Abstract
CdSe nanorods (NRs) with an average length of ≈120 nm were prepared by a solvothermal process and associated to TiO2 nanoparticles (Aeroxide® P25) by annealing at 300 °C for 1 h. The content of CdSe NRs in CdSe/TiO2 composites was varied from 0.5 to 5 wt %. The CdSe/TiO2 heterostructured materials were characterized by XRD, TEM, SEM, XPS, UV–visible spectroscopy and Raman spectroscopy. TEM images and XRD patterns show that CdSe NRs with wurtzite structure are associated to TiO2 particles. The UV–visible spectra demonstrate that the narrow bandgap of CdSe NRs serves to increase the photoresponse of CdSe/TiO2 composites until ≈725 nm. The CdSe (2 wt %)/TiO2 composite exhibits the highest photocatalytic activity for the degradation of rhodamine B in aqueous solution under simulated sunlight or visible light irradiation. The enhancement in photocatalytic activity likely originates from CdSe sensitization of TiO2 and the heterojunction between these materials which facilitates electron transfer from CdSe to TiO2. Due to its high stability (up to ten reuses without any significant loss in activity), the CdSe/TiO2 heterostructured catalysts show high potential for real water decontamination.
Introduction
The development of efficient photocatalysts to address environmental and energy needs, such as degradation of harmful organic compounds in water and in the air or the conversion of solar energy to chemical energy, for example, via water splitting to produce hydrogen, is the topic of numerous current research projects. Titanium dioxide (TiO2) has been widely investigated over the last three decades and has been demonstrated to be of high potential [1,2]. However, TiO2 suffers from two main drawbacks. First, due to its wide bandgap (Eg = 3.2 and 3.0 eV for anatase and rutile, respectively), TiO2 can only be activated by light with a wavelength of less than 390 nm to trigger the electron–hole separation. Second, TiO2 exhibits a low quantum efficiency due to the fast recombination of photogenerated charge carriers (electrons and holes).
To address these problems, a number of studies have been devoted to the improvement of light absorption and charge separation by hybridizing TiO2 with narrow bandgap semiconductors, doping with metal or nonmetal elements, association with noble metal particles, or constructing heterojunctions between TiO2 and graphene-based materials or carbon nitride (C3N4), acting as electron-transport materials [3-5]. All of these strategies have the common goal of decreasing the charge carrier recombination rate by increasing the spatial charge separation.
The interfacial electron transfer between two semiconductors has gained significant interest because the heterojunction improves both the optical absorption in the visible range and the charge separation yield and thus the charge carrier lifetime [3-8]. The photocatalytic activity is enhanced because oxidation of water by holes and reduction of oxygen by electrons are retained at two different sites.
Recently, many groups demonstrated that the creation of a heterojunction between TiO2 and CdS, CdSe or alloyed CdSeS nanoparticles allows the optical absorption of TiO2 to be extended into the visible light range and increases the photoconversion efficiency by improving the charge transfer [9-37]. CdSe is one of the most commonly used semiconductors due to its narrow bandgap (1.7 eV) and its band energies are located at relatively low potential. Despite the large number of reports describing the sensitization of TiO2 (used in the form of spherical particles, films, wires, tubes, etc.) with CdSe nanocrystals (generally quantum dots or spherical nanoclusters) [9-37], very little attention has been devoted to the influence of CdSe crystal morphology on the photocatalytic activity of the CdSe/TiO2 heterostructured photocatalysts. CdSe nanorods (NRs) and wires are of high interest for use as sensitizers for photocatalytic applications due to their high surface area, higher optical absorption cross section (as compared to spherical particles) and easier charge carrier separation [38]. Only a couple of reports describe the association of CdSe NRs with TiO2. Luo et al. used chemical vapor deposition to associate CdS, CdSe or CdSeS rods to TiO2 NRs arrays and demonstrated that the CdSeS/TiO2 heterostructure exhibits the highest performances as photoelectrode [39]. More recently, small CdSe NRs [40] or type II CdSe/CdSexTe1−x NRs [41] were also used as light harvesters to develop TiO2-based solar cells. Core/shell TiO2/CdSe NRs were also prepared by growing CdSe quantum dots onto TiO2 NRs at high temperature [42].
In this paper, we report an investigation on the synthesis of CdSe-NR-sensitized TiO2 nanoparticles and on the use of these materials for the degradation of rhodamine B (RhB) in aqueous solution. Our results demonstrate that the composite containing 2 wt % CdSe NRs exhibits the optimal photocatalytic activity under solar or visible light irradiation. Moreover, the photocatalytic response was maintained after ten cycles in simulated sunlight. A possible mechanism is also discussed.
Experimental
Chemicals
Cadmium acetate (Cd(OAc), 99.995%, Sigma), sodium selenite (Na2SeO3, 99%, Sigma), diethylenetriamine (99%, Sigma), TiO2 nanoparticles (Sigma-Aldrich, Aeroxide® P25, CAS: 13463-67-7), rhodamine B (RhB, >95%, Sigma), tert-butanol (t-BuOH, >99.5%, Sigma), p-benzoquinone (>98%, Sigma) and ethanol (>99%, anhydrous) were used as received without additional purification. All solutions were prepared using Milli-Q water (18.2 MΩ·cm, Millipore) as the solvent.
Synthesis of CdSe nanorods
CdSe NRs were synthetized via a solvothermal method according to the protocol described by Li et al. [43], with slight modifications. Briefly, diethylenetriamine (34.3 mL) and water (5.8 mL) were mixed in a reaction flask and the mixture stirred for 5 min. Next, Cd(OAc)2 (0.266 g, 1 mmol) and Na2SeO3 (0.173 g, 1 mmol) were added and the mixture further stirred for 30 min at room temperature to ensure complete dissolution of precursors. Next, the solution was transferred to a teflon-lined stainless steel autoclave with 100 mL capacity and heated to 180 °C for 12 h. After cooling to room temperature, the CdSe NRs were recovered by centrifugation (4000 rpm for 15 min), washed with water (5 × 10 mL), then with ethanol (2 × 10 mL) and finally dried at 50 °C for 12 h.
Preparation of CdSe/TiO2 composites
CdSe/TiO2 composites with a varied content of CdSe NRs (0.5, 1, 2 and 5 wt %) were prepared. For the CdSe (2 wt %)/TiO2 composite, commercial Aeroxide® P25 TiO2 nanoparticles (250 mg) and CdSe NRs (5 mg) were dispersed by sonication in 5 mL water for 30 min. Next, the mixture was heated to 70 °C to evaporate water. The solid obtained was washed with water (3 × 10 mL), with ethanol (3 × 5 mL) and then calcined under air at 300 °C for 1 h to couple the CdSe NRs with TiO2. CdSe/TiO2 composites with different wt % CdSe NRs were prepared using a similar synthetic procedure.
