Abstract
Using first principles calculations, we studied the stability and electronic properties of transition metal dichalcogenide monolayers of the type MX2 (M = Ti, Zr, Hf, V, Nb, Ta, Mo, Cr, W; X= S, Se, Te). The adsorption and diffusion of lithium on the stable MX2 phase was also investigated for potential application as an anode for lithium ion batteries. Some of these compounds were found to be stable in the 2H phase and some are in the 1T or 1T' phase, but only a few of them were stable in both 2H/1T or 2H/1T' phases. The results show that lithium is energetically favourable for adsorption on MX2 monolayers, which can be semiconductors with a narrow bandgap and metallic materials. Lithium cannot be adsorbed onto 2H-WS2 and 2H-WSe2, which have large bandgaps of 1.66 and 1.96 eV, respectively. The diffusion energy barrier is in the range between 0.17 and 0.64 eV for lithium on MX2 monolayers, while for most of the materials it was found to be around 0.25 eV. Therefore, this work illustrated that most of the MX2 monolayers explored in this work can be used as promising anode materials for lithium ion batteries.
Introduction
Lithium ion batteries (LIBs) have been widely used in portable electronic devices as power supplies, which have potential use in electrical vehicles (EVs) and smart grids. However, the energy and power density of current LIBs cannot satisfy the high demand of EVs. The development of new electrode materials is essential for improvement of the energy density. An ideal electrode material for LIBs should have good electronic conductivity, a lower Li diffusion energy barrier, as well as high energy and power densities. By reducing the bulk electrode materials to low-dimensional materials, a higher energy capacity and higher charge/discharge rate can be obtained as the low-dimensional materials have higher exposure to the electrolyte [1]. Two-dimensional materials, such as Co3O4, NiO, phosphorene, SnS and V2O5 all exhibit an excellent capacity retention, rate performance, lower energy barrier and long cycling life compared to their bulk counterparts used as electrode materials for LIBs [2-8].
Two-dimensional transition metal dichalcogenides, MX2 (where M and X correspond to transition metal and chalcogen atoms, respectively), have been synthesized using different strategies, such as exfoliation [9,10], physical vapour deposition [11] and chemical vapour deposition [12-14]. MX2 has received tremendous attention as an alternative to graphite for the anode material in LIBs [15,16]. In particular, MoS2 has been well-investigated as an anode material for LIBs both theoretically and experimentally. A graphene like-MoS2/graphene composite was shown to exhibit a high specific capacity of 1400 mA h/g and good rate performance as well as cycling ability [17]. It was reported that MoS2 zigzag nanoribbons are promising electrode materials for LIBs with a high power density and fast charge/discharge rates [18]. The presence of structural defects can enhance the adsorption of Li atoms onto two-dimensional materials. Different from the situation where Li atoms are trapped by the defects in graphene, the presence of structural defects does not affect the diffusion of lithium [19]. The main drawback of MoS2 is its poor electrical conductivity. Various strategies have been developed to improve the electrochemical properties of MoS2 as an anode for LIBs. Three-dimensional hierarchical structures constructed by assembling two-dimensional MoS2 nanosheets can deliver a capacity of 1009 mAh/g at 500 mA/g after 500 cycles [20]. The formation of composites of MoS2 with other materials, such as carbon-based materials and non-carbonaceous materials, can enhance the electromechanical properties of MoS2. Wang et al. [21] utilized a beneficial "bridging effect" of sulfur atoms to bind few-layered MoS2 with graphene, which provided fast electron conductivity and excellent cycling stability and superior rate performance. The composites exhibited a high discharge capacity of 1546 mAh/g after 300 cycles. The MoS2 composites grown on TiO2 nanotubes show better rate capability with a reversible capacity of 461 mAh/g at 1000 mA/g, compared with the capacity of pure MoS2 (129 mAh/g) at the same current density [22].
MX2 monolayers have three types of crystalline structures, hexagonal structure (2H), octahedral structure (1T) and distorted octahedral structure (1T') [23-25]. The structures depend on the arrangements of the M and X atoms. Phase transformation between the different phases occurs during the synthesis process and lithium/sodium intercalation [26-28]. Sun et al. [29] have studied the effect of electron doping on the stability of 2H- and 1T'-MoS2, and showed that electron doping can stabilize the crystal structure of 1T'-MoS2. The crystalline structure can also affect the energy conversion efficiency, for example in the hydrogen evolution reaction (HER). The basal plane of 2H-MoS2 is inert [30], where that of 1T'-MoS2 is catalytically active for HER [31]. Until now, there is no systematic study on the family of transition metal dichalcogenide monolayers used as anode for LIBs.
In this work, we studied the stability of MX2 monolayers, and the adsorption and diffusion of Li on the stable MX2 monolayers (M = Ti, Zr, Hf, V, Nb, Ta, Mo, Cr, W; X = S, Se, Te). These results are helpful for the design of two-dimensional transition metal dichalcogenide based electrodes for LIBs.
Results and Discussion
We systematically investigated the phase stability, Li adsorption and diffusion on MX2 monolayers (M = Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W; X = S, Se, Te). The combination of these elements have twenty seven possible binary compound materials. Three phases, including 2H, 1T and 1T' structures, were all considered for each of the binary monolayers. All the three structures can be viewed as a positively charged, two-dimensional M atoms, lattice-sandwiched by two hexagonal lattices of negatively charged X atoms. Each M atom is surrounded by six nearest X atoms, and each X atom is connected to three nearest M atoms with ionic M–X bonds. The side and cross-views of the ball and stick models of the MX2 monolayer are shown in Figure 1. The M atoms are located at the lattice positions of a hexagonal close-packed structure with a trigonal symmetry in the 2H-MX2 phase (Figure 1a), whereas M atoms are located at the octahedral/disordered octahedral centre of six S atoms in the 1T/1T' phase (Figure 1b,c). Some compounds are not stable in the 1T' phase, which will be relaxed to the 1T phase after relaxation.
![[2190-4286-8-270-1]](/bjnano/content/figures/2190-4286-8-270-1.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 1: Top and side views of ball and stick models of a MX2 monolayer in (a) 2H, (b) 1T and (c) 1T' phase. The M atoms have octahedral and trigonal prismatic coordination in the 1T/1T' and 2H phase, respectively.
Figure 1: Top and side views of ball and stick models of a MX2 monolayer in (a) 2H, (b) 1T and (c) 1T' phase....
The energy related to the 2H phase per formula unit (f.u.), E = E1T/1T'−E2H, is listed in Table 1. A negative value indicates that the 1T/1T' phase is more stable than the 2H phase. It can be seen from the table that some of these compound can be stable in the 2H phase, and some in the 1T or 1T' phase. Only a few of them are stable both in 2H/1T or 2H/1T' phases. The 2H phase is the minimum energy configuration for monolayers of NbX2 and TaX2, which agrees with previously reported results [32,33]. The energy of the 1T phase is smaller than the 2H phase for TiX2, VX2, VSe2, CrX2, ZrX2 and HfX2. 1T-VS2 monolayers are 0.02 eV/f.u. larger than the 2H phase, which indicates that the 1T phase is the energetically favourable one for these compounds, and VS2 monolayers maybe be stable both in the 2H/1T phase. The results agree with other calculations that imply that the 1T phase is more stable than 2H in TiX2 [32-34], CrS2 [35], ZrX2 [32,33] and HfX2 [33]. The 2H phase is the stable structure for MoS2, MoSe2, WS2 and WSe2 monolayers, which has also been predicted by other simulations [36-40]. 1T'-VTe2, 1T'-MoTe2 and 1T'-WTe2 are the energetically favourable phases. It is also can be seen from Table 1 that 1T'-MoTe2 is 0.06 eV/f.u. more energetically favourable than the 1T phase, which agrees with other simulations [36,39,40]. The energy difference between 2H-WTe2 and 1T'-WTe2 is 0.07 eV/f.u., which indicates MoTe2 and WTe2 maybe exist in two phases.
