Abstract
α-Fe2O3 nanomaterials with an elongated nanorod morphology exhibiting superior electrochemical performance were obtained through hydrothermal synthesis assisted by diamine derivatives as shape-controlling agents (SCAs) for application as anodes in lithium-ion batteries (LIBs). The physicochemical characteristics were investigated via XRD and FESEM, revealing well-crystallized α-Fe2O3 with adjustable nanorod lengths between 240 and 400 nm and aspect ratios in the range from 2.6 to 5.7. The electrochemical performance was evaluated by cyclic voltammetry and charge–discharge measurements. A SCA test series, including ethylenediamine, 1,2-diaminopropane, 2,3-diaminobutane, and N-methylethylenediamine, was implemented in terms of the impact on the nanorod aspect ratio. Varied substituents on the vicinal diamine structure were examined towards an optimized reaction center in terms of electron density and steric hindrance. Possible interaction mechanisms of the diamine derivatives with ferric species and the correlation between the aspect ratio and electrochemical performance are discussed. Intermediate-sized α-Fe2O3 nanorods with length/aspect ratios of ≈240 nm/≈2.6 and ≈280 nm/≈3.0 were found to have excellent electrochemical characteristics with reversible discharge capacities of 1086 and 1072 mAh g−1 at 0.1 C after 50 cycles.
Introduction
Since conventional transportation is seen as problematic in terms of fossil fuel consumption and human-induced greenhouse gas emissions [1], battery electric vehicles (BEVs) have moved into the focus of the automotive industry. As power sources, lithium-ion batteries (LIBs) are considered as the most promising candidates, since LIBs offer the highest energy density of all known rechargeable battery systems [2-4]. In order to address today’s challenges of electromobility (e.g., customer acceptance of BEVs by extending driving ranges, faster charging, and lower costs), the development and optimization of electrode materials are of great interest. Considering that a target driving range of 300 miles is required for BEVs to achieve a sustainable mass market penetration (as defined by the US Department of Energy), the energy density of the BEV power source must be increased by a factor of 2.5 by 2030 [5]. Since the commercialization of LIBs in 1991 by the Sony Cooperation [6,7], carbon-based materials such as graphite have predominantly been applied as negative electrodes. Although graphite is advantageous due to its high coulombic efficiency, its flat voltage curve and the low operating voltage of 0.1 V vs Li/Li+ it is limited to a lithium storage capacity of only 372 mAh g−1, given a stoichiometry of LiC6 [8]. In 2000, the Tarascon research group brought attention to transition metal oxides as a new class of possible anode materials that showed capacities in the range of twice as high as graphite [9], and some of them even show values higher than 1000 mAh g−1 with the interaction with lithium ions [10]. Therefore, transition metal oxides are candidates as new, high-capacity, electrode active materials in next generation LIBs. Among them, α-Fe2O3 (hematite) has attracted more and more attention due to its non-toxicity, high corrosion resistance, low processing costs, and especially, for its high theoretical gravimetric lithium-ion storage capacity of 1007 mAh g−1. However, α-Fe2O3 interacts with lithium ions via a conversion reaction, leading to finely dispersed metal nanocrystals in a Li2O matrix. This process is associated with large volume changes and also exhibits irreversible phase transformation (from a hexagonal anionic packing to a cubic phase [11]) that causes a poor long-time cycling behavior with severe capacity fading [12-14]. Possibilities to solve these problems include shape controlling towards nanometer-scale morphologies. It has been widely demonstrated that nanometer-scale α-Fe2O3 particles show excellent electrochemical performance in the reaction with lithium ions as compared to micrometer-scale crystals. This can be attributed to the comparatively shorter lithium-ion path ways and better ability to accommodate the strain during volume change caused by lithiation/delithiation [15]. The delithiation step, which is essential for the reaction reversibility, is considered thermodynamically impossible for micrometer-sized materials, but becomes feasible for their nanostructured derivatives [9,11,16-20]. Therefore, morphology control and nanostructuring of α-Fe2O3 is of great topical importance. Among several synthesis methods for α-Fe2O3 crystal phases, including precipitation and sol−gel approaches, the hydro/solvothermal synthesis route is known to be advantageous due to the homogenous nucleation/growth process and highly crystallized products [20-22]. In order to develop a synthesis method with morphology control aspects, inorganic ions as well as organic reagents (including phosphates, sulfates, chlorides, fluorides, benzenediols, nitrilotriacetic acid and diamine derivatives) have been successfully applied as shape-controlling agents (SCAs) to obtain α-Fe2O3 with various morphologies enclosed by different crystal facets [22-27]. The diamine-assisted hydrothermal synthesis of α-Fe2O3 nanoparticles is seen as beneficial, being a low-cost and time-efficient combination of an efficient SCA and a powerful synthesis tool. The resulting α-Fe2O3 is free from nitrogen (elemental analysis) [28], since the applied diamine derivatives are normally water-soluble and can be easily removed. With this technique, expensive, difficult to scale-up, structure-directing approaches involving sacrificial hard templates can be avoided. The aspect ratio of the resulting nanorod morphology is adjustable by the type of diamine derivative used and other reaction parameters. Li et al. applied a hydrothermal synthesis assisted by 1,2-diaminopropane as an SCA and obtained crystallized α-Fe2O3 nanorods with high and controllable aspect ratios [22]. Lin et al. demonstrated that such α-Fe2O3 nanorods outperform sub-micrometer and micrometer-sized particles in terms of electrochemical performance in LIBs [17]. Also, ethylenediamine is used in the hydrothermal synthesis of α-Fe2O3 nanoparticles, leading to a shuttle-like nanorod morphology [27]. Diamines increase the pH of the reaction mixture, supporting the phase transformation of FeOOH to α-Fe2O3. In addition, diamines form chelate complexes with ferric ions in solution, leading to a directional dependency of OH− attacks for subsequent hydrolysis processes, which results in a targeted growth direction of iron oxide products [29]. Furthermore, diamines directly coordinate to ferric lattice species and the complexes are released into solution. Since it is understood that this coordination will specifically take place on the α-Fe2O3 surface facets with only singly coordinated hydroxyl groups, this interaction mechanism can be applied to direct the crystal growth in a specific growth direction. It was found that 1,2-diaminopropane outperforms ethylenediamine as an SCA. Also, the vicinal position of the two amine groups seems to be crucial for shape controlling, since it was reported that 1,3-diaminopropane shows little effect on directing the crystal growth of α-Fe2O3. [22]. To gain a deeper understanding of the role of diamines in the shape-controlled synthesis of α-Fe2O3, a detailed test series, including ethylenediamine, 1,2-diaminopropane, 2,3-diaminobutane and N-methylethylenediamine, is presented. The effect of the increasing number of H3C-groups, contributing to a larger steric hindrance and an increase in the electron density (+M effect) at the reactive diamine unit, is investigated. N-methylethylenediamine was included since it is a constitutional isomer to 1,2-diaminopropane and allows investigation in terms of the position of the H3C-group. To the best of our knowledge, 2,3-diaminobutane and N-methylethylenediamine are presented for the first time as SCAs in α-Fe2O3 synthesis.
Experimental
In a typical procedure, 32 mL of 1,2-diaminopropane (99%, Sigma-Aldrich) was added drop wise into 18 mL of 1.5 M FeCl3·6H2O (99%, Riedel-de Haën) under constant magnetic stirring. Subsequently, the solution was transferred to a 100 mL autoclave with teflon-lined stainless steel for hydrothermal treatment at 180 °C for 16 h. The product was allowed to naturally cool down to room temperature after reaction and collected by centrifugation with deionized water and ethanol. The sample was dried overnight at 60 °C, and a red-colored product was obtained. For a systematic study, the concentration of FeCl3·6H2O was varied, while the ratio between iron ions and organic SCA was kept constant. The effectiveness of the SCA was compared by using ethylenediamine (99%, Sigma-Aldrich), 2,3-diaminobutane (95%, Otava) and N-methylethylenediamine (95%, Sigma-Aldrich) instead of 1,2-diaminopropane.
X-ray diffraction (XRD) analysis was performed on a Shimadzu XRD-6000 diffractometer operating at 40 kV and 40 mA using Cu Kα radiation (λ = 0.154 nm) with a copper target and a nickel filter. The surface morphology and microstructure of the samples was analyzed using a JEOL 6340F field emission scanning electron microscope (FESEM) in secondary electron imaging mode. The accelerating voltage was set to 5 kV.
The electrodes were prepared by mixing 40% of as-synthesized active iron oxide powder with 40% of conductive additives (Super P Li carbon, Timcal) and 20% of polyvinylidene difluoride (PVdF) binder (Kynar 2801) by weight in 1-methyl-2-pyrrolidinone (NMP, 99.5%, Sigma-Aldrich). This was coated on a copper foil with a doctor blade (gap height of 300 μm) then oven-dried at 60 °C overnight and roll-pressed with 20 tons. The dried electrode had a layer thickness of around 30 μm and a mass loading of around 2.5 mg cm−2. The porosity of the electrode was estimated to be around 30% by calculating the ratio between theoretical volume and the real volume of the electrode. The electrode was assembled into a coin cell (CR2016) with lithium foil (0.59 mm thickness, Hohsen Corp., Japan) as both a counter and a reference electrode in an argon-filled glove box (H2O, O2 < 1 ppm, Mbraun, Unilab, USA). 150 μL of LiPF6 (1 M) in ethylene carbonate/diethyl carbonate (1:1 w/w, Danvec) was used as the electrolyte and a Celgard 2400 membrane was used as the separator. The cycling performance tests were conducted with a multichannel battery tester (Neware Technology Limited) with discharge and charge current density of 0.1 C (100 mAh g−1) at room temperature. Rate performance tests were conducted using the same instrument with charge current of 0.1 C and discharge current varying from 0.1 to 3 C. Cycling voltammetry (CV) was performed in the range of 0.05 to 3.00 V with a sweep rate of 0.1 mV s−1 on a Solartron electrochemical workstation (1470E and SI 1255B impedance/gain-phase analyzer coupled with a potentiostat).