Photocatalytic activity measurements
The photocatalytic activity of CdSe/TiO2 composites was evaluated by the degradation of RhB in aqueous solution. Sylvania LuxLine FHO T5 neon tubes were used as the simulated solar light source (light intensity = 5 mW/cm2). A polycarbonate filter was added for experiments conducted under visible light irradiation (light intensity = 10 mW/cm2).
In a typical experiment, 50 mg of the CdSe/TiO2 composite were dispersed in 50 mL of aqueous solution of RhB (10 mg/L) in a 70 mL glass flask and the mixture was magnetically stirred in the dark under ambient conditions for 45 min to reach a thorough adsorption–desorption equilibrium. After that period, the light was turned on. Aliquots of 2 mL were taken at regular time intervals and centrifuged (4000 rpm for 2 min) to remove the CdSe/TiO2 photocatalyst. The relative concentration of RhB in the solution was determined by comparing its UV−visible absorption at 554 nm with that of the starting solution.
The mechanism of RhB photodegradation in aqueous solution was investigated through the use of scavengers. t-BuOH and p-benzoquinone, used as hydroxyl and superoxide scavengers, were used at concentrations of 1 and 10 mM, respectively.
Characterization
Transmission electron microscopy (TEM) investigations were performed with a JEOL ARM 200F-Cold FEG TEM/STEM (point resolution 0.19 nm in TEM mode and 0.078 nm in STEM mode) fitted with a GIF Quatum ER. For each sample, one drop of a dispersed solution was deposited on holey carbon grids and imaged.
Scanning electron microscopy (SEM) images were prepared using a JEOL SEM JSM-6490 LV.
The crystalline phase of the powders was determined by powder X-ray diffraction (XRD) on an X'Pert MPD diffractometer (Panalytical AXS) with a goniometer radius of 240 mm, fixed divergence slit module (1/2° divergence slit, 0.04 rd Sollers slits) and an X'Celerator as a detector. The samples were placed on a silicon zero-background sample holder and the XRD patterns were recorded at room temperature using Cu Kα radiation (λ = 0.15418 nm).
XPS analysis was conducted on a Gammadata Scienta (Uppsala, Sweden) SES 200-2 spectrometer under ultra-high vacuum conditions (P < 10−9 mbar). For all peak fitting procedures the CASA XPS software (Casa Software Ltd, Teignmouth, UK, http://www.casaxps.com) was used and the areas of each component were modified according to classical Scofield sensitivity factors.
All the optical measurements were performed at room temperature (20 ± 1 °C) under ambient conditions. The absorption spectra of liquid samples were recorded on a Thermo Scientific Evolution 220 UV–visible spectrophotometer. Diffuse reflectance spectra (DRS) were recorded on a Shimadzu 2600 UV−visible spectrophotometer. BaSO4 powder was used as a standard for baseline measurements and spectra were recorded in a range of 250–1400 nm. Raman spectra were recorded using an Xplora spectrometer from Horiba Scientific with 532 nm wavelength incident laser light.
The initial and final total organic carbon (TOC) content was determined using a Shimadzu TOC-VCSH analyzer to evaluate the degree of photomineralization.
Results and Discussion
Synthesis, morphological and structural characterization of CdSe/TiO2 composites
CdSe NRs were prepared by a solvothermal method using diethylenetriamine as structure directing agent [43]. The CdSe rods were associated to TiO2 nanoparticles by a heat treatment at 300 °C for 1 h to construct a heterojunction between these materials that favors the charge injection from CdSe to TiO2.
The microstructure of CdSe NRs and CdSe/TiO2 composites was first characterized by SEM (Figure S1, Supporting Information File 1). The small CdSe NRs were found to be strongly associated to each other to form larger particles with sizes varying between 1 and 4 µm. The associated energy dispersive X-ray (EDX) spectrum demonstrates that Cd and Se were the only elements detected in CdSe NRs. Due to the low content of CdSe NRs in CdSe/TiO2 composites, CdSe NRs could only hardly be observed by SEM. Ti, O, Cd and Se elements were detected for all composites, which indicates the successful association of CdSe NRs with TiO2. All CdSe/TiO2 composites exhibit a non-rigid texture and the pores observed should be beneficial for the diffusion of molecules and thus for an enhanced photocatalytic activity.
The morphology of the particles was further characterized by TEM. Commercial P25 TiO2 nanoparticles (Aeroxide® P25) are of spherical/ellipsoidal shape and their average diameter is ≈23 ± 6 nm (Figure 1a). CdSe NRs produced after the solvothermal synthesis have an average length and an average diameter of 120 and 12 nm, respectively (Figure 1b). Some nanowires with lengths up to 300 nm could also be detected. A typical TEM image of the CdSe (2 wt %)/TiO2 composite is given in Figure 1c and demonstrates the coexistence and the association of CdSe NRs and TiO2 particles. This could be further confirmed by HR-TEM (Figure 1d). The measured lattice spacing was 0.36 and 0.37 nm, corresponding to the (101) plane of anatase TiO2 and to the (100) plane of wurtzite CdSe, respectively.
![[2190-4286-8-273-1]](/bjnano/content/figures/2190-4286-8-273-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: TEM images of (a) TiO2, (b) CdSe nanorods and (c) the CdSe (2 wt %)/TiO2 composite. (d) HR-TEM image of CdSe nanorods associated to TiO2 nanoparticles.
Figure 1: TEM images of (a) TiO2, (b) CdSe nanorods and (c) the CdSe (2 wt %)/TiO2 composite. (d) HR-TEM imag...
XRD analysis of CdSe, TiO2 and CdSe-NR-sensitized TiO2 is presented in Figure 2. For pure CdSe rods, all peaks could be indexed to the hexagonal wurtzite structure (JCPDS No 77-2307), which is in accordance with the previously described HR-TEM results (Figure 2a). For TiO2, the diffraction peaks at 2θ = 25.41, 37.98, 48.18, 54.12, 55.24, 62.93, 69.03, 70.42 and 75.17° can be indexed to the (111), (004), (200) (105), (211), (204), (310), (116) and (220) crystal planes of anatase TiO2 (JCPDS No 21-1272), while those located at 2θ = 27.55, 36.23, 41.34 and 56.72° belong to rutile TiO2 (JCPDS No 21-1276) (Figure 2b). The anatase and rutile phases of TiO2 exhibit a higher catalytic activity than the brookite phase [44]. The anatase/rutile ratio was not affected by the heating at 300 °C (the phase transition occurs at 500 °C). The position of TiO2 diffraction peaks was not changed after association with CdSe, indicating that CdSe NRs do not affect the lattice structure of TiO2. The diffraction peaks of CdSe NRs could not be observed in all CdSe/TiO2 composites due to the low content of CdSe (from 0.5 to 5 wt %) and their dispersion in TiO2 particles.