Table 1: Energy (eV) referenced to the 2H phase per formula unit. A negative value indicates that the 1T/1T' phase is more stable than the 2H phase.
| M | S | Se | Te | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 2H | 1T | 1T' | 2H | 1T | 1T' | 2H | 1T | 1T' | |
| Ti | 0.00 | −0.44 | – | 0.00 | −0.33 | – | 0.00 | −0.26 | – |
| V | 0.00 | 0.02 | – | 0.00 | −0.16 | – | 0.00 | −0.06 | −0.15 |
| Cr | 0.00 | −0.45 | – | 0.00 | −0.39 | – | 0.00 | −0.17 | – |
| Zr | 0.00 | −0.54 | – | 0.00 | −0.40 | – | 0.00 | −0.26 | – |
| Nb | 0.00 | 0.21 | – | 0.00 | 0.22 | – | 0.00 | 0.15 | 0.10 |
| Mo | 0.00 | 0.68 | 0.60 | 0.00 | 0.35 | 0.29 | 0.00 | −0.20 | −0.26 |
| Hf | 0.00 | −0.62 | – | 0.00 | −0.50 | – | 0.00 | −0.35 | – |
| Ta | 0.00 | 0.18 | – | 0.00 | 0.23 | 0.21 | 0.00 | 0.15 | 0.54 |
| W | 0.00 | 0.91 | 0.61 | 0.00 | 0.81 | 0.35 | 0.00 | 0.61 | −0.07 |
The calculated lattice constants and bond length of the M–X bond in the stable phase is listed in Table 2 along with available values from other simulations. The values obtained in the present work agree well with other simulation results. It can be seen from Table 2 that lattice constants and bond lengths increase for the all the MX2 monolayers as the element X changes from S to Te in group VI for a given element M. The variation can be explained by the increasing atomic radius of elements X from S to Te.
Table 2: Lattice constants (a,b) and the bond length of the M–X bond (dM-X) in the stable phase as calculated in this work as compared to other values found in the literature from other simulations. The electronic conducting behaviour (ECB) of these compounds is also shown.
| MX2 | a/b (Å) | dM-X (Å) | a (Å) [Ref.] | dM-X (Å) [Ref.] | ECB |
|---|---|---|---|---|---|
| 1T-TiS2 | 3.40 | 2.42 | 3.39 [33] | 2.39 [41] | 0.59 |
| 1T-TiSe2 | 3.56 | 2.57 | 3.53 [33] | 2.51 [41] | 0.29 |
| 1T-TiTe2 | 3.72 | 2.77 | 3.74 [33] | 2.73 [41] | metal |
| 2H-VS2 | 3.19 | 2.39 | 3.17 [42-44] | 2.36 [42-44] | 0.58 |
| 1T-VS2 | 3.25 | 2.38 | 3.18 [43,45] | 2.35 [43,44,46] | metal |
| 1T-VSe2 | 3.37 | 2.55 | 3.24 [41] | 2.49 [46] | metal |
| 1T'-VTe2 |
3.80/
7.60 |
2.71/
2.76/ 2.79/ 2.81 |
– | – | metal |
| 1T-CrS2 | 3.33 | 2.41 | – | – | metal |
| 1T-CrSe2 | 3.47 | 2.56 | – | – | metal |
| 1T-CrTe2 | 3.67 | 2.81 | – | – | metal |
| 1T-ZrS2 | 3.57 | 2.55 | 3.68 [33,47] | 0.92 | |
| 1T-ZrSe2 | 3.70 | 2.68 | 3.79 [47] | 0.29 | |
| 1T-ZrTe2 | 3.89 | 2.90 | 3.98 [33] | metal | |
| 2H-NbS2 | 3.35 | 2.50 | 3.36 [48] | 2.49 [48] | 1.22 |
| 2H-NbSe2 | 3.49 | 2.64 | 3.48 [48] | 2.62 [48] | 1.00 |
| 2H-NbTe2 | 3.71 | 2.83 | 3.70 [48] | 2.82 [48] | 0.78 |
| 2H-MoS2 | 3.17 | 2.42 | 3.18 [47,49] | 2.42 [48] | 1.71 |
| 2H-MoSe2 | 3.32 | 2.55 | 3.32 [47] | 2.55 [48] | 1.41 |
| 1T-MoTe2 | 3.84 | 2.81 | – | – | metal |
| 1T'-MoTe2 |
3.89/
7.88 |
2.53/
2.55/ 2.60/ 2.61 |
– | – | 0.12 |
| 1T-HfS2 | 3.57 | 2.54 | 3.64 [33] | – | 1.09 |
| 1T-HfSe2 | 3.69 | 2.67 | 3.76 [33] | – | 0.50 |
| 1T-HfTe2 | 3.88 | 2.87 | 3.97 [33] | – | metal |
| 2H-TaS2 | 3.35 | 2.50 | 3.34 [48] | 2.48 [48] | 0.20 |
| 2H-TaSe2 | 3.47 | 2.63 | 3.48 [48] | 2.62 [48] | 0.46 |
| 2H-TaTe2 | 3.69 | 2.82 | 3.76 [48] | 2.82 [48] | 0.37 |
| 2H-WS2 | 3.18 | 2.44 | 3.18 [49] | 2.42 [48] | 1.96 |
| 2H-WSe2 | 3.32 | 2.57 | 3.32 [47,48] | 2.55 [48] | 1.66 |
| 2H-WTe2 | 3.56 | 2.76 | – | – | 1.22 |
| 1T'-WTe2 |
3.49/
6.98 |
2.74/
2.75/ 2.78/ 2.81 |
– | – | 0.38 |
The band structures of MX2 monolayers in the stable phase are shown in Figure 2. The MX2 monolayers can be semiconducting with a direct and indirect bandgap or metallic materials. The electronic conductive behaviour of these compounds are shown in Table 2. The 2H phase shows a semiconducting behaviour, such as 2H-WX2, 2H-NbX2, 2H-TaX2 and 2H-MoX2. The 1T phase can be metallic or semiconducting, such as in 1T-VX2 (X = S, Se), and 1T-CrX2 shows metallic behaviour, while 1T-TiX2, 1T-ZrX2 and 1T-HfX2 show semiconducting and metallic behaviour with X = S/Se and X = Te, respectively. 1T'-VTe2 and 1T'-MoTe2 show metallic behaviour and 1T'-WTe2 has a narrow bandgap of 0.50 eV. The obtained bandgap values are close to those previously reported for TiS2 [32], CrTe2 [40], TiX2 [32,33,47], MoX2 [33,40,47,50-55], HfX2 [33,47] and WX2 [33,40,47,50,55-58]. The metallic MX2 monolayers have good electrical conductivity, which may make them good anode materials.