Results and Discussion
Structural characterization
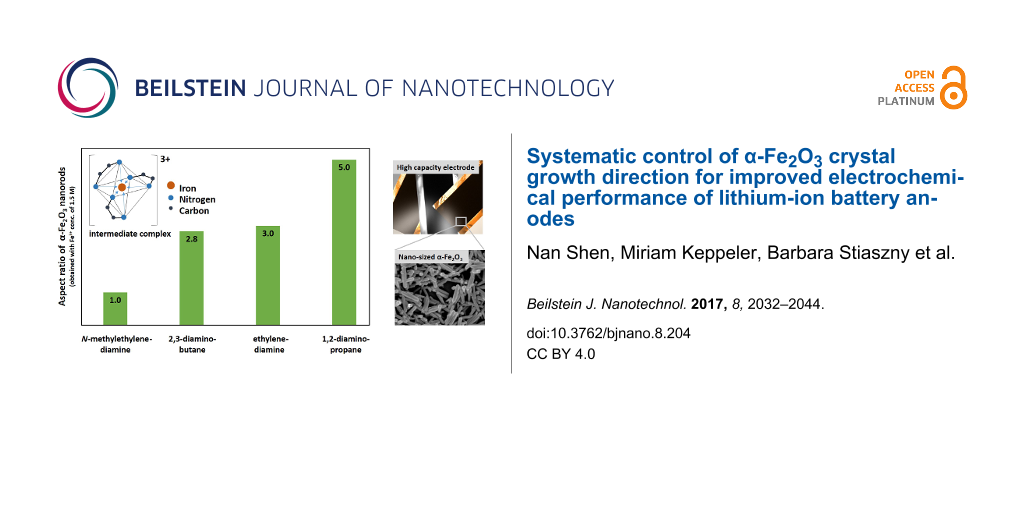
The sample nomenclature was set to α-Fe2O3-SX.X, in which S refers to the type of SCA (E for ethylenediamine, D for 1,2-diaminopropane, B for 2,3-diaminobutane, and N for N-methylethylenediamine) and X.X refers to the amount of Fe3+ in moles, which was varied over a range of 0.1 to 2.0. The hydrothermal synthesis parameters of all samples were set to a 1:13 molar ratio of FeCl3/SCA and the reaction temperature/time was 180 °C/16 h, which was optimized towards fully crystalized pure phase products in a preliminary test series and were found to be in good agreement with previously reported α-Fe2O3 synthesis [22,30]. A systematic study was conducted to analyze the effect of Fe3+ concentration and various SCAs on crystal phase formation and morphology of α-Fe2O3. All applied diamine derivatives are schematically illustrated in Figure 1.
![[2190-4286-8-204-1]](/bjnano/content/figures/2190-4286-8-204-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic illustration of applied SCAs containing two vicinal amine groups and different H3C-groups.
Figure 1: Schematic illustration of applied SCAs containing two vicinal amine groups and different H3C-groups....
For comparison, approaches without an SCA but using NaOH(aq) to adjust the pH value for the phase transformation of FeOOH to α-Fe2O3 were also conducted. These approaches result in micrometer-sized particles without a periodically repeating unit in the nanometer regime.
Figure 2a illustrates the XRD patterns of samples obtained from a test series with 1,2-diaminopropane as the SCA and a varied Fe3+ concentration (α-Fe2O3-D0.1, α-Fe2O3-D0.5, α-Fe2O3-D1.0, α-Fe2O3-D1.5, and α-Fe2O3-D2.0), whereas Figure 2b shows the diffractograms of samples obtained from a test series with a constant Fe3+ concentration (here 1.5 M) and varied SCA (α-Fe2O3-E1.5, α-Fe2O3-D1.5, α-Fe2O3-B1.5 and α-Fe2O3-N1.5).
![[2190-4286-8-204-2]](/bjnano/content/figures/2190-4286-8-204-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) XRD patterns of α-Fe2O3 nanoparticles synthesized with various Fe3+ concentrations and 1,2-diaminopropane as the SCA and b) different diamine derivatives as SCAs and a fixed Fe3+ concentration.
Figure 2: a) XRD patterns of α-Fe2O3 nanoparticles synthesized with various Fe3+ concentrations and 1,2-diami...
The reflections reported in Figure 2 indicate well-crystalized materials that can be indexed to α-Fe2O3 (JAPDS card No. 330664). The lattice constants were calculated using Rietveld refinement and found to be in the range of 5.030 to 5.033 Å for lattice constant a, and 13.748 to 13.757 Å for lattice constant c (see Table 1), which is in perfect agreement with reported values [31,32]. The obtained RBragg values vary from ≈3% to ≈7%, indicating a good reliability of the refined data. No reflections of detectable impurities were observed, indicating that diamines are useful to control the crystal growth direction, but do not have any observable impact on the crystal phase formation.
Table 1: Physicochemical characteristics of α-Fe2O3 synthesized with different SCAs and various Fe3+ concentrations. DAP: 1,2-diaminopropane, EDA: ethylenediamine, DAB: 2,3-diaminobutane, N-MED: N-methylethylenediamine.
| Test series | Sample ID |
Fe3+ conc.
(M)/SCA |
Lattice constant, a (Å) | Lattice constant, c (Å) |
RBragg value
(%) |
Rod length (nm) | Aspect ratioa |
|---|---|---|---|---|---|---|---|
| Varied Fe3+ conc., fixed SCA (EDA) | α-Fe2O3-E0.1 | 0.1/EDA | 5.031 | 13.752 | 6.406 | spherical particle | – |
| α-Fe2O3-E0.5 | 0.5/EDA | 5.033 | 13.753 | 5.158 | spherical particle | – | |
| α-Fe2O3-E0.8 | 0.8/EDA | 5.033 | 13.757 | 4.838 | 275 | 2.9 | |
| α-Fe2O3-E1.0 | 1.0/EDA | 5.033 | 13.755 | 5.184 | 280 | 3.0 | |
| α-Fe2O3-E1.5 | 1.5/EDA | 5.033 | 13.757 | 6.672 | 276 | 3.0 | |
| α-Fe2O3-E2.0 | 2.0/EDA | 5.030 | 13.749 | 5.819 | 277 | 2.9 | |
| Varied Fe3+ conc., fixed SCA (DAP) | α-Fe2O3-D0.1 | 0.1/DAP | 5.031 | 13.750 | 4.636 | spherical particle | – |
| α-Fe2O3-D0.5 | 0.5/DAP | 5.031 | 13.748 | 5.209 | 237 | 2.6 | |
| α-Fe2O3-D0.8 | 0.8/DAP | 5.030 | 13.749 | 2.997 | 320 | 4.7 | |
| α-Fe2O3-D1.0 | 1.0/DAP | 5.030 | 13.753 | 4.496 | 397 | 5.7 | |
| α-Fe2O3-D1.5 | 1.5/DAP | 5.031 | 13.750 | 3.645 | 394 | 5.0 | |
| α-Fe2O3-D2.0 | 2.0/DAP | 5.032 | 13.754 | 4.467 | 399 | 4.9 | |
| Varied SCA, fixed Fe3+ conc. (0.5M) | α-Fe2O3-E0.5 | 0.5/EDA | 5.033 | 13.753 | 5.158 | spherical particle | – |
| α-Fe2O3-D0.5 | 0.5/DAP | 5.031 | 13.748 | 5.209 | 237 | 2.6 | |
| α-Fe2O3-B0.5 | 0.5/DAB | 5.033 | 13.750 | 7.067 | particle | – | |
| α-Fe2O3-N0.5 | 0.5/N-MED | b | b | b | b | b | |
| Varied SCA, fixed Fe3+ conc. (1.5M) | α-Fe2O3-E1.5 | 1.5/EDA | 5.033 | 13.757 | 6.672 | 276 | 3.0 |
| α-Fe2O3-D1.5 | 1.5/DAP | 5.031 | 13.750 | 3.645 | 394 | 5.0 | |
| α-Fe2O3-B1.5 | 1.5/DAB | 5.032 | 13.751 | 5.073 | 256 | 2.8 | |
| α-Fe2O3-N1.5 | 1.5/N-MED | 5.031 | 13.748 | 7.019 | spherical particle | – | |
aAspect ratio is not calculated for samples without nanorod morphology. bNot synthesized, since an even higher Fe3+ concentration of 1.5 M did not lead to a nanorod morphology.