![[2190-4286-8-273-2]](/bjnano/content/figures/2190-4286-8-273-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray diffraction patterns of (a) CdSe and (b) TiO2 and CdSe/TiO2 composites with varying CdSe wt % from 0.5 to 5 wt %.
Figure 2: X-ray diffraction patterns of (a) CdSe and (b) TiO2 and CdSe/TiO2 composites with varying CdSe wt %...
Raman spectroscopy was further used to confirm the structures of CdSe/TiO2 composites. Two peaks located at 206 and 414 cm−1 corresponding to the longitudinal optical (LO) phonon and its overtone (2LO) can be observed for CdSe NRs (Figure S2, Supporting Information File 1) [45]. For TiO2, the Eg, B1g, A1g and Eg peaks located at 143, 397, 514 and 636 cm−1, respectively, are typical of the TiO2 anatase phase [46] (Figure 3). Due to the weak content in CdSe NRs, the LO signal of CdSe could only be detected for composites containing 2 and 5 wt % CdSe NRs. However, Raman analysis indicates that CdSe NRs are well-associated to TiO2 particles.
![[2190-4286-8-273-3]](/bjnano/content/figures/2190-4286-8-273-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Raman spectra of TiO2 and CdSe/TiO2 composites.
Figure 3: Raman spectra of TiO2 and CdSe/TiO2 composites.
The UV–visible absorption spectra of TiO2 and CdSe/TiO2 composites are shown in Figure 4a. The bandgap energies of TiO2, CdSe and CdSe/TiO2 composites were determined by plotting [F(R)hν]2 vs photon energy and extrapolating the plots at [F(R)hν]2 = 0 (F(R) is the Kubelka–Munk function, h is the Planck constant and ν is the frequency) (Figure 4b). No optical response in the visible region could be detected for TiO2 due to its wide bandgap (3.48 eV). As can be seen, the absorption increases with the increase of CdSe NRs in the composite from 400 to ≈725 nm, which corresponds to the energy bandgap of CdSe NRs (≈1.61 eV) – a value slightly lower than the standard bandgap of CdSe (1.78 eV) [47]. This enhanced visible light absorption originates from the excitation of CdSe NRs and indicates a decrease of the bandgap with increasing amount of CdSe (the bandgap of CdSe/TiO2 composites was found to decrease from 3.38 eV for 0.5 wt % CdSe to 2.88 eV for 5 wt % CdSe) (Figure 4b). Because the conduction band edge of CdSe NRs is higher than that of TiO2, the results shown in Figure 4 demonstrate that the excited electrons of CdSe NRs can be transferred to TiO2 nanoparticles due to the heterojunction between these materials (Figure 4c). This should contribute to enhance the photocatalytic activity of CdSe/TiO2 composites under visible light irradiation (vide infra).
![[2190-4286-8-273-4]](/bjnano/content/figures/2190-4286-8-273-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a) UV–visible absorption spectra of TiO2 and CdSe/TiO2 composites, (b) plots of transformed Kubelka–Munk functions [F(R)·hν]2 vs hν for TiO2 and CdSe/TiO2 composites, and (c) energy level diagram for CdSe NRs/TiO2 composites.
Figure 4: (a) UV–visible absorption spectra of TiO2 and CdSe/TiO2 composites, (b) plots of transformed Kubelk...
The chemical state of the elements composing the CdSe (2 wt %)/TiO2 composite was studied by XPS. The high-resolution spectra of the elements Cd 3d5/2, Se 3p3/2, Cd 3p1/2, Ti 2p and O 1s in the CdSe (2 wt %)/TiO2 composite are provided in Figure 5. The signals of Ti 2p3/2, Ti 2p1/2, O 1s (lattice oxygen atoms), and O 1s (surface hydroxyl groups) appear at 458.74, 464.56, 530.0, and 531.76 eV, respectively, which are values in good accordance with those measured for pure TiO2 [48]. The signals of Cd 3d5/2 and Se 3p3/2 and Se 3p1/2 can be observed at 405.46, 160.26 and 165.91 eV, respectively. These values are typical for CdSe [49] and further confirm that CdSe NRs are associated to TiO2. Both for CdSe and TiO2, no significant shift in binding energy was observed after building the heterojunction between these materials (see Supporting Information File 1, Figure S3 for the high-resolution XPS spectra of Cd 3d5/2 and Se 3p of CdSe NRs). Noteworthy is also that the surface of CdSe NRs was not oxidized into CdO during the calcination step. The signals of Cd 3d5/2 at ≈403.2 eV for CdO [50] and Se 3p3/2 at 165.1 eV for SeO2 [51] were not detected in the XPS spectra of the CdSe (2 wt %)/TiO2 composite.
![[2190-4286-8-273-5]](/bjnano/content/figures/2190-4286-8-273-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: High-resolution XPS spectra of the CdSe (2 wt %)/TiO2 composite: (a) Cd 3d, where Cd 3d5/2 (blue) at 405.46 eV comprised 45.18% of the area of the spectra and N 1s (pink) at 399.81 eV comprised 54.82% of the area of the spectra; (b) Se 3p, where Se 3p1/2 (blue) at 165.91 eV comprised 50.09% of the area of the spectra and Se 3p3/2 (pink) at 160.26 eV comprised 49.91% of the area of the spectra; (c) Ti 2p, where Ti 2p1/2 (blue) at 464.56 eV comprised 44.72% of the area of the spectra and Ti 2p3/2 (pink) at 458.74 eV comprised 55.28% of the area of the spectra and (d) O 1s, where C–O C=O (blue) at 531.72 eV comprised 24.81% of the area of the spectra and Ti–O (pink) at 530.00 eV comprised 75.19% of the area of the spectra. CPS = counts per second.
Figure 5: High-resolution XPS spectra of the CdSe (2 wt %)/TiO2 composite: (a) Cd 3d, where Cd 3d5/2 (blue) a...
Solar light photocatalytic activity of CdSe/TiO2 composites
The photocatalytic decomposition of RhB at pH 7 over TiO2 nanoparticles and CdSe/TiO2 composites under simulated solar light irradiation was first evaluated (light intensity: 5 mW/cm2). Before illumination, the adsorption/desorption equilibrium was established for 45 min. In the absence of the photocatalyst, the photodegradation of RhB was found to be negligible. As shown in Figure 6a, the highest photocatalytic activity was reached for the CdSe (2 wt %)/TiO2 material which fully bleached the dye in 150 min. Thereafter, the activity decreases when increasing the amount of CdSe NRs associated to TiO2. Because the photocatalytic activity originates from the spatial separation of photogenerated electrons and holes, it is likely that CdSe NRs are not properly associated to TiO2 and do not contribute to the photocatalytic process.