![[2190-4286-8-270-2]](/bjnano/content/figures/2190-4286-8-270-2.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 2: Band structures of MX2 monolayers in the stable phase. Fermi energy level is set to be 0.
Figure 2: Band structures of MX2 monolayers in the stable phase. Fermi energy level is set to be 0.
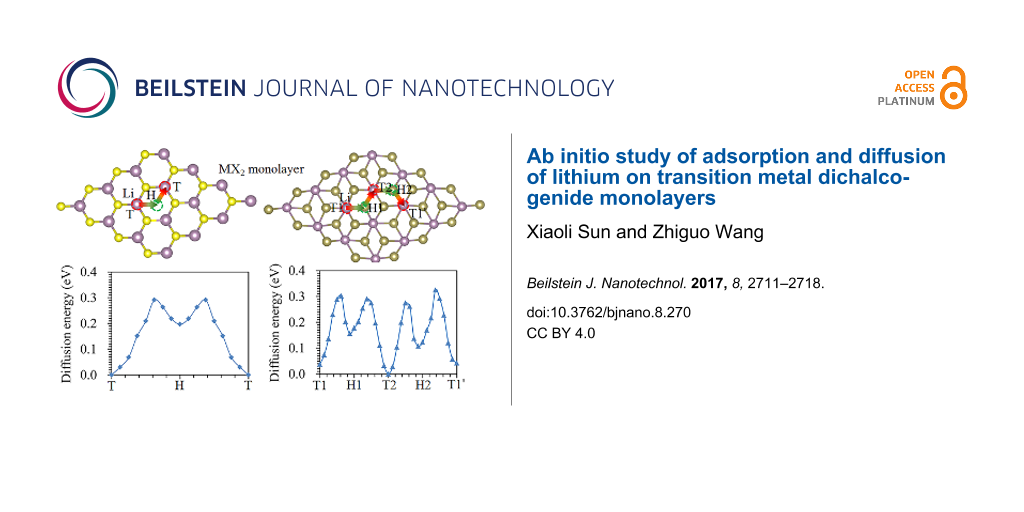
As shown in Figure 3a and Figure 3b, there are two stable adsorption sites, that is, the hollow site (H) and the top position above the M atom (T) for Li to be adsorbed on the 2H- and 1T-MX2 monolayers [18]. Four adsorption sites (T1, T2, H1, and H2) are considered for Li adsorption onto the 1T'-MX2 monolayer, as shown in Figure 3c. To analyse the stability of Li adsorbed on the MX2 monolayers, the adsorption energy, Ead(Li), is calculated using Equation 1:
![[2190-4286-8-270-i1]](/bjnano/content/inline/2190-4286-8-270-i1.svg?max-width=590&scale=1.18182)
where EMX2+Li and EMX2 are the total energy of the MX2 monolayer with and without Li adsorption, respectively. ELi is the energy of a Li atom in bulk material. The calculated adsorption energy of Li on the stable phase of the MX2 monolayers is shown in Figure 4. The adsorption energy has positive values for Li adsorbed on 2H-WS2 and 2H-WSe2, which indicates that Li cannot be adsorbed on these two compounds and they are not ideal anodes for LIBs. The other compounds have negative values of adsorption energy. The adsorption energy of Li on 2H-MoS2 is −0.05 and −0.25 eV for H and T sites, respectively. The materials will have a large energy storage capacity if they have a large exothermic reaction energy with Li [19]. Previous studies have shown that the 2H-MoS2 monolayer is a good anode material for LIBs [26-28]. The absolute value of the adsorption energy for Li adsorbed on other compounds is larger than that of 2H-MoS2, so other MX2 compounds are also good anode candidates for LIBs. The adsorption energy as a function of the bandgap of the MX2 monolayer is show in Figure 4. It can also be seen from the figure that MX2 is a semiconducting material with a narrow bandgap, and for metallic materials, the adsorption energy has larger negative values. The materials with a large bandgap have smaller adsorption energy, even those with positive values. For example, the bandgap energies are 1.96, 1.71, 0.78, 0.58 and 0.29 eV and the adsorption energies for Li adsorbed at H/T sites are 0.37/0.24, −0.05/−0.25, −0.51/−0.65, −1.79/1.87 and −2.08/2.07 eV on 2H-WS2, 2H-MoS2, 2H-NbTe2, 2H-VS2 and 1T-TiSe2 monolayers, respectively.
![[2190-4286-8-270-3]](/bjnano/content/figures/2190-4286-8-270-3.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 3: Possible adsorption sites and diffusion paths for Li on a monolayer of (a) 2H-, (b) 1T- and (c) 1T'-MX2. Diffusion energy profiles for Li on (d) 2H-MoS2, (e) 1T-TiS2, and (f) 1T'-MoTe2.
Figure 3: Possible adsorption sites and diffusion paths for Li on a monolayer of (a) 2H-, (b) 1T- and (c) 1T'...
![[2190-4286-8-270-4]](/bjnano/content/figures/2190-4286-8-270-4.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 4: Adsorption energy and diffusion energy barrier for Li on MX2 monolayers in the stable phase.
Figure 4: Adsorption energy and diffusion energy barrier for Li on MX2 monolayers in the stable phase.
The diffusion of Li on the MX2 monolayers is through the T→H→T and T1→H1→T2→H2→T1 paths for the 2H/1T and 1T' phases, respectively [59], as shown in Figure 3a–c. The typical diffusion energy profiles are shown in Figure 3d, Figure 3e and Figure 3f for Li on 2H-MoS2, 1T-TiS2, and 1T'-MoTe2 monolayers, respectively. The constrained method was used to evaluate the diffusion behaviour of Li on MX2 monolayers – this method is more simple and intuitive compared to the nudged elastic band method and dimer method [60]. The diffusion energy barriers are 0.29, 0.25 and 0.28 eV for Li on 2H-MoS2, 1T-TiS2, and 1T'-MoTe2 monolayers, respectively. These values are reasonable for use as anodes for LIBs. The Li diffusion energy barrier on a MX2 monolayer is shown in Figure 4. Our calculated values agree well with those reported by other researchers. The diffusion energy barrier for Li on the 2H phase of monolayer WS2, WSe2 and VS2 is 0.21 eV, 0.18 eV and 0.20 eV, respectively, which is consistent with the previously reported values of 0.22, 0.23 eV [61], and 0.22 eV [42] respectively. The diffusion energy barrier of Li on 2H-MoS2 monolayer is 0.29 eV, which is consistent with the previously reported value of 0.25 eV [18,42].
A good anode material should have a high electron and Li mobility and a large exothermic reaction energy with lithium. High electronic and ion mobility determine the rate capability and cycling performance, and a large exothermic reaction energy indicates the anode materials have a large energy storage capacity. The diffusion energy barrier is in the range between 0.17 and 0.63 eV, and most are around 0.25 eV, which indicates that all the MX2 monolayers have a reasonable diffusion energy barrier for lithium. The metallic MX2 monolayers and those with small bandgaps have a large adsorption energy for Li, which indicates that they are good anode materials for LIBs with high electronic and ion mobility and large energy storage capacity.