FESEM was applied to examine the morphology of the final α-Fe2O3 products. Figure 3a–f displays the morphology evolution with increased Fe3+ concentration from 0.1 to 2.0 M and 1,2-diaminopropane used as an SCA. At an Fe3+ concentration of 0.1 M, nanoparticles with a diameter of ≈100 nm are obtained. When the Fe3+ concentration is increased to 0.5 M, a 1-D rod-like morphology starts to form with rod length of ≈240 nm, resulting in an aspect ratio of ≈2.6. Increasing the Fe3+ concentration to 0.8 M leads to an elongated nanorod shape with rod length of ≈320 nm and an aspect ratio of ≈4.7. At higher Fe3+ concentrations from 1.0 to 2.0 M, pronounced elongated nanorods with a length of ≈400 nm and aspect ratio in the range of 4.9 to 5.7 were obtained.
![[2190-4286-8-204-3]](/bjnano/content/figures/2190-4286-8-204-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: FESEM images of α-Fe2O3 nanoparticles obtained with 1,2-diaminopropane as the SCA and varied Fe3+ concentration of a) 0.1 M, b) 0.5 M, c) 0.8 M, d) 1.0 M, e) 1.5 M and f) 2.0 M; and α-Fe2O3 nanoparticles obtained with ethylenediamine as the SCA and varied Fe3+ concentration of g) 0.1 M, h) 0.5 M, i) 0.8 M, j) 1.0 M, k) 1.5 M and l) 2.0 M; magnification: 50,000.
Figure 3: FESEM images of α-Fe2O3 nanoparticles obtained with 1,2-diaminopropane as the SCA and varied Fe3+ c...
To verify the impact of the additional H3C-group at the vicinal diamine unit, 1,2-diaminopropane is substituted by ethylenediamine, while all further reaction parameters are kept constant. In general, the Fe3+ concentration vs aspect ratio dependence shows an analogical profile shape as obtained for 1,2-diaminopropane, but shorter rod length for each tested Fe3+ concentration (Figure 3g–l and Figure 4). In detail, at lower Fe3+ concentrations of 0.1 M and 0.5 M, nanoparticles with a diameter of ≈100 nm are obtained with ethylenediamine as the SCA. When the Fe3+ concentration is increased to 0.8 M, nanorods with a length of ≈275 nm and aspect ratio of ≈2.9 are achieved. A further increase of the Fe3+ concentration up to 2.0 M leads to a comparable rod-like morphology with a rod length of ≈280 nm and aspect ratio of ≈3.0.
![[2190-4286-8-204-4]](/bjnano/content/figures/2190-4286-8-204-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Aspect ratio vs Fe3+ concentration for α-Fe2O3 nanoparticles obtained with 1,2-diaminopropane and ethylenediamine as the SCA (the aspect ratio for spherical particles is set to 1; 20 nanorods were taken for the estimation of the aspect ratio for each Fe3+ concentration; a certain fluctuation margin cannot be excluded especially due to a natural variation in the rod length of about 20%).
Figure 4: Aspect ratio vs Fe3+ concentration for α-Fe2O3 nanoparticles obtained with 1,2-diaminopropane and e...
Diamines have several substantial effects in the α-Fe2O3 synthesis that in general involves a two-step phase transformation, formally described as Fe(OH)3 → FeOOH → α-Fe2O3. The pKb is decreased in the presence of diamines, which accelerates the conversion of FeOOH to α-Fe2O3 [28]. In addition, the crystal growth direction of iron oxide polymorphs like FeOOH, Fe2O3 or Fe3O4 is controlled by diamines. Among other synthesis parameters (concentration, reaction time, temperature and solvents) [27,29,33], the shape controlling ability can be adjusted by substituents located at the reactive diamine unit that affects electronic density (+M effect) and steric interactions, which is verified by results of our test series. Diamines are typical chelate ligands that interact with appropriate coordination centers. First, octahedral-shaped complexes are formed [29], formally described as (Fe[C2H2N2R1R2]3)3+. Diamine derivatives, in which the two amine groups are located at adjacent C atoms (vicinal position) are assumed to be supportive, since a thermodynamically favored 5-membered ring can be formed. This possibly explains the superior shape-controlling ability of 1,2-diaminopropane over 1,3-diaminopropane. Besides shape control, the complex formation has a second effect: the free-iron concentration in the solution is reduced, which separates nucleation and growth processes and supports high quality crystallization [27].
The complex stability is expected to decrease during synthesis with increasing temperature and pressure until the Fe3+ in complex formation can be successfully attacked by OH−. This will result in the gradual loss of diamine ligands, leading to complexes with two OH− groups and two diamines that form a quasi-square-plane perpendicular to the OH− direction [29]. Therefore, the following hydrolysis process is inhibited in the planar direction, but favored in the normal direction, supporting the growth of FeOOH along the [110] direction [22,29]. In addition, the growth direction of α-Fe2O3 is dictated since diamines not only interact with free-iron ions in solution, but also coordinate directly to the ferric lattice, and the complexes are released into solution [28]. The diamine adsorption capacity of α-Fe2O3 surfaces varies for differently indexed crystal facets depending on their loading with singly, doubly, triply or geminal coordinated hydroxyl groups [34]. Only singly coordinated hydroxyl groups are assumed to be involved in the interaction with the diamines [35]. According to Barrón et al., singly coordinated hydroxyl groups are located at the [100], [110], [012], [104] and [113] facets of natural and artificial α-Fe2O3 [36]. Hence the crystal growth in these directions is minimized, leading to α-Fe2O3 products with elongated shapes along the [001] direction.
All supposed reaction mechanisms involve the direct interaction with ferric species and the freely accessible diamine unit. To support this assumption, N-methylethylenediamine was also tested as an SCA, since it is a constitutional isomer to 1,2-diaminopropane and allows investigation in terms of the position of the H3C-group. Consistent with other tested diamines, N-methylethylenediamine leads to fully crystallized α-Fe2O3 products (Figure 2). However, even when following these shape-controlling concepts, N-methylethylenediamine-assisted hydrothermal approaches did not lead to a nanorod morphology, but rather spherically shaped particles with a diameter of ≈100 nm (Fe3+ conc.: 1.5 M, Figure 5), indicating that N-methylethylenediamine substantially supports the α-Fe2O3 synthesis by providing an appropriate alkaline reaction medium for the phase transformation of FeOOH to α-Fe2O3. It can be concluded that the H3C-shift from C to N suppresses the ferric complex formation due to steric hindrance, and therefore, the directional dependency of the crystal growth is inhibited.
![[2190-4286-8-204-5]](/bjnano/content/figures/2190-4286-8-204-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: FESEM images of α-Fe2O3 nanoparticles obtained with a) ethylenediamine, b) 1,2-diaminopropane, c) N-methylethylenediamine and d) 2,3-diaminobutane, where the Fe3+ concentration was kept at 1.5 M, magnification: 50,000.
Figure 5: FESEM images of α-Fe2O3 nanoparticles obtained with a) ethylenediamine, b) 1,2-diaminopropane, c) N...
Since 1,2-diaminopropane shows better shape-controlling ability compared to ethylenediamine (possibly attributed to the additional H3C-group that supports complex formation by increased electron density (+M effect)), 2,3-diaminobutane was applied to investigate the impact of a second H3C-group. Besides an increased electron density, the steric hindrance caused by the exchange of H with the comparative bulky H3C-group (especially for the direct interaction with surface lattice ferric species in the later formation process) needs to be investigated. Fully crystallized α-Fe2O3 nanorods were obtained with 2,3-diaminobutane with a length of ≈260 nm and aspect ratio of ≈2.8 (Fe3+ conc.: 1.5 M). As discussed above, equivalent synthesis conditions led to slightly longer nanorods of ≈280 nm with an aspect ratio of ≈3.0 when ethylenediamine was used as the SCA. The use of 1,2-diaminopropane leads to products with longer lengths of ≈390 nm and aspect ratios of ≈5.0, indicating a marginal negative impact on the second H3C group on the complex formation with Fe3+ ions in solution and/or the interaction with the ferric species of the α-Fe2O3 surface due to steric hindrance. In general, the shape-controlling ability decreases in the following order: 1,2-diaminopropane > ethylenediamine ≥ 2,3-diaminobutane >> N-methylethylenediamine, which is in perfect agreement with the electronic and steric conditions of the applied diamines with respect to the above mentioned Fe-diamine interaction concepts.
The improved shape-controlling ability of 1,2-diaminopropane is also confirmed in the Fe3+ concentration test series. For 1,2-diaminopropane-assisted synthesis, the nanorod formation can already be observed with comparatively low Fe3+ concentrations of 0.5 M leading to products with ≈240 nm in length and aspect ratios of ≈2.6. The application of the other discussed diamine derivatives results in spherical particles of ≈100 nm diameter under the same conditions (Figure 6).
![[2190-4286-8-204-6]](/bjnano/content/figures/2190-4286-8-204-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: FESEM images of α-Fe2O3 nanoparticles obtained with a) ethylenediamine, b) 1,2-diaminopropane and c) 2,3-diaminobutane, where the Fe3+ concentration was kept at 0.5 M, magnification: 50,000.
Figure 6: FESEM images of α-Fe2O3 nanoparticles obtained with a) ethylenediamine, b) 1,2-diaminopropane and c...