![[2190-4286-8-273-6]](/bjnano/content/figures/2190-4286-8-273-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a) Photocatalytic activity of TiO2 and CdSe/TiO2 composites for the degradation of RhB under simulated solar light irradiation, (b) UV–visible absorption spectra of RhB during the photodegradation using the CdSe (2 wt %)/TiO2 composite (the inset shows the chemical structure of RhB), and (c) a digital photograph of the RhB solution over the course of the photocatalytic degradation.
Figure 6: (a) Photocatalytic activity of TiO2 and CdSe/TiO2 composites for the degradation of RhB under simul...
Total organic carbon (TOC) measurements showed that the RhB solution was discolored during photocatalytic experiments (Figure 6c) and the dye was also partially decomposed (the TOC value decreased from 6.9 ± 0.3 to 2.4 ± 0.2 mg/L after the 150 min of irradiation).
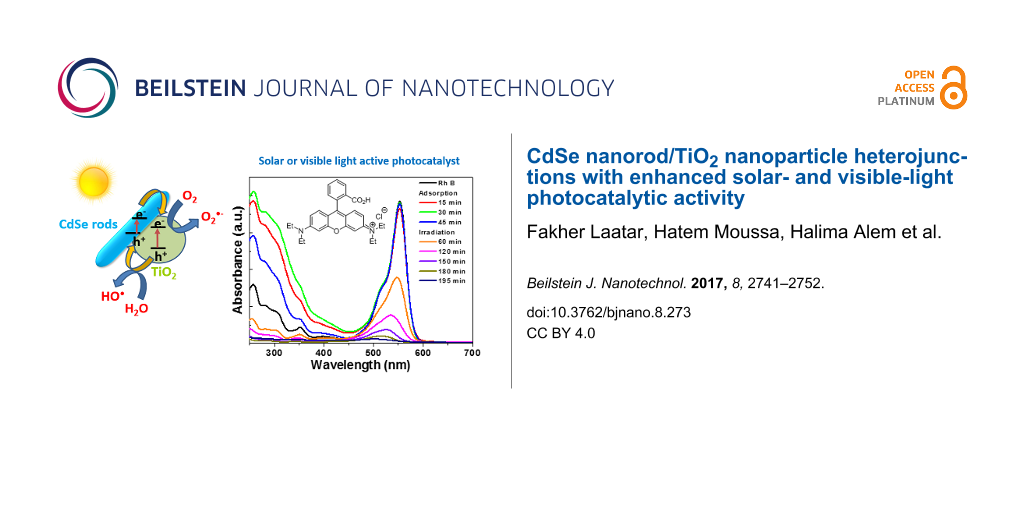
The temporal evolution of the UV–vis absorption spectra is displayed in Figure 6b. The intensity of the RhB absorption from 250 to 600 nm decreases over time. A blue shift (from 553 to 503 nm) of the main absorption peak is also observed, which is indicative of stepwise N-de-ethylation of the dye during the photocatalytic process [52]. The plots of ln(C0/C) of RhB vs irradiation time indicate that the photodegradation of RhB over CdSe/TiO2 composites follows a pseudo-first-order kinetic model (C0 is the initial concentration of RhB and C is the concentration of the dye at time t). The rate constants k determined from the slopes of ln(C0/C) vs time were found to be 0.017, 0.012, 0.013, 0.019, and 0.014 min−1 for TiO2 and CdSe/TiO2 composites loaded with 0.5, 1, 2, and 5 wt % CdSe, respectively (Figure S4, Supporting Information File 1).
Influence of the photocatalyst dosage and rhodamine B concentration
The photodegradation efficiency is generally affected by the amount of catalyst used and by the concentration of the pollutant. First, we investigated the influence of the catalyst concentration (25, 50 or 100 mg in 50 mL of RhB solution) (Figure 7a). Using 25 mg of the CdSe (2 wt %)/TiO2 photocatalyst, the dye decomposition required 255 min while the degradation could be achieved within 75 min using 100 mg of photocatalyst. As expected, the photocatalytic degradation rates increased with the catalyst dosage, which is linked to the concentration of adsorption sites and photocatalytically active sites (k = 0.010, 0.019 and 0.034 min−1 for reactions conducted with 25, 50 and 100 mg of photocatalyst, respectively).
![[2190-4286-8-273-7]](/bjnano/content/figures/2190-4286-8-273-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: (a) Effect of the catalyst concentration on the photodegradation efficiency (25, 50 or 100 mg of photocatalyst were dispersed in 50 mL of a 10 mg/L RhB solution). (b) Influence of RhB concentration on the photocatalytic efficiency (50 mg of photocatalyst and 50 mL of the dye solution were used).
Figure 7: (a) Effect of the catalyst concentration on the photodegradation efficiency (25, 50 or 100 mg of ph...
Next, the concentration of the RhB solution was varied from 5, 10 to 25 mg/L while the amount of catalyst was fixed at 50 mg (Figure 7b). The photodegradation rate decreases with increasing RhB concentration (k = 0.028, 0.019 and 0.002 min−1 for RhB concentrations of 5, 10 and 25 mg/L, respectively). This originates (i) from the high amount of dye and/or the photodegradation intermediates adsorbed at the photocatalyst surface and (ii) from the decrease of the light penetration in the reactor due to the high absorption of RhB and thus to the decreased amount of reactive oxygen species produced.
Influence of the pH of the rhodamine B solution
The pH of the aqueous solution may play a crucial role on the kinetic of the photodegradation since it influences the surface charge of the catalyst and the structure of the dye in solution. Figure 8 shows that the CdSe (2 wt %)/TiO2 photocatalyst exhibits the highest activity at pH values varying from 5 to 8 (k values are varying from 0.017 to 0.031 min−1). At pH 3, the photodegradation kinetic is reduced (k = 0.011 min-1) but the highest inhibition was observed in basic media (k = 0.009 and 0.006 min−1 at pH 9 and 11, respectively). The point of zero charge of the TiO2 nanoparticles is at ≈6.5 [53] and the pKa of the acid function of RhB is 3.7. At pH ≥ 9, the weak photocatalytic activity probably originates from the strong repellency between the carboxylate function of RhB and the negatively charged catalyst. At pH 3, the positively charged RhB exhibits only a modest affinity for the positively charged catalyst. At pH values ranging from 5 to 8, RhB may associate to the photocatalyst either via interaction involving the carboxylate function or the positively charged diethylamino groups.
![[2190-4286-8-273-8]](/bjnano/content/figures/2190-4286-8-273-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Influence of pH on the degradation of rhodamine B using the CdSe (2 wt %)/TiO2 photocatalyst.