Conclusion
Using density functional theory (DFT) simulations, the stability and electronic properties of MX2 monolayers were investigated. TiX2, VSe2, CrX2, ZrX2 and HfX2 are energetically favourable in the 1T phase, and 1T-VS2 can be stable both in the 2H/1T phase. The 2H phase is the stable structure for MoS2, MoSe2, WS2 and WSe2. The 1T' phase is the most energetically favourable for VTe2, MoTe2 and WTe2. The 2H phase shows a semiconducting behaviour, for example, 2H-WX2, 2H-NbX2, 2H-TaX2 and 2H-MoX2. The 1T phase can be metallic or semiconducting, for example 1T-CrX2 shows a metallic behaviour while 1T-TiX2, 1T-ZrX2 and 1T-HfX2 show semiconducting and metallic behaviour with X = S/Se and X = Te, respectively. 1T'-VTe2 and 1T'-MoTe2 show metallic behaviour and 1T'-WTe2 has a narrow bandgap of 0.50 eV. The adsorption and diffusion of lithium on the stable MX2 phase were also investigated. The results show that lithium is energetically able to adsorb on MX2 monolayers, which are semiconductors with a narrow bandgap, and on metallic materials. Lithium cannot be adsorbed on 2H-WS2 and 2H-WSe2, which have a large bandgap of 1.66 and 1.96 eV, respectively. The diffusion energy barrier is in the range between 0.17 and 0.63 eV for lithium on MX2 monolayers, and most of the materials are around 0.25 eV. It is therefore concluded that most of the MX2 monolayers can be used as promising anode materials for lithium ion batteries.
Simulation Details
All the spin-polarized DFT calculations were performed with SIESTA code [62], in which norm-conserving pseudopotentials and a Perdew–Burcke–Ernzerhof functional was used to describe the electron–ion interaction and electron exchange correlation, respectively. Numerical atomic orbits were represented as double zeta basis sets plus polarization, and a cut-off energy of 250 Ry was chosen to calculate the Hamiltonian element. The Monkhorst–Pack scheme with 11 × 11 × 1 k-point meshes were used for integration in the irreducible Brillouin zone for the relaxation of the primitive cell. A 2 × 2 × 1 k-point mesh was used for the calculation of adsorption and diffusion of Li on a 6 × 6 × 1 supercell. As the electrochemical process involves insertion of Li ions into anode materials with a concurrent flow of electrons to compensate charge balance, and therefore, the neutral state of Li was considered in this work. The atomic positions were relaxed by using a conjugate gradient minimization until the Hellmann–Feynman force is less than 0.02 eV/Å on each atom. A vacuum spacing between the slabs and its image of greater than 20 Å is given to avoid the periodic image interactions. As the radii are different for different elements of X in MX2 monolayers (i.e., the radius increases from S to Te in group VI), the lattice constants and bond length of the M–X bond will change for MX2 monolayers with different X elements, which can affect the adsorption and diffusion of Li on MX2 monolayers.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (11474047) and the Fundamental Research Funds for the Central Universities (ZYGX2016J202). This work was carried out at National Supercomputer Center in Tianjin, and the calculations were performed on TianHe-1(A).
References
-
Goodenough, J. B.; Kim, Y. Chem. Mater. 2010, 22, 587–603. doi:10.1021/cm901452z
Return to citation in text: [1] -
Cheng, C.; Zhou, G.; Du, J.; Zhang, H.; Guo, D.; Li, Q.; Wei, W.; Chen, L. New J. Chem. 2014, 38, 2250. doi:10.1039/c3nj01642k
Return to citation in text: [1] -
Chen, G.; Fu, E.; Zhou, M.; Xu, Y.; Fei, L.; Deng, S.; Chaitanya, V.; Wang, Y.; Luo, H. J. Alloys Compd. 2013, 578, 349–354. doi:10.1016/j.jallcom.2013.06.042
Return to citation in text: [1] -
Wang, L.; Dong, Z.; Wang, D.; Zhang, F.; Jin, J. Nano Lett. 2013, 13, 6244–6250. doi:10.1021/nl403715h
Return to citation in text: [1] -
Vasilyeva, E.; Nasibulin, A.; Tolochko, O.; Rudskoy, A.; Sachdev, A.; Xiao, X. Z. Phys. Chem. 2015, 229, 1429–1437. doi:10.1515/zpch-2015-0573
Return to citation in text: [1] -
Yao, Q.; Huang, C.; Yuan, Y.; Liu, Y.; Liu, S.; Deng, K.; Kan, E. J. Phys. Chem. C 2015, 119, 6923–6928. doi:10.1021/acs.jpcc.5b02130
Return to citation in text: [1] -
Kang, J.-G.; Park, J.-G.; Kim, D.-W. Electrochem. Commun. 2010, 12, 307–310. doi:10.1016/j.elecom.2009.12.025
Return to citation in text: [1] -
Wang, Z.; Su, Q.; Deng, H. Phys. Chem. Chem. Phys. 2013, 15, 8705–8709. doi:10.1039/C3CP51167G
Return to citation in text: [1] -
Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Nat. Nanotechnol. 2011, 6, 147–150. doi:10.1038/nnano.2010.279
Return to citation in text: [1] -
Lee, C.; Yan, H.; Brus, L. E.; Heinz, T. F.; Hone, J.; Ryu, S. ACS Nano 2010, 4, 2695–2700. doi:10.1021/nn1003937
Return to citation in text: [1] -
Lauritsen, J. V.; Kibsgaard, J.; Helveg, S.; Topsøe, H.; Clausen, B. S.; Lægsgaard, E.; Besenbacher, F. Nat. Nanotechnol. 2007, 2, 53–58. doi:10.1038/nnano.2006.171
Return to citation in text: [1] -
Liu, K.-K.; Zhang, W. J.; Lee, Y.-H.; Lin, Y.-C.; Chang, M.-T.; Su, C.-Y.; Chang, C.-S.; Li, H.; Shi, Y.; Zhang, H.; Lai, C.-S.; Li, L.-J. Nano Lett. 2012, 12, 1538–1544. doi:10.1021/nl2043612
