nmat1368 |
| 41. | Bruce, P. G.; Scrosati, B.; Tarascon, J.-M. Angew. Chem., Int. Ed. 2008, 47, 2930–2946. doi:10.1002/anie.200702505 |
| 42. | Ma, H.; Zhang, S.; Ji, W.; Tao, Z.; Chen, J. J. Am. Chem. Soc. 2008, 130, 5361–5367. doi:10.1021/ja800109u |
| 38. | Wang, J.; Polleux, J.; Lim, J.; Dunn, B. J. Phys. Chem. C 2007, 111, 14925–14931. doi:10.1021/jp074464w |
| 30. | Tang, K.; Yu, Y.; Mu, X.; van Aken, P. A.; Maier, J. Electrochem. Commun. 2013, 28, 54–57. doi:10.1016/j.elecom.2012.12.005 |
| 2. | Luo, L.; Qiao, H.; Chen, K.; Fei, Y.; Wei, Q. Electrochim. Acta 2015, 177, 283–289. doi:10.1016/j.electacta.2015.01.100 |
| 34. | Qiao, H.; Li, J.; Fu, J.; Kumar, D.; Wei, Q.; Cai, Y.; Huang, F. ACS Appl. Mater. Interfaces 2011, 3, 3704–3708. doi:10.1021/am200884k |
| 35. | Cherian, C. T.; Sundaramurthy, J.; Reddy, M. V.; Kumar, P. S.; Mani, K.; Pliszka, D.; Sow, C. H.; Ramakrishna, S.; Chowdari, B. V. R. ACS Appl. Mater. Interfaces 2013, 5, 9957–9963. doi:10.1021/am401779p |
| 36. | Morgan, B. J.; Madden, P. A. Phys. Rev. B 2012, 86, 035147. doi:10.1103/PhysRevB.86.035147 |
| 37. | Moriguchi, I.; Hidaka, R.; Yamada, H.; Kudo, T.; Murakami, H.; Nakashima, N. Adv. Mater. 2006, 18, 69–73. doi:10.1002/adma.200501366 |
© 2017 Zhang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)