Figure 8: Influence of pH on the degradation of rhodamine B using the CdSe (2 wt %)/TiO2 photocatalyst.
Visible light photocatalytic activity of the CdSe (2 wt %)/TiO2 composite
The CdSe (2 wt %)/TiO2 composite is also photocatalytically active when the incident light wavelength is greater than 400 nm. The spectral evolution of RhB and the degradation profile are shown in Figure 9. As can be seen, the photodegradation is slower than that initiated by simulated solar light and ≈70% of RhB is bleached after 6.25 h of irradiation. A similar hypsochromic shift of the absorption band at 553 nm to that observed under simulated sunlight irradiation was observed, indicating that photooxidative N-de-ethylation of RhB occurred in the early stages of its photodegradation. Finally, although the operating conditions and the structure of the pollutant were different from those used in this study, the CdSe NR/TiO2 nanoparticle photocatalyst favorably compares with catalysts engineered from CdSe quantum dots and TiO2 nanoparticles or nanotubes [9,11,12,22,26,31,33,34].
![[2190-4286-8-273-9]](/bjnano/content/figures/2190-4286-8-273-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: (a) UV–visible spectral evolution of rhodamine B as a function of irradiation time using the CdSe (2 wt %)/TiO2 catalyst under visible light irradiation (light intensity: 10 mW/cm2) and (b) evolution of C/C0 vs irradiation time.
Figure 9: (a) UV–visible spectral evolution of rhodamine B as a function of irradiation time using the CdSe (...
Reusability of the CdSe (2 wt %)/TiO2 photocatalyst
The CdSe (2 wt %)/TiO2 photocatalyst was subjected to reuse under simulated solar light irradiation. After the first run, the photocatalyst was recovered by centrifugation (4000 rpm for 15 min) and reused without any treatment. As can be seen from Figure 10, no marked decrease in photocatalytic activity was observed after ten reuses, indicating that byproducts originating from RhB decomposition do not block the active sites on the photocatalyst surface and that the photocatalyst is not photocorroded. Furthermore, XRD patterns of the CdSe (2 wt %)/TiO2 composite before and after ten reuses and the SEM analysis clearly demonstrate that the photocatalyst is stable during the reaction (Supporting Information File 1, Figure S5). The stability of the CdSe/TiO2 catalyst is of high importance for real photocatalytic applications.
![[2190-4286-8-273-10]](/bjnano/content/figures/2190-4286-8-273-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Recycling of the CdSe (2 wt %)/TiO2 catalyst in the degradation of RhB under simulated solar light irradiation.
Figure 10: Recycling of the CdSe (2 wt %)/TiO2 catalyst in the degradation of RhB under simulated solar light ...
Photodegradation mechanism
Under simulated solar light irradiation, electron (e−)/hole (h+) pairs are generated both in CdSe NRs and TiO2 nanoparticles. The holes in the valence band (VB) of TiO2 are transferred to the VB of CdSe NRs while electrons are transferred from the conduction band (CB) of CdSe NRs to the CB of TiO2 (Figure 11a). Under visible light illumination, only CdSe can be activated. A CdSe NR electron is promoted from the VB to the CB, leaving a hole in the VB. Then, the electron is transferred to TiO2 (Figure 11b). All these transfer processes are thermodynamically favorable since both the CB and VB of CdSe NRs lie above that of TiO2. These charge separations may effectively reduce the probability of recombination and increase the lifetime of charge carriers (e− and h+).
![[2190-4286-8-273-11]](/bjnano/content/figures/2190-4286-8-273-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Schematic of the charge separation in the CdSe/TiO2 photocatalyst under (a) solar light and (b) visible light irradiation. (c) The influence of O2•− and •OH scavengers on the photocatalytic degradation of rhodamine B under visible light irradiation (p-benzoquinone and t-BuOH were used at concentrations of 1 mM and 10 mM, respectively).
Figure 11: Schematic of the charge separation in the CdSe/TiO2 photocatalyst under (a) solar light and (b) vis...
Under visible light irradiation, holes in the VB of CdSe may directly oxidize the dye. Meanwhile, the electrons accumulated in TiO2 can be trapped by dissolved oxygen molecules and generate superoxide O2•− radicals which are strong oxidants able to decompose organic substances. These O2•− radicals may also react with an electron and protons to form hydrogen peroxide which is further decomposed into hydroxyl •OH radicals able to oxidize RhB.
To estimate which of these reactive oxygen species plays a key role in the photodegradation of RhB under visible light irradiation, experiments were carried out by adding t-BuOH and p-benzoquinone, used as •OH and O2•− radicals scavengers, respectively (Figure 11c). As can be seen, RhB could not be photodegraded in the presence of p-benzoquinone, even when used at a low concentration (1 mM), indicating that O2•− radicals play a key role in the photodegradation. In the presence of t-BuOH (10 mM), the reaction rate was markedly decreased (k = 0.004 min−1 vs 0.012 min−1 in the absence of t-BuOH) but RhB can still be photodegraded. On the basis of these results, the photodegradation catalyzed by CdSe/TiO2 composites under visible light irradiation occurs mainly via O2•− radicals and to a lesser extend via •OH radicals. It is therefore likely that the heterojunction between CdSe and TiO2 favors the transfer of electrons in the CB of TiO2 followed by the production of O2•− radicals (Figure 11b).
Conclusion
In this study, CdSe NRs with an average length of ≈120 nm were successfully associated to TiO2 particles to form heterostructured CdSe/TiO2 photocatalysts. Due to their structure, the CdSe/TiO2 composites exhibit full visible and near-infrared light absorption and improved photocatalytic activity via charge separation between CdSe and TiO2 materials. The CdSe (2 wt %)/TiO2 composite was demonstrated to be the more efficient for the degradation of rhodamine B both under simulated sunlight or visible light irradiation. This facile approach for the preparation of a CdSe NRs/TiO2 photocatalyst, along with the high stability and low sensitivity to pH changes, demonstrates the high potential of this material for practical photocatalytic applications. The results described in this work stimulate the idea that CdSe/TiO2 composites may be of high potential for other applications, such as CdSe-sensitized TiO2 films for solar cells.