Electrochemical studies
The interaction of lithium with α-Fe2O3 is assumed to occur via a multistep reaction with a conversion mechanism according to the following equations [37]:
![[2190-4286-8-204-i1]](/bjnano/content/inline/2190-4286-8-204-i1.svg?max-width=590&scale=1.18182)
![[2190-4286-8-204-i2]](/bjnano/content/inline/2190-4286-8-204-i2.svg?max-width=590&scale=1.18182)
![[2190-4286-8-204-i3]](/bjnano/content/inline/2190-4286-8-204-i3.svg?max-width=590&scale=1.18182)
To investigate the reaction mechanism of nanometer-scale α-Fe2O3-SX.X with lithium ions, cycling voltammetry (CV) tests were applied. The obtained data for all samples is shown in Figure 7 where the cyclic voltammetry curve for the first three cycles of α-Fe2O3-E1.5 in the voltage range of 3.00 to 0.05 V at a scan rate of 0.1 mV s−1 is given.
![[2190-4286-8-204-7]](/bjnano/content/figures/2190-4286-8-204-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Cyclic voltamogramm profile of nanorod-shaped α-Fe2O3 (obtained with a 1.5 M Fe3+ concentration and ethylenediamine as the SCA).
Figure 7: Cyclic voltamogramm profile of nanorod-shaped α-Fe2O3 (obtained with a 1.5 M Fe3+ concentration and...
For the first cathodic sweep, peaks located at 1.5 to 1.3 V and 1.1 V are observed, which correspond to lithium-ion intercalation into the α-Fe2O3 matrix with a multistep electrochemical reaction associated with the phase transformation from LixFe2O3 to cubic Li2Fe2O3 [38]. Subsequently, an intense peak at 0.78 V is presented, indicating the reduction to Fe0, associated with the conversion reaction leading to metallic particles finely dispersed in Li2O and electrolyte decomposition with solid–electrolyte interface (SEI) formation [39]. From the second cathodic cycle onwards, the peaks from 1.5 to 1.3 V and at 1.1 V do not appear and also the peak at 0.78 V shifts slightly into the higher voltage range with decreased intensity, most likely due to the irreversibility of the redox reaction as stated in the literature [14]. For the first anodic sweep, broad oxidation peaks at 1.6 and 1.8 V are observed, corresponding to the oxidation of Fe0 to Fe2+/Fe3+ [40]. The galvanostatic delithiation/lithiation profiles of α-Fe2O3 with nanorod morphology at 0.1 C between the voltage window of 0.05 to 3.00 V are shown in Figure 8a,b.
![[2190-4286-8-204-8]](/bjnano/content/figures/2190-4286-8-204-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: a) Galvanostatic delithiation/lithiation profiles (1st, 2nd and 5th cycle) for α-Fe2O3-E1.5, b) 1st and 2nd cycle comparison, c) cycling performance, and d) rate testing of α-Fe2O3-E0.5, α-Fe2O3-E1.5, α-Fe2O3-D0.5, α-Fe2O3-D0.8, α-Fe2O3-D1.5 and α-Fe2O3-B1.5.
Figure 8: a) Galvanostatic delithiation/lithiation profiles (1st, 2nd and 5th cycle) for α-Fe2O3-E1.5, b) 1st...
The obtained data are in good agreement with the CV profiles. In general, all α-Fe2O3 samples exhibit two slopes for the first charge curve from the starting voltage to around 1.2 V and from around 1.1 V to 0.8 V, which corresponds to the intercalation of lithium into α-Fe2O3, resulting in LixFe2O3 and cubic Li2Fe2O3, respectively [11,37]. A strong, pronounced plateau at around 0.8 V indicates the reduction of Fe2+ to Fe0 and formation of amorphous Li2O [14]. As illustrated for α-Fe2O3-E1.5 (Figure 8a), all samples exhibit a significant capacity loss during the first lithiation cycle. The first lithiation capacities for α-Fe2O3-E0.5, α-Fe2O3-E1.5, α-Fe2O3-D0.5, α-Fe2O3-D0.8, α-Fe2O3-D1.5 and α-Fe2O3-B1.5 are around 1400–1500 mAh g−1, and the capacity of the 1st delithiation cycle is about 1100 mAh g−1 with the respective coulombic efficiency of about 75%. The exact values are stated in Table 2. The initial capacity loss is possibly attributed to an incomplete conversion reaction and irreversible loss of lithium due to SEI formation [41]. The average 1st cycle lithiation voltages are calculated in the range of 0.69 to 0.75 V, while the average 1st cycle delithiation voltages are calculated from 1.43 to 1.49 V. Details for the delithiation capacity, relative capacity, error bars for cycling performance, and rate capability of α-Fe2O3-E0.5, α-Fe2O3-E1.5, α-Fe2O3-D0.5, α-Fe2O3-D0.8, α-Fe2O3-D1.5 and α-Fe2O3-B1.5 can be found in Supporting Information File 1.
Table 2: Electrochemical data of α-Fe2O3 synthesized with different SCAs and various Fe3+ concentrations.
| Sample ID | 1st cycle lithiation capacity (mAh g−1) | 1st cycle delithiation capacity (mAh g−1) | 1st cycle columbic efficiency (%) | 1st cycle average lithiation voltage (V) | 1st cycle average delithiation voltage (V) | 1st cycle ∆E (V) | 50th cycle lithiation capacity (mAh g−1) | 50th cycle delithiation capacity (mAh g−1) |
|---|---|---|---|---|---|---|---|---|
| α-Fe2O3-E0.5 | 1465 | 1066 | 73 | 0.739 | 1.432 | 0.693 | 1001 | 986 |
| α-Fe2O3-E1.5 | 1545 | 1132 | 73 | 0.746 | 1.436 | 0.690 | 1072 | 1053 |
| α-Fe2O3-D0.5 | 1479 | 1087 | 73 | 0.745 | 1.433 | 0.688 | 1086 | 1062 |
| α-Fe2O3-D0.8 | 1494 | 1143 | 77 | 0.743 | 1.494 | 0.751 | 957 | 935 |
| α-Fe2O3-D1.5 | 1526 | 1121 | 73 | 0.760 | 1.491 | 0.731 | 848 | 829 |
| α-Fe2O3-B1.5 | 1422 | 1020 | 72 | 0.726 | 1.447 | 0.721 | 1034 | 1017 |
The cycling performance of α-Fe2O3-E0.5, α-Fe2O3-E1.5, α-Fe2O3-D0.5, α-Fe2O3-D0.8, α-Fe2O3-D1.5 and α-Fe2O3-B1.5 at 0.1 C is illustrated in Figure 8c. Exceptional cycling stability at around 1000 mAh g−1 for 50 cycles was observed. In particular, samples with intermediate rod lengths (α-Fe2O3-E1.5, α-Fe2O3-D0.5 and α-Fe2O3-B1.5) in the range of ≈240 nm up to ≈280 nm show a higher capacity over spherical α-Fe2O3 nanoparticles (α-Fe2O3-E0.5) or samples with longer rod lengths of approximately ≈320 nm (α-Fe2O3-D0.8) and ≈390 nm (α-Fe2O3-D1.5). In general, the cycling behavior of the presented nanometer-scale α-Fe2O3 samples is improved over their micrometer-sized counterparts, whose capacity is reported to decrease below 200 mAh g−1 after a few cycles [17]. This can be attributed to the specific nanometer-scale dimensions of the material, which have several impacts: (1) improved ability to accommodate the strain during volume changes caused by lithiation/delithiation. (2) The rate of lithium insertion and removal is increased. The time constant t = L2/D (L: diffusion length, D: diffusion constant) is minimized by reducing the diffusion length L of the nanostructures [42]. (3) The extraction of lithium from Li2O becomes thermodynamically possible for nanometer-sized materials [18].
All nanometer-scale designed α-Fe2O3 samples exhibit capacity values that are far higher than those of commercialized graphite anode material at low rates (372 mAh g−1 for stoichiometry of LiC6) even in the higher C regions [8]. The samples α-Fe2O3-E1.5, α-Fe2O3-D0.5 and α-Fe2O3-B1.5 with intermediate nanorod lengths of ≈280, ≈240 and ≈260 nm show charge capacities of 746, 770 and 740 mAh g−1 at a charge current density of 3 C. This exhibits the superior electrode kinetics over nanoparticles (590 mAh g−1) and longer rods with ≈320 nm (696 mAh g−1) and ≈390 nm (655 mAh g−1) lengths. The sample α-Fe2O3-B1.5, synthesized using 2,3-diaminobutane, exhibited a nanorod length of ≈260 nm with an aspect ratio of ≈2.8 and showed the highest ability to recover the initial capacity range by providing 1018 mAh g−1 when the current rate was returned to its starting value of 0.1 C (Figure 8d).
A comparison of the presented α-Fe2O3 nanorods with other elongated α-Fe2O3 nanoparticles (e.g. nanorods with hexagonal structure, electrospun nanorods, and nanowires) or other morphologies (e.g. nanotubes, flower-like nanostructure, spindles, and hollow spheres) showed a notable electrochemical performance in the interaction with lithium. The results are also comparable to other iron oxides or iron oxide–carbon composites, such as Fe3O4, Fe2O3/C or Fe3O4/C, which can be also obtained by facile synthesis approaches (Table 3).