Return to citation in text: [1] -
Lee, H. S.; Min, S.-W.; Chang, Y.-G.; Park, M. K.; Nam, T.; Kim, H.; Kim, J. H.; Ryu, S.; Im, S. Nano Lett. 2012, 12, 3695–3700. doi:10.1021/nl301485q
Return to citation in text: [1] -
Lee, Y.-H.; Zhang, X.-Q.; Zhang, W.; Chang, M.-T.; Lin, C.-T.; Chang, K.-D.; Yu, Y.-C.; Wang, J. T.-W.; Chang, C.-S.; Li, L.-J.; Lin, T.-W. Adv. Mater. 2012, 24, 2320–2325. doi:10.1002/adma.201104798
Return to citation in text: [1] -
Cai, Y.; Yang, X.; Liang, T.; Dai, L.; Ma, L.; Huang, G.; Chen, W.; Chen, H.; Su, H.; Xu, M. Nanotechnology 2014, 25, 465401. doi:10.1088/0957-4484/25/46/465401
Return to citation in text: [1] -
Xiao, J.; Choi, D.; Cosimbescu, L.; Koech, P.; Liu, J.; Lemmon, J. P. Chem. Mater. 2010, 22, 4522–4524. doi:10.1021/cm101254j
Return to citation in text: [1] -
Liu, Y.; Zhao, Y.; Jiao, L.; Chen, J. J. Mater. Chem. A 2014, 2, 13109–13115. doi:10.1039/c4ta01644k
Return to citation in text: [1] -
Li, Y.; Wu, D.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. Lett. 2012, 3, 2221–2227. doi:10.1021/jz300792n
Return to citation in text: [1] [2] [3] -
Sun, X.; Wang, Z.; Fu, Y. Q. Sci. Rep. 2015, 5, 18712. doi:10.1038/srep18712
Return to citation in text: [1] [2] -
Xie, X.; Chen, S.; Sun, B.; Wang, C.; Wang, G. ChemSusChem 2015, 8, 2948–2955. doi:10.1002/cssc.201500149
Return to citation in text: [1] -
Wang, X.; Li, G.; Seo, M. H.; Hassan, F. M.; Hoque, M. A.; Chen, Z. Adv. Energy Mater. 2015, 5, 1501106. doi:10.1002/aenm.201501106
Return to citation in text: [1] -
Jian, Z.; Zhao, B.; Liu, P.; Li, F.; Zheng, M.; Chen, M.; Shi, Y.; Zhou, H. Chem. Commun. 2014, 50, 1215–1217. doi:10.1039/C3CC47977C
Return to citation in text: [1] -
Hu, T.; Li, R.; Dong, J. J. Chem. Phys. 2013, 139, 174702. doi:10.1063/1.4827082
Return to citation in text: [1] -
Song, I.; Park, C.; Choi, H. C. RSC Adv. 2015, 5, 7495–7514. doi:10.1039/C4RA11852A
Return to citation in text: [1] -
Wypych, F.; Schöllhorn, R. J. Chem. Soc., Chem. Commun. 1992, 1386–1388. doi:10.1039/C39920001386
Return to citation in text: [1] -
Wang, X.; Shen, X.; Wang, Z.; Yu, R.; Chen, L. ACS Nano 2014, 8, 11394–11400. doi:10.1021/nn505501v
Return to citation in text: [1] [2] -
Wang, L.; Xu, Z.; Wang, W.; Bai, X. J. Am. Chem. Soc. 2014, 136, 6693–6697. doi:10.1021/ja501686w
Return to citation in text: [1] [2] -
Cheng, Y.; Nie, A.; Zhang, Q.; Gan, L.-Y.; Shahbazian-Yassar, R.; Schwingenschlogl, U. ACS Nano 2014, 8, 11447–11453. doi:10.1021/nn505668c
Return to citation in text: [1] [2] -
Sun, X.; Wang, Z.; Li, Z.; Fu, Y.-Q. Sci. Rep. 2016, 6, 26666. doi:10.1038/srep26666
Return to citation in text: [1] -
Lee, J. H.; Jang, W. S.; Han, S. W.; Baik, H. K. Langmuir 2014, 30, 9866–9873. doi:10.1021/la501349k
Return to citation in text: [1] -
Tang, Q.; Jiang, D.-e. ACS Catal. 2016, 6, 4953–4961. doi:10.1021/acscatal.6b01211
Return to citation in text: [1] -
Yang, E.; Ji, H.; Jung, Y. J. Phys. Chem. C 2015, 119, 26374–26380. doi:10.1021/acs.jpcc.5b09935
Return to citation in text: [1] [2] [3] [4] [5] -
Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] -
Tibbetts, K.; Miranda, C. R.; Meng, Y. S.; Ceder, G. Chem. Mater. 2007, 19, 5302–5308. doi:10.1021/cm0715242
Return to citation in text: [1] -
Ambrosi, A.; Sofer, Z.; Pumera, M. Chem. Commun. 2015, 51, 8450–8453. doi:10.1039/c5cc00803d
Return to citation in text: [1] -
Li, Y.; Duerloo, K.-A. N.; Wauson, K.; Reed, E. J. Nat. Commun. 2016, 7, 10671. doi:10.1038/ncomms10671
Return to citation in text: [1] [2] -
Enyashin, A. N.; Yadgarov, L.; Houben, L.; Popov, I.; Weidenbach, M.; Tenne, R.; Bar-Sadan, M.; Seifert, G. J. Phys. Chem. C 2011, 115, 24586–24591. doi:10.1021/jp2076325
Return to citation in text: [1] -
Sun, Y.; Wang, Y.; Sun, D.; Carvalho, B. R.; Read, C. G.; Lee, C.-h.; Lin, Z.; Fujisawa, K.; Robinson, J. A.; Crespi, V. H.; Terrones, M.; Schaak, R. E. Angew. Chem., Int. Ed. 2016, 55, 2830–2834. doi:10.1002/anie.201510029
Return to citation in text: [1] -
Duerloo, K.-A. N.; Li, Y.; Reed, E. J. Nat. Commun. 2014, 5, 4214. doi:10.1038/ncomms5214
Return to citation in text: [1] [2] -
Reyes-Retana, J. A.; Cervantes-Sodi, F. Sci. Rep. 2016, 6, 24093. doi:10.1038/srep24093
Return to citation in text: [1] [2] [3] [4] [5] -
Ataca, C.; Şahin, H.; Ciraci, S. J. Phys. Chem. C 2012, 116, 8983–8999. doi:10.1021/jp212558p
Return to citation in text: [1] [2] [3] [4] -
Jing, Y.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. C 2013, 117, 25409–25413. doi:10.1021/jp410969u
Return to citation in text: [1] [2] [3] [4] -
Zhang, H.; Liu, L.-M.; Lau, W.-M. J. Mater. Chem. A 2013, 1, 10821. doi:10.1039/c3ta12098h
Return to citation in text: [1] [2] [3] [4] -
Kan, M.; Wang, B.; Lee, Y.-H.; Sun, Q. Nano Res. 2015, 8, 1348–1356. doi:10.1007/s12274-014-0626-5
Return to citation in text: [1] [2] [3] -
Zhuang, H. L.; Hennig, R. G. Phys. Rev. B 2016, 93, 054429. doi:10.1103/PhysRevB.93.054429
Return to citation in text: [1] -
Ma, Y.; Dai, Y.; Guo, M.; Niu, C.; Zhu, Y.; Huang, B. ACS Nano 2012, 6, 1695–1701. doi:10.1021/nn204667z
Return to citation in text: [1] [2] -
Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] -