Supporting Information
| Supporting Information File 1: Additional figures. | ||
| Format: PDF | Size: 530.6 KB | Download |
References
-
Nakata, K.; Fujishima, A. J. Photochem. Photobiol., C: Photochem. Rev. 2012, 13, 169–189. doi:10.1016/j.jphotochemrev.2012.06.001
Return to citation in text: [1] -
Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D. W. Chem. Rev. 2014, 114, 9919–9986. doi:10.1021/cr5001892
Return to citation in text: [1] -
Kumar, S. G.; Den, L. G. J. Phys. Chem. A 2011, 115, 13211–13241. doi:10.1021/jp204364a
Return to citation in text: [1] [2] -
Huang, Q.; Tian, S.; Zeng, D.; Wang, X.; Song, W.; Li, Y.; Xiao, W.; Xie, C. ACS Catal. 2013, 3, 1477–1485. doi:10.1021/cs400080w
Return to citation in text: [1] [2] -
Chen, H.; Xie, Y.; Sun, X.; Lv, M.; Wu, F.; Zhang, L.; Li, L.; Xu, X. Dalton Trans. 2015, 44, 13030–13039. doi:10.1039/C5DT01757B
Return to citation in text: [1] [2] -
Wang, H.; Zhang, L.; Chen, Z.; Hu, J.; Li, S.; Wang, Z.; Liu, J.; Wang, X. Chem. Soc. Rev. 2014, 43, 5234–5244. doi:10.1039/C4CS00126E
Return to citation in text: [1] -
Labiadh, H.; Ben Chaabane, T.; Balan, L.; Becheik, N.; Corbel, S.; Medjahdi, G.; Schneider, R. Appl. Catal., B: Environ. 2014, 144, 29–35. doi:10.1016/j.apcatb.2013.07.004
Return to citation in text: [1] -
Donat, F.; Corbel, S.; Alem, H.; Pontvianne, S.; Balan, L.; Medjahdi, G.; Schneider, R. Beilstein J. Nanotechnol. 2017, 8, 1080–1093. doi:10.3762/bjnano.8.110
Return to citation in text: [1] -
Lo, S.-C.; Lin, C.-F.; Wu, C.-H.; Hsieh, P.-H. J. Hazard. Mater. 2004, 114, 183–190. doi:10.1016/j.jhazmat.2004.08.007
Return to citation in text: [1] [2] [3] -
Shen, Q.; Arae, D.; Toyoda, T. J. Photochem. Photobiol., A: Chem. 2004, 164, 75–80. doi:10.1016/j.jphotochem.2003.12.027
Return to citation in text: [1] [2] -
Liu, H.; Gao, L. J. Am. Ceram. Soc. 2005, 88, 1020–1022. doi:10.1111/j.1551-2916.2005.00196.x
Return to citation in text: [1] [2] [3] -
Ho, W.; Yu, J. C. J. Mol. Catal. A: Chem. 2006, 247, 268–274. doi:10.1016/j.molcata.2005.11.057
Return to citation in text: [1] [2] [3] -
Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2006, 128, 2385–2393. doi:10.1021/ja056494n
Return to citation in text: [1] [2] -
Lee, J.-C.; Kim, T. G.; Choi, H.-J.; Sung, Y.-M. Cryst. Growth Des. 2007, 7, 2588–2593. doi:10.1021/cg070588m
Return to citation in text: [1] [2] -
Robel, I.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2007, 129, 4136–4137. doi:10.1021/ja070099a
Return to citation in text: [1] [2] -
Shen, X.-C.; Zhang, Z.-L.; Zhou, B.; Peng, J.; Xie, M.; Zhang, M.; Pang, D.-W. Environ. Sci. Technol. 2008, 42, 5049–5054. doi:10.1021/es800668g
Return to citation in text: [1] [2] -
Kongkanand, A.; Tvrdy, K.; Takechi, K.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2008, 130, 4007–4015. doi:10.1021/ja0782706
Return to citation in text: [1] [2] -
López-Luke, T.; Wolcott, A.; Xu, L.-p.; Chen, S.; Wen, Z.; Li, J.; De La Rosa, E.; Zhang, J. Z. J. Phys. Chem. C 2008, 112, 1282–1292. doi:10.1021/jp077345p
Return to citation in text: [1] [2] -
Lee, H.; Wang, M.; Chen, P.; Gamelin, D. R.; Zakeeruddin, S. M.; Grätzel, M.; Nazeeruddin, M. K. Nano Lett. 2009, 9, 4221–4227. doi:10.1021/nl902438d
Return to citation in text: [1] [2] -
Zhang, H.; Quan, X.; Chen, S.; Yu, H.; Ma, N. Chem. Mater. 2009, 21, 3090–3095. doi:10.1021/cm900100k
Return to citation in text: [1] [2] -
Hensel, J.; Wang, G.; Li, Y.; Zhang, J. Z. Nano Lett. 2010, 10, 478–483. doi:10.1021/nl903217w
Return to citation in text: [1] [2] -
Yang, L.; Luo, S.; Liu, R.; Cai, Q.; Xiao, Y.; Liu, S.; Su, F.; Wen, L. J. Phys. Chem. C 2010, 114, 4783–4789. doi:10.1021/jp910489h
Return to citation in text: [1] [2] [3] -
Wang, C.; Thompson, R. L.; Baltrus, J.; Matranga, C. J. Phys. Chem. Lett. 2010, 1, 48–53. doi:10.1021/jz9000032
Return to citation in text: [1] [2] -
Chi, C.-F.; Liau, S.-Y.; Lee, Y.-L. Nanotechnology 2010, 21, 025202. doi:10.1088/0957-4484/21/2/025202
Return to citation in text: [1] [2] -
Xie, Y.; Ali, G.; Yoo, S. H.; Cho, S. O. ACS Appl. Mater. Interfaces 2010, 2, 2910–2914. doi:10.1021/am100605a
Return to citation in text: [1] [2] -
Lim, C.-S.; Chen, M.-L.; Oh, W.-C. Bull. Korean Chem. Soc. 2011, 32, 1657–1661. doi:10.5012/bkcs.2011.32.5.1657
Return to citation in text: [1] [2] [3] -
Liu, L.; Wang, G.; Li, Y.; Li, Y.; Zhang, J. Z. Nano Res. 2011, 4, 249–258. doi:10.1007/s12274-010-0076-7
Return to citation in text: [1] [2] -