Table 3: Electrochemical performance of several α-Fe2O3 nanoparticles with different morphologies.
| Active material | Particle size |
Surface area
(m2 g−1) |
Current rate |
Initial
capacity (mAh g−1) |
Reversible capacity
(mAh g−1)/Cycle no. |
Ref. |
|---|---|---|---|---|---|---|
| α-Fe2O3 nanorods | length: ≈276 nm | not specified | 0.1 C |
1545
(discharge) |
1072/50 | this work |
| α-Fe2O3 nanorods with hexagonal structure |
length: ≈400 nm
diameter: ≈40 nm |
not specified | 0.2 C |
908
(charge) |
900/30 | [17] |
|
electrospun α-Fe2O3 nanorods
(sample includes 8% γ-Fe2O3) |
average diameter: 150 nm | 27.6 (±0.2) | 50 mA g−1 |
1515 (±20)
(discharge) |
1095/50 | [31] |
| α-Fe2O3 nanotubes | length: 60 µm | 45 | 100 mA g−1 |
1415
(discharge) |
510/100 | [43] |
| α-Fe2O3 nanowires | width: ≈200 nm length/diameter ratio: 500 | 152 | 0.1 C | 1303 (discharge) | 456/100 | [44] |
| α-Fe2O3 flower-like nanostructures |
1–2 µm
(composed of ≈20 nm thin petals) |
71.9 | 0.2 mA cm−2 | 974.43 (discharge) | 548.47/30 | [45] |
| spindle-like mesoporous α-Fe2O3 |
length: ≈0.8 μm
width: ≈0.4 μm, composed of clustered Fe2O3 nanoparticles <20 nm |
75 | 0.2 C |
1372
(discharge) |
911/50 | [46] |
| α-Fe2O3 hollow spheres | 1 µm spherical particles composed of nanosheet subunits | 103.3 | 200 mA g−1 |
1219
(discharge) |
710/100 | [47] |
| Fe2O3/graphene composite |
carbon content: ≈20%
diameter: 50–200 nm anchored on graphene |
77 | 100 mA g−1 |
1515
(discharge) |
995/50 | [48] |
| carbon coated Fe3O4 nanospindle |
carbon content: 21.5%
spindle particle with length: ≈400 nm diameter: ≈100 nm |
35.1 | 0.5 C |
749
(charge, at C/5) |
530/80 | [49] |
| Fe3O4 submicron particle |
diameter:
200–300 nm |
not specified | 0.2 C |
1332
(discharge) |
900/60 | [50] |
|
carbon-encapsulated Fe3O4
nanocomposite |
carbon content: 30%, diameter: ≈30 nm for Fe3O4 encapsulated in carbon | 110.6 | 93 mA g−1 | 1480 (discharge) | 920/50 | [51] |
| Fe3O4 anchored onto helical carbon nanofibers | carbon content: 50%, diameter: 10–50 nm for Fe3O4, anchored onto helical carbon nanofibers | 126 | 80 mA g−1 | not specified | 1220/20 (fully recovered at 85 cycles after higher rate testing) | [52] |
| Fe3O4/graphene oxide composites |
carbon content: 19%
diameter 500 nm in graphene sheet |
57.1 | 100 mA g−1 | 1233 | 1039/170 | [53] |
It should be mentioned that capacity profiles for all samples show an increase after a minimum at ≈25 cycles (Figure 8c). Especially for the shorter α-Fe2O3 nanorods (≈240 nm up to ≈280 nm) the capacity even reaches values beyond the theoretical value of 1007 mAh g−1 (based on the classical conversion reaction) over the monitored area. Such additional capacity occurrences and/or capacity increases in the reversible capacity regime were reported for several iron-oxide-based electrodes, including porous α-Fe2O3 nanorods [13], macroporous α-Fe2O3 submicrometer spheres [54], and 1D hollow α-Fe2O3 electrospun nanofibers [55], however its origin remains speculative. Several hypotheses are stated in the literature, among others, such as an interfacial lithium accommodation along with charge separation at phase boundaries proposed by Jamnik and Maier [56]. After conversion of α-Fe2O3, resulting in Fe finely dispersed in a Li2O matrix, additional lithium is supposed to be accommodated on the Li2O surfaces, whereas the electrons are transferred to the metal surface [57]. This mechanism strongly depends on the particle size and becomes significant in the nanometer regime [56]. Further explanations include processes related to the interfacial film formation, for instance, the possible gradual release of excessive Li+, previously captured by an initially inhomogeneous SEI that had sequentially developed and evolved towards a steady state over repetitive charge/discharge [58].
Conclusion
In this work, the shape-controlling effect of various vicinal diamine derivatives, including ethylenediamine, 1,2-diaminopropane, 2,3-diaminobutane, and N-methylethylenediamine, towards nanorod formation of crystalline α-Fe2O3 under hydrothermal conditions was systematically studied. The position and amount of substituents (H, H3C) show mutual effects on the structure-directing complex formation with the ferric species (solvated, and on the surface of α-Fe2O3) due to electron density enhancement (+M effect) and steric hindrance. The shape-controlling ability was found to decrease according to: 1,2-diaminopropane > ethylenediamine ≥ 2,3-diaminobutane >> N-methylethylenediamine. H3C groups, directly located at the nitrogen (N-methylethylenediamine), inhibit the ferric complex formation due to steric hindrance, and therefore, the directional dependence of the crystal growth is minimized. This confirms the assumption that the structure-directing process involves the direct interaction of the free vicinal diamine unit with the ferric species. Superior shape-controlling ability is presented for 1,2-diaminopropane, resulting in nanorod formation with ≈390 nm in length and aspect ratios of ≈5.0 (1.5 M Fe3+ concentration), indicating that one H3C-group located at the same C as an amine group supports chelating of ferric species by increased electron density (+M effect) but provides less steric hindrance. However, electrochemical investigation of α-Fe2O3 nanorods as anode materials in LIBs suggests that intermediate rod lengths in the range of 240 to 280 nm show superior electrochemical performance over longer rods and in addition to spherical particles or commercial powders. This suggests that the mechanical strength against deformation during lithiation and delithiation seems to become lower for a more elongated rod shape. The highest electrochemical performance within the current test series was achieved by the samples synthesized with 1,2-diaminopropane/0.5 M Fe3+ (α-Fe2O3-D0.5), resulting in a nanorod length and aspect ratio of ≈240 nm and ≈2.6, respectively. The obtained reversible capacities at 0.1 C were 1086 mAh g−1 for α-Fe2O3-D0.5. At 50 cycles and also at a higher current rate of 3 C, this sample showed a high capacity of 1062 mAh g−1 and 770 mAh g−1, respectively, suggesting that it is a promising material that should be considered for further energy storage R&D applications.
Supporting Information
After-cycling FESEM images for intermediate-sized and elongated nanorods, galvanostatic delithiation/lithiation and rate testing with error bars, ex situ XRD reflections for α-Fe2O3-E1.5 before and after cycling, and galvanostatic delithiation/lithiation for a half-cell composed of pure carbon black and PVdF (80% carbon black), cycled at 0.1 C.
| Supporting Information File 1: Additional experimental results. | ||