Huang, W.; Da, H.; Liang, G. J. Appl. Phys. 2013, 113, 104304. doi:10.1063/1.4794363
Return to citation in text: [1] [2] -
Qian, X.; Lu, J.; Fu, L.; Li, J. Science 2014, 346, 1344–1347. doi:10.1126/science.1256815
Return to citation in text: [1] [2] -
Mak, K. F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T. F. Phys. Rev. Lett. 2010, 105, 136805. doi:10.1103/PhysRevLett.105.136805
Return to citation in text: [1] -
Tongay, S.; Zhou, J.; Ataca, C.; Lo, K.; Matthews, T. S.; Li, J.; Grossman, J. C.; Wu, J. Nano Lett. 2012, 12, 5576–5580. doi:10.1021/nl302584w
Return to citation in text: [1] -
Conley, H. J.; Wang, B.; Ziegler, J. I.; Haglund, R. F., Jr.; Pantelides, S. T.; Bolotin, K. I. Nano Lett. 2013, 13, 3626–3630. doi:10.1021/nl4014748
Return to citation in text: [1] -
Ji, Q.; Zhang, Y.; Gao, T.; Zhang, Y.; Ma, D.; Liu, M.; Chen, Y.; Qiao, X.; Tan, P.-H.; Kan, M.; Feng, J.; Sun, Q.; Liu, Z. Nano Lett. 2013, 13, 3870–3877. doi:10.1021/nl401938t
Return to citation in text: [1] -
Huang, H. H.; Fan, X.; Singh, D. J.; Chen, H.; Jiang, Q.; Zheng, W. T. Phys. Chem. Chem. Phys. 2016, 18, 4086–4094. doi:10.1039/c5cp06706e
Return to citation in text: [1] [2] -
Sik Hwang, W. S.; Remskar, M.; Yan, R.; Protasenko, V.; Tahy, K.; Doo Chae, S.; Zhao, P.; Konar, A.; Xing, H.; Seabaugh, A.; Jena, D. Appl. Phys. Lett. 2012, 101, 013107. doi:10.1063/1.4732522
Return to citation in text: [1] -
Gutiérrez, H. R.; Perea-López, N.; Elias, A. L.; Berkdemir, A.; Wang, B.; Lv, R.; López-Urías, F.; Crespi, V. H.; Terrones, H.; Terrones, M. Nano Lett. 2013, 13, 3447–3454. doi:10.1021/nl3026357
Return to citation in text: [1] -
Kozawa, D.; Kumar, R.; Carvalho, A.; Kumar Amara, K.; Zhao, W.; Wang, S.; Toh, M.; Ribeiro, R. M.; Castro Neto, A. H.; Matsuda, K.; Eda, G. Nat. Commun. 2014, 5, 4543. doi:10.1038/ncomms5543
Return to citation in text: [1] -
Nasr Esfahani, D.; Leenaerts, O.; Sahin, H.; Partoens, B.; Peeters, F. M. J. Phys. Chem. C 2015, 119, 10602–10609. doi:10.1021/jp510083w
Return to citation in text: [1] -
Henkelman, G.; Jóhannesson, G.; Jónsson, H. Methods for Finding Saddle Points and Minimum Energy Path. In Theoretical Methods in Condensed Phase Chemistry; Schwartz, S. D., Ed.; Springer: Amsterdam, Netherlands, 2002; pp 269–302.
Return to citation in text: [1] -
Wang, D.; Liu, L. M.; Zhao, S. J.; Hu, Z. Y.; Liu, H. J. Phys. Chem. C 2016, 120, 4779–4788. doi:10.1021/acs.jpcc.5b11677
Return to citation in text: [1] -
Soler, J. M.; Artacho, E.; Gale, J. D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. J. Phys.: Condens. Matter 2002, 14, 2745–2779. doi:10.1088/0953-8984/14/11/302
Return to citation in text: [1]
| 41. | Ataca, C.; Şahin, H.; Ciraci, S. J. Phys. Chem. C 2012, 116, 8983–8999. doi:10.1021/jp212558p |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 60. | Henkelman, G.; Jóhannesson, G.; Jónsson, H. Methods for Finding Saddle Points and Minimum Energy Path. In Theoretical Methods in Condensed Phase Chemistry; Schwartz, S. D., Ed.; Springer: Amsterdam, Netherlands, 2002; pp 269–302. |
| 41. | Ataca, C.; Şahin, H.; Ciraci, S. J. Phys. Chem. C 2012, 116, 8983–8999. doi:10.1021/jp212558p |
| 61. | Wang, D.; Liu, L. M.; Zhao, S. J.; Hu, Z. Y.; Liu, H. J. Phys. Chem. C 2016, 120, 4779–4788. doi:10.1021/acs.jpcc.5b11677 |
| 26. | Wang, X.; Shen, X.; Wang, Z.; Yu, R.; Chen, L. ACS Nano 2014, 8, 11394–11400. doi:10.1021/nn505501v |
| 27. | Wang, L.; Xu, Z.; Wang, W.; Bai, X. J. Am. Chem. Soc. 2014, 136, 6693–6697. doi:10.1021/ja501686w |
| 28. | Cheng, Y.; Nie, A.; Zhang, Q.; Gan, L.-Y.; Shahbazian-Yassar, R.; Schwingenschlogl, U. ACS Nano 2014, 8, 11447–11453. doi:10.1021/nn505668c |
| 59. | Nasr Esfahani, D.; Leenaerts, O.; Sahin, H.; Partoens, B.; Peeters, F. M. J. Phys. Chem. C 2015, 119, 10602–10609. doi:10.1021/jp510083w |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 41. | Ataca, C.; Şahin, H.; Ciraci, S. J. Phys. Chem. C 2012, 116, 8983–8999. doi:10.1021/jp212558p |
| 46. | Ma, Y.; Dai, Y.; Guo, M.; Niu, C.; Zhu, Y.; Huang, B. ACS Nano 2012, 6, 1695–1701. doi:10.1021/nn204667z |
| 43. | Zhang, H.; Liu, L.-M.; Lau, W.-M. J. Mater. Chem. A 2013, 1, 10821. doi:10.1039/c3ta12098h |
| 45. | Zhuang, H. L.; Hennig, R. G. Phys. Rev. B 2016, 93, 054429. doi:10.1103/PhysRevB.93.054429 |
| 62. | Soler, J. M.; Artacho, E.; Gale, J. D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. J. Phys.: Condens. Matter 2002, 14, 2745–2779. doi:10.1088/0953-8984/14/11/302 |
| 43. | Zhang, H.; Liu, L.-M.; Lau, W.-M. J. Mater. Chem. A 2013, 1, 10821. doi:10.1039/c3ta12098h |
| 44. | Kan, M.; Wang, B.; Lee, Y.-H.; Sun, Q. Nano Res. 2015, 8, 1348–1356. doi:10.1007/s12274-014-0626-5 |
| 46. | Ma, Y.; Dai, Y.; Guo, M.; Niu, C.; Zhu, Y.; Huang, B. ACS Nano 2012, 6, 1695–1701. doi:10.1021/nn204667z |
| 42. | Jing, Y.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. C 2013, 117, 25409–25413. doi:10.1021/jp410969u |
| 43. | Zhang, H.; Liu, L.-M.; Lau, W.-M. J. Mater. Chem. A 2013, 1, 10821. doi:10.1039/c3ta12098h |
| 44. | Kan, M.; Wang, B.; Lee, Y.-H.; Sun, Q. Nano Res. 2015, 8, 1348–1356. doi:10.1007/s12274-014-0626-5 |