Cheng, S.; Fu, W.; Yang, H.; Zhang, L.; Ma, J.; Zhao, H.; Sun, M.; Yang, L. J. Phys. Chem. C 2012, 116, 2615–2621. doi:10.1021/jp209258r
Return to citation in text: [1] [2] -
Meng, Z.-D.; Zhu, L.; Ye, S.; Sun, Q.; Ullah, K.; Cho, K.-Y.; Oh, W.-C. Nanoscale Res. Lett. 2013, 8, 189. doi:10.1186/1556-276X-8-189
Return to citation in text: [1] [2] -
Lee, S.; Lee, K.; Kim, W. D.; Lee, S.; Shin, D. J.; Lee, D. C. J. Phys. Chem. C 2014, 118, 23627–23634. doi:10.1021/jp508315m
Return to citation in text: [1] [2] -
Wang, P.; Li, D.; Chen, J.; Zhang, X.; Xian, J.; Yang, X.; Zheng, X.; Li, X.; Shao, Y. Appl. Catal., B: Environ. 2014, 160–161, 217–226. doi:10.1016/j.apcatb.2014.05.032
Return to citation in text: [1] [2] [3] -
Wang, H.; Zhu, W.; Chong, B.; Qin, K. Int. J. Hydrogen Energy 2014, 39, 90–99. doi:10.1016/j.ijhydene.2013.10.048
Return to citation in text: [1] [2] -
Zhang, M.; Xu, Y.; Lv, J.; Yang, L.; Jiang, X.; He, G.; Song, X.; Sun, Z. Nanoscale Res. Lett. 2014, 9, 636. doi:10.1186/1556-276X-9-636
Return to citation in text: [1] [2] [3] -
Lv, J.; Wang, H.; Gao, H.; Xu, G.; Wang, D.; Chen, Z.; Zhang, X.; Zheng, Z.; Wu, Y. Surf. Coat. Technol. 2015, 261, 356–363. doi:10.1016/j.surfcoat.2014.10.066
Return to citation in text: [1] [2] [3] -
Zhou, R.; Niu, H.; Zhang, Q.; Uchaker, E.; Guo, Z.; Wan, L.; Miao, S.; Xu, J.; Cao, G. J. Mater. Chem. A 2015, 3, 12539–12549. doi:10.1039/C5TA01461A
Return to citation in text: [1] [2] -
Wang, P.; Li, X.; Fang, J.; Li, D.; Chen, J.; Zhang, X.; Shao, Y.; He, Y. Appl. Catal., B: Environ. 2016, 181, 838–847. doi:10.1016/j.apcatb.2015.08.046
Return to citation in text: [1] [2] -
Zhang, G.; Zhou, Y.; Fan, X.; Zou, J.; Dong, W.; Xu, X. Int. J. Hydrogen Energy 2017, 42, 19877–19884. doi:10.1016/j.ijhydene.2017.06.153
Return to citation in text: [1] [2] -
Huynh, W. U.; Dittmer, J. J.; Alivisatos, A. P. Science 2002, 295, 2425–2457. doi:10.1126/science.1069156
Return to citation in text: [1] -
Luo, J.; Ma, L.; He, T.; Ng, C. F.; Wang, S.; Sun, H.; Fan, H. J. J. Phys. Chem. C 2012, 116, 11956–11963. doi:10.1021/jp3031754
Return to citation in text: [1] -
Esparza, D.; Zarazùa, I.; López-Luke, T.; Cerdán-Pasarán, A.; Sánchez-Solis, A.; Torres-Castro, A.; Mora-Sero, I.; De La Rosa, E. J. Phys. Chem. C 2015, 119, 13394–13403. doi:10.1021/acs.jpcc.5b01525
Return to citation in text: [1] -
Lee, S.; Flanagan, J. C.; Kang, J.; Kim, J.; Shim, M.; Park, B. Sci. Rep. 2015, 5, 17472. doi:10.1038/srep17472
Return to citation in text: [1] -
Hassan, Y.; Chuang, C.-H.; Kobayashi, Y.; Coombs, N.; Gorantla, S.; Botton, G. A.; Winnik, M. A.; Burda, C.; Scholes, G. D. J. Phys. Chem. C 2014, 118, 3347–3358. doi:10.1021/jp411830u
Return to citation in text: [1] -
Li, Y.; Hu, Y.; Peng, S.; Lu, G.; Li, S. J. Phys. Chem. C 2009, 113, 9352–9358. doi:10.1021/jp901505j
Return to citation in text: [1] [2] -
Mills, A.; Davies, R. H.; Worksky, D. Chem. Soc. Rev. 1993, 22, 417–423. doi:10.1039/cs9932200417
Return to citation in text: [1] -
Dzhagan, V. M.; Valakh, M. Ya.; Raevskaya, A. E.; Stroyuk, A. L.; Kuchmiy, S. Ya.; Zahn, D. R. T. Nanotechnology 2007, 18, 285701. doi:10.1088/0957-4484/18/28/285701
Return to citation in text: [1] -
Tian, F.; Zhang, Y.; Zhang, J.; Pan, C. J. Phys. Chem. C 2012, 116, 7515–7519. doi:10.1021/jp301256h
Return to citation in text: [1] -
Mastai, Y.; Polsky, R.; Koltypin, Yu.; Gedanken, A.; Hodes, G. J. Am. Chem. Soc. 1999, 121, 10047–10052. doi:10.1021/ja9908772
Return to citation in text: [1] -
Nawawi, W. I.; Nawi, M. A. J. Mol. Catal. A: Chem. 2013, 374–375, 39–45. doi:10.1016/j.molcata.2013.03.024
Return to citation in text: [1] -
Aldeek, F.; Mustin, C.; Balan, L.; Medjahdi, G.; Roques-Carmes, T.; Arnoux, P.; Schneider, R. Eur. J. Inorg. Chem. 2011, 794–801. doi:10.1002/ejic.201000790
Return to citation in text: [1] -
Vargas-Hernández, C.; Lara, V. C.; Vallejo, J. E.; Jurado, J. F.; Giraldo, O. Phys. Status Solidi A 2005, 242, 1897–1901. doi:10.1002/pssb.200461717
Return to citation in text: [1] -
Wagner, C. D.; Gale, L. H.; Raymond, R. H. Anal. Chem. 1979, 51, 466–482. doi:10.1021/ac50040a005
Return to citation in text: [1] -
Fu, H.; Pan, C.; Yao, W.; Zhu, Y. J. Phys. Chem. B 2005, 109, 22432–22439. doi:10.1021/jp052995j
Return to citation in text: [1] -
Zhao, J.; Hidaka, H.; Takamura, A.; Pelizzetti, E.; Serpone, N. Langmuir 1993, 3, 1646–1650. doi:10.1021/la00031a008
Return to citation in text: [1]
| 50. | Vargas-Hernández, C.; Lara, V. C.; Vallejo, J. E.; Jurado, J. F.; Giraldo, O. Phys. Status Solidi A 2005, 242, 1897–1901. doi:10.1002/pssb.200461717 |