| Format: PDF | Size: 741.7 KB | Download |
References
-
Lam, L. T.; Louey, R. J. Power Sources 2006, 158, 1140–1148. doi:10.1016/j.jpowsour.2006.03.022
Return to citation in text: [1] -
Goodenough, J. B.; Park, K.-S. J. Am. Chem. Soc. 2013, 135, 1167–1176. doi:10.1021/ja3091438
Return to citation in text: [1] -
Manthiram, A. J. Phys. Chem. Lett. 2011, 2, 176–184. doi:10.1021/jz1015422
Return to citation in text: [1] -
Manthiram, A.; Chemelewski, K.; Lee, E.-S. Energy Environ. Sci. 2014, 7, 1339–1350. doi:10.1039/c3ee42981d
Return to citation in text: [1] -
Andre, D.; Kim, S.-J.; Lamp, P.; Lux, S. F.; Maglia, F.; Paschos, O.; Stiaszny, B. J. Mater. Chem. A 2015, 3, 6709–6732. doi:10.1039/C5TA00361J
Return to citation in text: [1] -
Thackeray, M. M.; Kang, S.-H.; Johnson, C. S.; Vaughey, J. T.; Benedek, R.; Hackney, S. J. Mater. Chem. 2007, 17, 3112–3125. doi:10.1039/b702425h
Return to citation in text: [1] -
Yoshino, A. Angew. Chem., Int. Ed. 2012, 51, 5798–5800. doi:10.1002/anie.201105006
Return to citation in text: [1] -
Yoshio, M.; Wang, H.; Fukuda, K. Angew. Chem. 2003, 115, 4335–4338. doi:10.1002/ange.200351203
Return to citation in text: [1] [2] -
Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.-M. Nature 2000, 407, 496–499. doi:10.1038/35035045
Return to citation in text: [1] [2] -
Yu, S.-H.; Lee, S. H.; Lee, D. J.; Sung, Y.-E.; Hyeon, T. Small 2016, 12, 2146–2172. doi:10.1002/smll.201502299
Return to citation in text: [1] -
Larcher, D.; Bonnin, D.; Cortes, R.; Rivals, I.; Personnaz, L.; Tarascon, J.-M. J. Electrochem. Soc. 2003, 150, A1643–A1650. doi:10.1149/1.1622959
Return to citation in text: [1] [2] [3] -
Wang, P. C.; Ding, H. P.; Bark, T.; Chen, C. H. Electrochim. Acta 2007, 52, 6650–6655. doi:10.1016/j.electacta.2007.04.072
Return to citation in text: [1] -
Yao, X.; Tang, C.; Yuan, G.; Cui, P.; Xu, X.; Liu, Z. Electrochem. Commun. 2011, 13, 1439–1442. doi:10.1016/j.elecom.2011.09.015
Return to citation in text: [1] [2] -
Wang, J.; Li, L.; Wong, C. L.; Sun, L.; Shen, Z.; Madhavi, S. RSC Adv. 2013, 3, 15316–15326. doi:10.1039/c3ra41886c
Return to citation in text: [1] [2] [3] -
Keppeler, M.; Shen, N.; Nageswaran, S.; Srinivasan, M. J. Mater. Chem. A 2016, 4, 18223–18239. doi:10.1039/C6TA08456G
Return to citation in text: [1] -
Larcher, D.; Masquelier, C.; Bonnin, D.; Chabre, Y.; Masson, V.; Leriche, J.-B.; Tarascon, J.-M. J. Electrochem. Soc. 2003, 150, A133–A139. doi:10.1149/1.1528941
Return to citation in text: [1] -
Lin, Y.-M.; Abel, P. R.; Heller, A.; Mullins, C. B. J. Phys. Chem. Lett. 2011, 2, 2885–2891. doi:10.1021/jz201363j
Return to citation in text: [1] [2] [3] [4] -
Liu, H.; Wang, G.; Park, J.; Wang, J.; Liu, H.; Zhang, C. Electrochim. Acta 2009, 54, 1733–1736. doi:10.1016/j.electacta.2008.09.071
Return to citation in text: [1] [2] -
Matsumura, T.; Sonoyama, N.; Kanno, R.; Takano, M. Solid State Ionics 2003, 158, 253–260. doi:10.1016/S0167-2738(02)00913-X
Return to citation in text: [1] -
NuLi, Y.; Zhang, P.; Guo, Z.; Liu, H. J. Electrochem. Soc. 2008, 155, A196–A200. doi:10.1149/1.2826278
Return to citation in text: [1] [2] -
Byrappa, K.; Adschiri, T. Prog. Cryst. Growth Charact. Mater. 2007, 53, 117–166. doi:10.1016/j.pcrysgrow.2007.04.001
Return to citation in text: [1] -
Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Liu, Z.; Lv, B.; Xu, Y.; Wu, D. J. Mater. Chem. A 2013, 1, 3040–3046. doi:10.1039/c2ta00987k
Return to citation in text: [1] -
Lv, B.; Liu, Z.; Tian, H.; Xu, Y.; Wu, D.; Sun, Y. Adv. Funct. Mater. 2010, 20, 3987–3996. doi:10.1002/adfm.201001021
Return to citation in text: [1] -
Sugimoto, T.; Itoh, H.; Mochida, T. J. Colloid Interface Sci. 1998, 205, 42–52. doi:10.1006/jcis.1998.5588
Return to citation in text: [1] -
Yang, Y.; Ma, H.; Zhuang, J.; Wang, X. Inorg. Chem. 2011, 50, 10143–10151. doi:10.1021/ic201104w
Return to citation in text: [1] -
Zhu, L.-P.; Liao, G.-H.; Bing, N.-C.; Zhao, X.; Gu, Y.-Y. Mater. Lett. 2011, 65, 1287–1290. doi:10.1016/j.matlet.2011.01.053
Return to citation in text: [1] [2] [3] [4] -
Kandori, K.; Yasukawa, A.; Ishikawa, T. J. Colloid Interface Sci. 1996, 180, 446–452. doi:10.1006/jcis.1996.0324
Return to citation in text: [1] [2] [3] -
Zhang, D.-E.; Zhang, X.-J.; Ni, X.-M.; Zheng, H.-G. Mater. Lett. 2006, 60, 1915–1917. doi:10.1016/j.matlet.2005.12.053
Return to citation in text: [1] [2] [3] [4] [5] -
Aravindan, V.; Nan, S.; Keppeler, M.; Madhavi, S. Electrochim. Acta 2016, 208, 225–230. doi:10.1016/j.electacta.2016.05.035
Return to citation in text: [1] -
Cherian, C. T.; Sundaramurthy, J.; Kalaivani, M.; Ragupathy, P.; Kumar, P. S.; Thavasi, V.; Reddy, M. V.; Sow, C. H.; Mhaisalkar, S.; Ramakrishna, S.; Chowdari, B. V. R. J. Mater. Chem. 2012, 22, 12198–12204. doi:10.1039/c2jm31053h
Return to citation in text: [1] [2] -
Pourghahramani, P.; Forssberg, E. Int. J. Miner. Process. 2006, 79, 106–119. doi:10.1016/j.minpro.2006.02.001
Return to citation in text: [1] -
Zhu, L.-P.; Xiao, H.-M.; Zhang, W.-D.; Yang, G.; Fu, S.-Y. Cryst. Growth Des. 2008, 8, 957–963. doi:10.1021/cg700861a
Return to citation in text: [1] -
Cornell, R. M.; Schwertmann, U. The iron oxides: structure, properties, reactions, occurrences and uses; Wiley-VCH: Weinheim, 2003. doi:10.1002/3527602097
Return to citation in text: [1] -
Jia, C.-J.; Sun, L.-D.; Yan, Z.-G.; You, L.-P.; Luo, F.; Han, X.-D.; Pang, Y.-C.; Zhang, Z.; Yan, C.-H. Angew. Chem. 2005, 117, 4402–4407. doi:10.1002/ange.200463038
Return to citation in text: [1] -
Barrón, V.; Torrent, J. J. Colloid Interface Sci. 1996, 177, 407–410. doi:10.1006/jcis.1996.0051
Return to citation in text: [1] -
Liu, X.; Si, W.; Zhang, J.; Sun, X.; Deng, J.; Baunack, S.; Oswald, S.; Liu, L.; Yan, C.; Schmidt, O. G. Sci. Rep. 2014, 4, 7452. doi:10.1038/srep07452
Return to citation in text: [1] [2] -
Wang, Z.; Luan, D.; Madhavi, S.; Li, C. M.; Lou, X. W. Chem. Commun. 2011, 47, 8061–8063. doi:10.1039/c1cc12111a
Return to citation in text: [1] -
Zhang, X.; Liu, H.; Petnikota, S.; Ramakrishna, S.; Fan, H. J. J. Mater. Chem. A 2014, 2, 10835–10841. doi:10.1039/c3ta15123a
Return to citation in text: [1] -
Xiao, W.; Wang, Z.; Guo, H.; Li, X.; Wang, J.; Huang, S.; Gan, L. Appl. Surf. Sci. 2013, 266, 148–154. doi:10.1016/j.apsusc.2012.11.118
Return to citation in text: [1] -
Xue, X.-Y.; Ma, C.-H.; Cui, C.-X.; Xing, L.-L. Solid State Sci. 2011, 13, 1526–1530. doi:10.1016/j.solidstatesciences.2011.05.015
Return to citation in text: [1] -
Bruce, P. G.; Scrosati, B.; Tarascon, J.-M. Angew. Chem., Int. Ed. 2008, 47, 2930–2946. doi:10.1002/anie.200702505
Return to citation in text: [1] -
Chen, J.; Xu, L.; Li, W.; Gou, X. Adv. Mater. 2005, 17, 582–586. doi:10.1002/adma.200401101