| 42. | Jing, Y.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. C 2013, 117, 25409–25413. doi:10.1021/jp410969u |
| 42. | Jing, Y.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. C 2013, 117, 25409–25413. doi:10.1021/jp410969u |
| 43. | Zhang, H.; Liu, L.-M.; Lau, W.-M. J. Mater. Chem. A 2013, 1, 10821. doi:10.1039/c3ta12098h |
| 44. | Kan, M.; Wang, B.; Lee, Y.-H.; Sun, Q. Nano Res. 2015, 8, 1348–1356. doi:10.1007/s12274-014-0626-5 |
| 18. | Li, Y.; Wu, D.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. Lett. 2012, 3, 2221–2227. doi:10.1021/jz300792n |
| 42. | Jing, Y.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. C 2013, 117, 25409–25413. doi:10.1021/jp410969u |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 49. | Huang, W.; Da, H.; Liang, G. J. Appl. Phys. 2013, 113, 104304. doi:10.1063/1.4794363 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 1. | Goodenough, J. B.; Kim, Y. Chem. Mater. 2010, 22, 587–603. doi:10.1021/cm901452z |
| 12. | Liu, K.-K.; Zhang, W. J.; Lee, Y.-H.; Lin, Y.-C.; Chang, M.-T.; Su, C.-Y.; Chang, C.-S.; Li, H.; Shi, Y.; Zhang, H.; Lai, C.-S.; Li, L.-J. Nano Lett. 2012, 12, 1538–1544. doi:10.1021/nl2043612 |
| 13. | Lee, H. S.; Min, S.-W.; Chang, Y.-G.; Park, M. K.; Nam, T.; Kim, H.; Kim, J. H.; Ryu, S.; Im, S. Nano Lett. 2012, 12, 3695–3700. doi:10.1021/nl301485q |
| 14. | Lee, Y.-H.; Zhang, X.-Q.; Zhang, W.; Chang, M.-T.; Lin, C.-T.; Chang, K.-D.; Yu, Y.-C.; Wang, J. T.-W.; Chang, C.-S.; Li, L.-J.; Lin, T.-W. Adv. Mater. 2012, 24, 2320–2325. doi:10.1002/adma.201104798 |
| 29. | Sun, X.; Wang, Z.; Li, Z.; Fu, Y.-Q. Sci. Rep. 2016, 6, 26666. doi:10.1038/srep26666 |
| 49. | Huang, W.; Da, H.; Liang, G. J. Appl. Phys. 2013, 113, 104304. doi:10.1063/1.4794363 |
| 11. | Lauritsen, J. V.; Kibsgaard, J.; Helveg, S.; Topsøe, H.; Clausen, B. S.; Lægsgaard, E.; Besenbacher, F. Nat. Nanotechnol. 2007, 2, 53–58. doi:10.1038/nnano.2006.171 |
| 30. | Lee, J. H.; Jang, W. S.; Han, S. W.; Baik, H. K. Langmuir 2014, 30, 9866–9873. doi:10.1021/la501349k |
| 9. | Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Nat. Nanotechnol. 2011, 6, 147–150. doi:10.1038/nnano.2010.279 |
| 10. | Lee, C.; Yan, H.; Brus, L. E.; Heinz, T. F.; Hone, J.; Ryu, S. ACS Nano 2010, 4, 2695–2700. doi:10.1021/nn1003937 |
| 23. | Hu, T.; Li, R.; Dong, J. J. Chem. Phys. 2013, 139, 174702. doi:10.1063/1.4827082 |
| 24. | Song, I.; Park, C.; Choi, H. C. RSC Adv. 2015, 5, 7495–7514. doi:10.1039/C4RA11852A |
| 25. | Wypych, F.; Schöllhorn, R. J. Chem. Soc., Chem. Commun. 1992, 1386–1388. doi:10.1039/C39920001386 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 2. | Cheng, C.; Zhou, G.; Du, J.; Zhang, H.; Guo, D.; Li, Q.; Wei, W.; Chen, L. New J. Chem. 2014, 38, 2250. doi:10.1039/c3nj01642k |
| 3. | Chen, G.; Fu, E.; Zhou, M.; Xu, Y.; Fei, L.; Deng, S.; Chaitanya, V.; Wang, Y.; Luo, H. J. Alloys Compd. 2013, 578, 349–354. doi:10.1016/j.jallcom.2013.06.042 |
| 4. | Wang, L.; Dong, Z.; Wang, D.; Zhang, F.; Jin, J. Nano Lett. 2013, 13, 6244–6250. doi:10.1021/nl403715h |
| 5. | Vasilyeva, E.; Nasibulin, A.; Tolochko, O.; Rudskoy, A.; Sachdev, A.; Xiao, X. Z. Phys. Chem. 2015, 229, 1429–1437. doi:10.1515/zpch-2015-0573 |
| 6. | Yao, Q.; Huang, C.; Yuan, Y.; Liu, Y.; Liu, S.; Deng, K.; Kan, E. J. Phys. Chem. C 2015, 119, 6923–6928. doi:10.1021/acs.jpcc.5b02130 |
| 7. | Kang, J.-G.; Park, J.-G.; Kim, D.-W. Electrochem. Commun. 2010, 12, 307–310. doi:10.1016/j.elecom.2009.12.025 |
| 8. | Wang, Z.; Su, Q.; Deng, H. Phys. Chem. Chem. Phys. 2013, 15, 8705–8709. doi:10.1039/C3CP51167G |
| 26. | Wang, X.; Shen, X.; Wang, Z.; Yu, R.; Chen, L. ACS Nano 2014, 8, 11394–11400. doi:10.1021/nn505501v |
| 27. | Wang, L.; Xu, Z.; Wang, W.; Bai, X. J. Am. Chem. Soc. 2014, 136, 6693–6697. doi:10.1021/ja501686w |
| 28. | Cheng, Y.; Nie, A.; Zhang, Q.; Gan, L.-Y.; Shahbazian-Yassar, R.; Schwingenschlogl, U. ACS Nano 2014, 8, 11447–11453. doi:10.1021/nn505668c |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 21. | Wang, X.; Li, G.; Seo, M. H.; Hassan, F. M.; Hoque, M. A.; Chen, Z. Adv. Energy Mater. 2015, 5, 1501106. doi:10.1002/aenm.201501106 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 18. | Li, Y.; Wu, D.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. Lett. 2012, 3, 2221–2227. doi:10.1021/jz300792n |
| 22. | Jian, Z.; Zhao, B.; Liu, P.; Li, F.; Zheng, M.; Chen, M.; Shi, Y.; Zhou, H. Chem. Commun. 2014, 50, 1215–1217. doi:10.1039/C3CC47977C |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 17. | Liu, Y.; Zhao, Y.; Jiao, L.; Chen, J. J. Mater. Chem. A 2014, 2, 13109–13115. doi:10.1039/c4ta01644k |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 15. | Cai, Y.; Yang, X.; Liang, T.; Dai, L.; Ma, L.; Huang, G.; Chen, W.; Chen, H.; Su, H.; Xu, M. Nanotechnology 2014, 25, 465401. doi:10.1088/0957-4484/25/46/465401 |
| 16. | Xiao, J.; Choi, D.; Cosimbescu, L.; Koech, P.; Liu, J.; Lemmon, J. P. Chem. Mater. 2010, 22, 4522–4524. doi:10.1021/cm101254j |