Return to citation in text: [1] -
Liu, H.; Wexler, D.; Wang, G. J. Alloys Compd. 2009, 487, L24–L27. doi:10.1016/j.jallcom.2009.08.043
Return to citation in text: [1] -
Zeng, S.; Tang, K.; Li, T.; Liang, Z.; Wang, D.; Wang, Y.; Qi, Y.; Zhou, W. J. Phys. Chem. C 2008, 112, 4836–4843. doi:10.1021/jp0768773
Return to citation in text: [1] -
Xu, X.; Cao, R.; Jeong, S.; Cho, J. Nano Lett. 2012, 12, 4988–4991. doi:10.1021/nl302618s
Return to citation in text: [1] -
Wang, B.; Chen, J. S.; Wu, H. B.; Wang, Z.; Lou, X. W. J. Am. Chem. Soc. 2011, 133, 17146–17148. doi:10.1021/ja208346s
Return to citation in text: [1] -
Xiao, L.; Wu, D.; Han, S.; Huang, Y.; Li, S.; He, M.; Zhang, F.; Feng, X. ACS Appl. Mater. Interfaces 2013, 5, 3764–3769. doi:10.1021/am400387t
Return to citation in text: [1] -
Zhang, W.-M.; Wu, X.-L.; Hu, J.-S.; Guo, Y.-G.; Wan, L.-J. Adv. Funct. Mater. 2008, 18, 3941–3946. doi:10.1002/adfm.200801386
Return to citation in text: [1] -
Wang, S.; Zhang, J.; Chen, C. J. Power Sources 2010, 195, 5379–5381. doi:10.1016/j.jpowsour.2010.03.035
Return to citation in text: [1] -
Prakash, R.; Fanselau, K.; Ren, S.; Mandal, T. K.; Kübel, C.; Hahn, H.; Fichtner, M. Beilstein J. Nanotechnol. 2013, 4, 699–704. doi:10.3762/bjnano.4.79
Return to citation in text: [1] -
Ren, S.; Prakash, R.; Wang, D.; Chakravadhanula, V. S. K.; Fichtner, M. ChemSusChem 2012, 5, 1397–1400. doi:10.1002/cssc.201200139
Return to citation in text: [1] -
Chen, Y.; Song, B.; Tang, X.; Lu, L.; Xue, J. J. Mater. Chem. 2012, 22, 17656–17662. doi:10.1039/c2jm32057f
Return to citation in text: [1] -
Kwon, K.-A.; Lim, H.-S.; Sun, Y.-K.; Suh, K.-D. J. Phys. Chem. C 2014, 118, 2897–2907. doi:10.1021/jp5000057
Return to citation in text: [1] -
Chaudhari, S.; Srinivasan, M. J. Mater. Chem. 2012, 22, 23049–23056. doi:10.1039/c2jm32989a
Return to citation in text: [1] -
Jamnik, J.; Maier, J. Phys. Chem. Chem. Phys. 2003, 5, 5215–5220. doi:10.1039/b309130a
Return to citation in text: [1] [2] -
Bekaert, E.; Balaya, P.; Murugavel, S.; Maier, J.; Ménétrier, M. Chem. Mater. 2009, 21, 856–861. doi:10.1021/cm8028005
Return to citation in text: [1] -
Guo, B.; Li, C.; Yuan, Z.-Y. J. Phys. Chem. C 2010, 114, 12805–12817. doi:10.1021/jp103705q
Return to citation in text: [1]
| 28. | Kandori, K.; Yasukawa, A.; Ishikawa, T. J. Colloid Interface Sci. 1996, 180, 446–452. doi:10.1006/jcis.1996.0324 |
| 34. | Cornell, R. M.; Schwertmann, U. The iron oxides: structure, properties, reactions, occurrences and uses; Wiley-VCH: Weinheim, 2003. doi:10.1002/3527602097 |
| 35. | Jia, C.-J.; Sun, L.-D.; Yan, Z.-G.; You, L.-P.; Luo, F.; Han, X.-D.; Pang, Y.-C.; Zhang, Z.; Yan, C.-H. Angew. Chem. 2005, 117, 4402–4407. doi:10.1002/ange.200463038 |
| 11. | Larcher, D.; Bonnin, D.; Cortes, R.; Rivals, I.; Personnaz, L.; Tarascon, J.-M. J. Electrochem. Soc. 2003, 150, A1643–A1650. doi:10.1149/1.1622959 |
| 37. | Liu, X.; Si, W.; Zhang, J.; Sun, X.; Deng, J.; Baunack, S.; Oswald, S.; Liu, L.; Yan, C.; Schmidt, O. G. Sci. Rep. 2014, 4, 7452. doi:10.1038/srep07452 |
| 14. | Wang, J.; Li, L.; Wong, C. L.; Sun, L.; Shen, Z.; Madhavi, S. RSC Adv. 2013, 3, 15316–15326. doi:10.1039/c3ra41886c |
| 14. | Wang, J.; Li, L.; Wong, C. L.; Sun, L.; Shen, Z.; Madhavi, S. RSC Adv. 2013, 3, 15316–15326. doi:10.1039/c3ra41886c |
| 40. | Xiao, W.; Wang, Z.; Guo, H.; Li, X.; Wang, J.; Huang, S.; Gan, L. Appl. Surf. Sci. 2013, 266, 148–154. doi:10.1016/j.apsusc.2012.11.118 |
| 38. | Wang, Z.; Luan, D.; Madhavi, S.; Li, C. M.; Lou, X. W. Chem. Commun. 2011, 47, 8061–8063. doi:10.1039/c1cc12111a |
| 39. | Zhang, X.; Liu, H.; Petnikota, S.; Ramakrishna, S.; Fan, H. J. J. Mater. Chem. A 2014, 2, 10835–10841. doi:10.1039/c3ta15123a |
| 36. | Barrón, V.; Torrent, J. J. Colloid Interface Sci. 1996, 177, 407–410. doi:10.1006/jcis.1996.0051 |
| 37. | Liu, X.; Si, W.; Zhang, J.; Sun, X.; Deng, J.; Baunack, S.; Oswald, S.; Liu, L.; Yan, C.; Schmidt, O. G. Sci. Rep. 2014, 4, 7452. doi:10.1038/srep07452 |
| 41. | Xue, X.-Y.; Ma, C.-H.; Cui, C.-X.; Xing, L.-L. Solid State Sci. 2011, 13, 1526–1530. doi:10.1016/j.solidstatesciences.2011.05.015 |
| 17. | Lin, Y.-M.; Abel, P. R.; Heller, A.; Mullins, C. B. J. Phys. Chem. Lett. 2011, 2, 2885–2891. doi:10.1021/jz201363j |
| 42. | Bruce, P. G.; Scrosati, B.; Tarascon, J.-M. Angew. Chem., Int. Ed. 2008, 47, 2930–2946. doi:10.1002/anie.200702505 |
| 45. | Zeng, S.; Tang, K.; Li, T.; Liang, Z.; Wang, D.; Wang, Y.; Qi, Y.; Zhou, W. J. Phys. Chem. C 2008, 112, 4836–4843. doi:10.1021/jp0768773 |
| 46. | Xu, X.; Cao, R.; Jeong, S.; Cho, J. Nano Lett. 2012, 12, 4988–4991. doi:10.1021/nl302618s |
| 43. | Chen, J.; Xu, L.; Li, W.; Gou, X. Adv. Mater. 2005, 17, 582–586. doi:10.1002/adma.200401101 |
| 44. | Liu, H.; Wexler, D.; Wang, G. J. Alloys Compd. 2009, 487, L24–L27. doi:10.1016/j.jallcom.2009.08.043 |
| 17. | Lin, Y.-M.; Abel, P. R.; Heller, A.; Mullins, C. B. J. Phys. Chem. Lett. 2011, 2, 2885–2891. doi:10.1021/jz201363j |
| 31. | Cherian, C. T.; Sundaramurthy, J.; Kalaivani, M.; Ragupathy, P.; Kumar, P. S.; Thavasi, V.; Reddy, M. V.; Sow, C. H.; Mhaisalkar, S.; Ramakrishna, S.; Chowdari, B. V. R. J. Mater. Chem. 2012, 22, 12198–12204. doi:10.1039/c2jm31053h |
| 18. | Liu, H.; Wang, G.; Park, J.; Wang, J.; Liu, H.; Zhang, C. Electrochim. Acta 2009, 54, 1733–1736. doi:10.1016/j.electacta.2008.09.071 |
| 8. | Yoshio, M.; Wang, H.; Fukuda, K. Angew. Chem. 2003, 115, 4335–4338. doi:10.1002/ange.200351203 |
| 48. | Xiao, L.; Wu, D.; Han, S.; Huang, Y.; Li, S.; He, M.; Zhang, F.; Feng, X. ACS Appl. Mater. Interfaces 2013, 5, 3764–3769. doi:10.1021/am400387t |
| 49. | Zhang, W.-M.; Wu, X.-L.; Hu, J.-S.; Guo, Y.-G.; Wan, L.-J. Adv. Funct. Mater. 2008, 18, 3941–3946. doi:10.1002/adfm.200801386 |
| 47. | Wang, B.; Chen, J. S.; Wu, H. B.; Wang, Z.; Lou, X. W. J. Am. Chem. Soc. 2011, 133, 17146–17148. doi:10.1021/ja208346s |
| 1. | Lam, L. T.; Louey, R. J. Power Sources 2006, 158, 1140–1148. doi:10.1016/j.jpowsour.2006.03.022 |
| 8. | Yoshio, M.; Wang, H.; Fukuda, K. Angew. Chem. 2003, 115, 4335–4338. doi:10.1002/ange.200351203 |
| 22. | Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603 |
| 55. | Chaudhari, S.; Srinivasan, M. J. Mater. Chem. 2012, 22, 23049–23056. doi:10.1039/c2jm32989a |
| 6. | Thackeray, M. M.; Kang, S.-H.; Johnson, C. S.; Vaughey, J. T.; Benedek, R.; Hackney, S. J. Mater. Chem. 2007, 17, 3112–3125. doi:10.1039/b702425h |
| 7. | Yoshino, A. Angew. Chem., Int. Ed. 2012, 51, 5798–5800. doi:10.1002/anie.201105006 |
| 17. | Lin, Y.-M.; Abel, P. R.; Heller, A.; Mullins, C. B. J. Phys. Chem. Lett. 2011, 2, 2885–2891. doi:10.1021/jz201363j |
| 5. | Andre, D.; Kim, S.-J.; Lamp, P.; Lux, S. F.; Maglia, F.; Paschos, O.; Stiaszny, B. J. Mater. Chem. A 2015, 3, 6709–6732. doi:10.1039/C5TA00361J |
| 22. | Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603 |
| 23. | Liu, Z.; Lv, B.; Xu, Y.; Wu, D. J. Mater. Chem. A 2013, 1, 3040–3046. doi:10.1039/c2ta00987k |
| 24. | Lv, B.; Liu, Z.; Tian, H.; Xu, Y.; Wu, D.; Sun, Y. Adv. Funct. Mater. 2010, 20, 3987–3996. doi:10.1002/adfm.201001021 |
| 25. | Sugimoto, T.; Itoh, H.; Mochida, T. J. Colloid Interface Sci. 1998, 205, 42–52. doi:10.1006/jcis.1998.5588 |
| 26. | Yang, Y.; Ma, H.; Zhuang, J.; Wang, X. Inorg. Chem. 2011, 50, 10143–10151. doi:10.1021/ic201104w |
| 27. | Zhu, L.-P.; Liao, G.-H.; Bing, N.-C.; Zhao, X.; Gu, Y.-Y. Mater. Lett. 2011, 65, 1287–1290. doi:10.1016/j.matlet.2011.01.053 |
| 13. | Yao, X.; Tang, C.; Yuan, G.; Cui, P.; Xu, X.; Liu, Z. Electrochem. Commun. 2011, 13, 1439–1442. doi:10.1016/j.elecom.2011.09.015 |
| 2. | Goodenough, J. B.; Park, K.-S. J. Am. Chem. Soc. 2013, 135, 1167–1176. doi:10.1021/ja3091438 |
| 3. | Manthiram, A. J. Phys. Chem. Lett. 2011, 2, 176–184. doi:10.1021/jz1015422 |
| 4. | Manthiram, A.; Chemelewski, K.; Lee, E.-S. Energy Environ. Sci. 2014, 7, 1339–1350. doi:10.1039/c3ee42981d |
| 28. | Kandori, K.; Yasukawa, A.; Ishikawa, T. J. Colloid Interface Sci. 1996, 180, 446–452. doi:10.1006/jcis.1996.0324 |
| 54. | Kwon, K.-A.; Lim, H.-S.; Sun, Y.-K.; Suh, K.-D. J. Phys. Chem. C 2014, 118, 2897–2907. doi:10.1021/jp5000057 |
| 12. | Wang, P. C.; Ding, H. P.; Bark, T.; Chen, C. H. Electrochim. Acta 2007, 52, 6650–6655. doi:10.1016/j.electacta.2007.04.072 |
| 13. | Yao, X.; Tang, C.; Yuan, G.; Cui, P.; Xu, X.; Liu, Z. Electrochem. Commun. 2011, 13, 1439–1442. doi:10.1016/j.elecom.2011.09.015 |
| 14. | Wang, J.; Li, L.; Wong, C. L.; Sun, L.; Shen, Z.; Madhavi, S. RSC Adv. 2013, 3, 15316–15326. doi:10.1039/c3ra41886c |
| 9. | Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.-M. Nature 2000, 407, 496–499. doi:10.1038/35035045 |
| 11. | Larcher, D.; Bonnin, D.; Cortes, R.; Rivals, I.; Personnaz, L.; Tarascon, J.-M. J. Electrochem. Soc. 2003, 150, A1643–A1650. doi:10.1149/1.1622959 |
| 16. | Larcher, D.; Masquelier, C.; Bonnin, D.; Chabre, Y.; Masson, V.; Leriche, J.-B.; Tarascon, J.-M. J. Electrochem. Soc. 2003, 150, A133–A139. doi:10.1149/1.1528941 |
| 17. | Lin, Y.-M.; Abel, P. R.; Heller, A.; Mullins, C. B. J. Phys. Chem. Lett. 2011, 2, 2885–2891. doi:10.1021/jz201363j |
| 18. | Liu, H.; Wang, G.; Park, J.; Wang, J.; Liu, H.; Zhang, C. Electrochim. Acta 2009, 54, 1733–1736. doi:10.1016/j.electacta.2008.09.071 |
| 19. | Matsumura, T.; Sonoyama, N.; Kanno, R.; Takano, M. Solid State Ionics 2003, 158, 253–260. doi:10.1016/S0167-2738(02)00913-X |
| 20. | NuLi, Y.; Zhang, P.; Guo, Z.; Liu, H. J. Electrochem. Soc. 2008, 155, A196–A200. doi:10.1149/1.2826278 |
| 52. | Ren, S.; Prakash, R.; Wang, D.; Chakravadhanula, V. S. K.; Fichtner, M. ChemSusChem 2012, 5, 1397–1400. doi:10.1002/cssc.201200139 |
| 11. | Larcher, D.; Bonnin, D.; Cortes, R.; Rivals, I.; Personnaz, L.; Tarascon, J.-M. J. Electrochem. Soc. 2003, 150, A1643–A1650. doi:10.1149/1.1622959 |
| 20. | NuLi, Y.; Zhang, P.; Guo, Z.; Liu, H. J. Electrochem. Soc. 2008, 155, A196–A200. doi:10.1149/1.2826278 |
| 21. | Byrappa, K.; Adschiri, T. Prog. Cryst. Growth Charact. Mater. 2007, 53, 117–166. doi:10.1016/j.pcrysgrow.2007.04.001 |
| 22. | Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603 |
| 53. | Chen, Y.; Song, B.; Tang, X.; Lu, L.; Xue, J. J. Mater. Chem. 2012, 22, 17656–17662. doi:10.1039/c2jm32057f |
| 10. | Yu, S.-H.; Lee, S. H.; Lee, D. J.; Sung, Y.-E.; Hyeon, T. Small 2016, 12, 2146–2172. doi:10.1002/smll.201502299 |
| 50. | Wang, S.; Zhang, J.; Chen, C. J. Power Sources 2010, 195, 5379–5381. doi:10.1016/j.jpowsour.2010.03.035 |
| 9. | Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.-M. Nature 2000, 407, 496–499. doi:10.1038/35035045 |
| 15. | Keppeler, M.; Shen, N.; Nageswaran, S.; Srinivasan, M. J. Mater. Chem. A 2016, 4, 18223–18239. doi:10.1039/C6TA08456G |
| 51. | Prakash, R.; Fanselau, K.; Ren, S.; Mandal, T. K.; Kübel, C.; Hahn, H.; Fichtner, M. Beilstein J. Nanotechnol. 2013, 4, 699–704. doi:10.3762/bjnano.4.79 |
| 22. | Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603 |
| 27. | Zhu, L.-P.; Liao, G.-H.; Bing, N.-C.; Zhao, X.; Gu, Y.-Y. Mater. Lett. 2011, 65, 1287–1290. doi:10.1016/j.matlet.2011.01.053 |
| 29. | Zhang, D.-E.; Zhang, X.-J.; Ni, X.-M.; Zheng, H.-G. Mater. Lett. 2006, 60, 1915–1917. doi:10.1016/j.matlet.2005.12.053 |
| 56. | Jamnik, J.; Maier, J. Phys. Chem. Chem. Phys. 2003, 5, 5215–5220. doi:10.1039/b309130a |
| 58. | Guo, B.; Li, C.; Yuan, Z.-Y. J. Phys. Chem. C 2010, 114, 12805–12817. doi:10.1021/jp103705q |
| 56. | Jamnik, J.; Maier, J. Phys. Chem. Chem. Phys. 2003, 5, 5215–5220. doi:10.1039/b309130a |
| 57. | Bekaert, E.; Balaya, P.; Murugavel, S.; Maier, J.; Ménétrier, M. Chem. Mater. 2009, 21, 856–861. doi:10.1021/cm8028005 |
| 29. | Zhang, D.-E.; Zhang, X.-J.; Ni, X.-M.; Zheng, H.-G. Mater. Lett. 2006, 60, 1915–1917. doi:10.1016/j.matlet.2005.12.053 |
| 22. | Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603 |
| 29. | Zhang, D.-E.; Zhang, X.-J.; Ni, X.-M.; Zheng, H.-G. Mater. Lett. 2006, 60, 1915–1917. doi:10.1016/j.matlet.2005.12.053 |
| 29. | Zhang, D.-E.; Zhang, X.-J.; Ni, X.-M.; Zheng, H.-G. Mater. Lett. 2006, 60, 1915–1917. doi:10.1016/j.matlet.2005.12.053 |
| 27. | Zhu, L.-P.; Liao, G.-H.; Bing, N.-C.; Zhao, X.; Gu, Y.-Y. Mater. Lett. 2011, 65, 1287–1290. doi:10.1016/j.matlet.2011.01.053 |
| 28. | Kandori, K.; Yasukawa, A.; Ishikawa, T. J. Colloid Interface Sci. 1996, 180, 446–452. doi:10.1006/jcis.1996.0324 |
| 27. | Zhu, L.-P.; Liao, G.-H.; Bing, N.-C.; Zhao, X.; Gu, Y.-Y. Mater. Lett. 2011, 65, 1287–1290. doi:10.1016/j.matlet.2011.01.053 |
| 29. | Zhang, D.-E.; Zhang, X.-J.; Ni, X.-M.; Zheng, H.-G. Mater. Lett. 2006, 60, 1915–1917. doi:10.1016/j.matlet.2005.12.053 |
| 33. | Zhu, L.-P.; Xiao, H.-M.; Zhang, W.-D.; Yang, G.; Fu, S.-Y. Cryst. Growth Des. 2008, 8, 957–963. doi:10.1021/cg700861a |
| 22. | Li, Z.; Lai, X.; Wang, H.; Mao, D.; Xing, C.; Wang, D. Nanotechnology 2009, 20, 245603. doi:10.1088/0957-4484/20/24/245603 |
| 30. | Aravindan, V.; Nan, S.; Keppeler, M.; Madhavi, S. Electrochim. Acta 2016, 208, 225–230. doi:10.1016/j.electacta.2016.05.035 |
| 31. | Cherian, C. T.; Sundaramurthy, J.; Kalaivani, M.; Ragupathy, P.; Kumar, P. S.; Thavasi, V.; Reddy, M. V.; Sow, C. H.; Mhaisalkar, S.; Ramakrishna, S.; Chowdari, B. V. R. J. Mater. Chem. 2012, 22, 12198–12204. doi:10.1039/c2jm31053h |
| 32. | Pourghahramani, P.; Forssberg, E. Int. J. Miner. Process. 2006, 79, 106–119. doi:10.1016/j.minpro.2006.02.001 |
© 2017 Shen et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)