| 20. | Xie, X.; Chen, S.; Sun, B.; Wang, C.; Wang, G. ChemSusChem 2015, 8, 2948–2955. doi:10.1002/cssc.201500149 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 32. | Yang, E.; Ji, H.; Jung, Y. J. Phys. Chem. C 2015, 119, 26374–26380. doi:10.1021/acs.jpcc.5b09935 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 34. | Tibbetts, K.; Miranda, C. R.; Meng, Y. S.; Ceder, G. Chem. Mater. 2007, 19, 5302–5308. doi:10.1021/cm0715242 |
| 31. | Tang, Q.; Jiang, D.-e. ACS Catal. 2016, 6, 4953–4961. doi:10.1021/acscatal.6b01211 |
| 32. | Yang, E.; Ji, H.; Jung, Y. J. Phys. Chem. C 2015, 119, 26374–26380. doi:10.1021/acs.jpcc.5b09935 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 32. | Yang, E.; Ji, H.; Jung, Y. J. Phys. Chem. C 2015, 119, 26374–26380. doi:10.1021/acs.jpcc.5b09935 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 48. | Ding, Y.; Wang, Y.; Ni, J.; Shi, L.; Shi, S.; Tang, W. Physica B 2011, 406, 2254–2260. doi:10.1016/j.physb.2011.03.044 |
| 41. | Ataca, C.; Şahin, H.; Ciraci, S. J. Phys. Chem. C 2012, 116, 8983–8999. doi:10.1021/jp212558p |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 36. | Li, Y.; Duerloo, K.-A. N.; Wauson, K.; Reed, E. J. Nat. Commun. 2016, 7, 10671. doi:10.1038/ncomms10671 |
| 39. | Duerloo, K.-A. N.; Li, Y.; Reed, E. J. Nat. Commun. 2014, 5, 4214. doi:10.1038/ncomms5214 |
| 40. | Reyes-Retana, J. A.; Cervantes-Sodi, F. Sci. Rep. 2016, 6, 24093. doi:10.1038/srep24093 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 40. | Reyes-Retana, J. A.; Cervantes-Sodi, F. Sci. Rep. 2016, 6, 24093. doi:10.1038/srep24093 |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 50. | Qian, X.; Lu, J.; Fu, L.; Li, J. Science 2014, 346, 1344–1347. doi:10.1126/science.1256815 |
| 55. | Huang, H. H.; Fan, X.; Singh, D. J.; Chen, H.; Jiang, Q.; Zheng, W. T. Phys. Chem. Chem. Phys. 2016, 18, 4086–4094. doi:10.1039/c5cp06706e |
| 56. | Sik Hwang, W. S.; Remskar, M.; Yan, R.; Protasenko, V.; Tahy, K.; Doo Chae, S.; Zhao, P.; Konar, A.; Xing, H.; Seabaugh, A.; Jena, D. Appl. Phys. Lett. 2012, 101, 013107. doi:10.1063/1.4732522 |
| 57. | Gutiérrez, H. R.; Perea-López, N.; Elias, A. L.; Berkdemir, A.; Wang, B.; Lv, R.; López-Urías, F.; Crespi, V. H.; Terrones, H.; Terrones, M. Nano Lett. 2013, 13, 3447–3454. doi:10.1021/nl3026357 |
| 58. | Kozawa, D.; Kumar, R.; Carvalho, A.; Kumar Amara, K.; Zhao, W.; Wang, S.; Toh, M.; Ribeiro, R. M.; Castro Neto, A. H.; Matsuda, K.; Eda, G. Nat. Commun. 2014, 5, 4543. doi:10.1038/ncomms5543 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 18. | Li, Y.; Wu, D.; Zhou, Z.; Cabrera, C. R.; Chen, Z. J. Phys. Chem. Lett. 2012, 3, 2221–2227. doi:10.1021/jz300792n |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 40. | Reyes-Retana, J. A.; Cervantes-Sodi, F. Sci. Rep. 2016, 6, 24093. doi:10.1038/srep24093 |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 50. | Qian, X.; Lu, J.; Fu, L.; Li, J. Science 2014, 346, 1344–1347. doi:10.1126/science.1256815 |
| 51. | Mak, K. F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T. F. Phys. Rev. Lett. 2010, 105, 136805. doi:10.1103/PhysRevLett.105.136805 |
| 52. | Tongay, S.; Zhou, J.; Ataca, C.; Lo, K.; Matthews, T. S.; Li, J.; Grossman, J. C.; Wu, J. Nano Lett. 2012, 12, 5576–5580. doi:10.1021/nl302584w |
| 53. | Conley, H. J.; Wang, B.; Ziegler, J. I.; Haglund, R. F., Jr.; Pantelides, S. T.; Bolotin, K. I. Nano Lett. 2013, 13, 3626–3630. doi:10.1021/nl4014748 |
| 54. | Ji, Q.; Zhang, Y.; Gao, T.; Zhang, Y.; Ma, D.; Liu, M.; Chen, Y.; Qiao, X.; Tan, P.-H.; Kan, M.; Feng, J.; Sun, Q.; Liu, Z. Nano Lett. 2013, 13, 3870–3877. doi:10.1021/nl401938t |
| 55. | Huang, H. H.; Fan, X.; Singh, D. J.; Chen, H.; Jiang, Q.; Zheng, W. T. Phys. Chem. Chem. Phys. 2016, 18, 4086–4094. doi:10.1039/c5cp06706e |
| 36. | Li, Y.; Duerloo, K.-A. N.; Wauson, K.; Reed, E. J. Nat. Commun. 2016, 7, 10671. doi:10.1038/ncomms10671 |
| 37. | Enyashin, A. N.; Yadgarov, L.; Houben, L.; Popov, I.; Weidenbach, M.; Tenne, R.; Bar-Sadan, M.; Seifert, G. J. Phys. Chem. C 2011, 115, 24586–24591. doi:10.1021/jp2076325 |
| 38. | Sun, Y.; Wang, Y.; Sun, D.; Carvalho, B. R.; Read, C. G.; Lee, C.-h.; Lin, Z.; Fujisawa, K.; Robinson, J. A.; Crespi, V. H.; Terrones, M.; Schaak, R. E. Angew. Chem., Int. Ed. 2016, 55, 2830–2834. doi:10.1002/anie.201510029 |
| 39. | Duerloo, K.-A. N.; Li, Y.; Reed, E. J. Nat. Commun. 2014, 5, 4214. doi:10.1038/ncomms5214 |
| 40. | Reyes-Retana, J. A.; Cervantes-Sodi, F. Sci. Rep. 2016, 6, 24093. doi:10.1038/srep24093 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
| 35. | Ambrosi, A.; Sofer, Z.; Pumera, M. Chem. Commun. 2015, 51, 8450–8453. doi:10.1039/c5cc00803d |
| 40. | Reyes-Retana, J. A.; Cervantes-Sodi, F. Sci. Rep. 2016, 6, 24093. doi:10.1038/srep24093 |
| 32. | Yang, E.; Ji, H.; Jung, Y. J. Phys. Chem. C 2015, 119, 26374–26380. doi:10.1021/acs.jpcc.5b09935 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 32. | Yang, E.; Ji, H.; Jung, Y. J. Phys. Chem. C 2015, 119, 26374–26380. doi:10.1021/acs.jpcc.5b09935 |
| 33. | Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X. C. J. Phys. Chem. C 2014, 118, 7242–7249. doi:10.1021/jp501734s |
| 47. | Rasmussen, F. A.; Thygesen, K. S. J. Phys. Chem. C 2015, 119, 13169–13183. doi:10.1021/acs.jpcc.5b02950 |
© 2017 Sun and Wang; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)