| 48. | Nawawi, W. I.; Nawi, M. A. J. Mol. Catal. A: Chem. 2013, 374–375, 39–45. doi:10.1016/j.molcata.2013.03.024 |
| 49. | Aldeek, F.; Mustin, C.; Balan, L.; Medjahdi, G.; Roques-Carmes, T.; Arnoux, P.; Schneider, R. Eur. J. Inorg. Chem. 2011, 794–801. doi:10.1002/ejic.201000790 |
| 1. | Nakata, K.; Fujishima, A. J. Photochem. Photobiol., C: Photochem. Rev. 2012, 13, 169–189. doi:10.1016/j.jphotochemrev.2012.06.001 |
| 2. | Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D. W. Chem. Rev. 2014, 114, 9919–9986. doi:10.1021/cr5001892 |
| 9. | Lo, S.-C.; Lin, C.-F.; Wu, C.-H.; Hsieh, P.-H. J. Hazard. Mater. 2004, 114, 183–190. doi:10.1016/j.jhazmat.2004.08.007 |
| 10. | Shen, Q.; Arae, D.; Toyoda, T. J. Photochem. Photobiol., A: Chem. 2004, 164, 75–80. doi:10.1016/j.jphotochem.2003.12.027 |
| 11. | Liu, H.; Gao, L. J. Am. Ceram. Soc. 2005, 88, 1020–1022. doi:10.1111/j.1551-2916.2005.00196.x |
| 12. | Ho, W.; Yu, J. C. J. Mol. Catal. A: Chem. 2006, 247, 268–274. doi:10.1016/j.molcata.2005.11.057 |
| 13. | Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2006, 128, 2385–2393. doi:10.1021/ja056494n |
| 14. | Lee, J.-C.; Kim, T. G.; Choi, H.-J.; Sung, Y.-M. Cryst. Growth Des. 2007, 7, 2588–2593. doi:10.1021/cg070588m |
| 15. | Robel, I.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2007, 129, 4136–4137. doi:10.1021/ja070099a |
| 16. | Shen, X.-C.; Zhang, Z.-L.; Zhou, B.; Peng, J.; Xie, M.; Zhang, M.; Pang, D.-W. Environ. Sci. Technol. 2008, 42, 5049–5054. doi:10.1021/es800668g |
| 17. | Kongkanand, A.; Tvrdy, K.; Takechi, K.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2008, 130, 4007–4015. doi:10.1021/ja0782706 |
| 18. | López-Luke, T.; Wolcott, A.; Xu, L.-p.; Chen, S.; Wen, Z.; Li, J.; De La Rosa, E.; Zhang, J. Z. J. Phys. Chem. C 2008, 112, 1282–1292. doi:10.1021/jp077345p |
| 19. | Lee, H.; Wang, M.; Chen, P.; Gamelin, D. R.; Zakeeruddin, S. M.; Grätzel, M.; Nazeeruddin, M. K. Nano Lett. 2009, 9, 4221–4227. doi:10.1021/nl902438d |
| 20. | Zhang, H.; Quan, X.; Chen, S.; Yu, H.; Ma, N. Chem. Mater. 2009, 21, 3090–3095. doi:10.1021/cm900100k |
| 21. | Hensel, J.; Wang, G.; Li, Y.; Zhang, J. Z. Nano Lett. 2010, 10, 478–483. doi:10.1021/nl903217w |
| 22. | Yang, L.; Luo, S.; Liu, R.; Cai, Q.; Xiao, Y.; Liu, S.; Su, F.; Wen, L. J. Phys. Chem. C 2010, 114, 4783–4789. doi:10.1021/jp910489h |
| 23. | Wang, C.; Thompson, R. L.; Baltrus, J.; Matranga, C. J. Phys. Chem. Lett. 2010, 1, 48–53. doi:10.1021/jz9000032 |
| 24. | Chi, C.-F.; Liau, S.-Y.; Lee, Y.-L. Nanotechnology 2010, 21, 025202. doi:10.1088/0957-4484/21/2/025202 |
| 25. | Xie, Y.; Ali, G.; Yoo, S. H.; Cho, S. O. ACS Appl. Mater. Interfaces 2010, 2, 2910–2914. doi:10.1021/am100605a |
| 26. | Lim, C.-S.; Chen, M.-L.; Oh, W.-C. Bull. Korean Chem. Soc. 2011, 32, 1657–1661. doi:10.5012/bkcs.2011.32.5.1657 |
| 27. | Liu, L.; Wang, G.; Li, Y.; Li, Y.; Zhang, J. Z. Nano Res. 2011, 4, 249–258. doi:10.1007/s12274-010-0076-7 |
| 28. | Cheng, S.; Fu, W.; Yang, H.; Zhang, L.; Ma, J.; Zhao, H.; Sun, M.; Yang, L. J. Phys. Chem. C 2012, 116, 2615–2621. doi:10.1021/jp209258r |
| 29. | Meng, Z.-D.; Zhu, L.; Ye, S.; Sun, Q.; Ullah, K.; Cho, K.-Y.; Oh, W.-C. Nanoscale Res. Lett. 2013, 8, 189. doi:10.1186/1556-276X-8-189 |
| 30. | Lee, S.; Lee, K.; Kim, W. D.; Lee, S.; Shin, D. J.; Lee, D. C. J. Phys. Chem. C 2014, 118, 23627–23634. doi:10.1021/jp508315m |
| 31. | Wang, P.; Li, D.; Chen, J.; Zhang, X.; Xian, J.; Yang, X.; Zheng, X.; Li, X.; Shao, Y. Appl. Catal., B: Environ. 2014, 160–161, 217–226. doi:10.1016/j.apcatb.2014.05.032 |
| 32. | Wang, H.; Zhu, W.; Chong, B.; Qin, K. Int. J. Hydrogen Energy 2014, 39, 90–99. doi:10.1016/j.ijhydene.2013.10.048 |
| 33. | Zhang, M.; Xu, Y.; Lv, J.; Yang, L.; Jiang, X.; He, G.; Song, X.; Sun, Z. Nanoscale Res. Lett. 2014, 9, 636. doi:10.1186/1556-276X-9-636 |
| 34. | Lv, J.; Wang, H.; Gao, H.; Xu, G.; Wang, D.; Chen, Z.; Zhang, X.; Zheng, Z.; Wu, Y. Surf. Coat. Technol. 2015, 261, 356–363. doi:10.1016/j.surfcoat.2014.10.066 |
| 35. | Zhou, R.; Niu, H.; Zhang, Q.; Uchaker, E.; Guo, Z.; Wan, L.; Miao, S.; Xu, J.; Cao, G. J. Mater. Chem. A 2015, 3, 12539–12549. doi:10.1039/C5TA01461A |
| 36. | Wang, P.; Li, X.; Fang, J.; Li, D.; Chen, J.; Zhang, X.; Shao, Y.; He, Y. Appl. Catal., B: Environ. 2016, 181, 838–847. doi:10.1016/j.apcatb.2015.08.046 |
| 37. | Zhang, G.; Zhou, Y.; Fan, X.; Zou, J.; Dong, W.; Xu, X. Int. J. Hydrogen Energy 2017, 42, 19877–19884. doi:10.1016/j.ijhydene.2017.06.153 |
| 46. | Tian, F.; Zhang, Y.; Zhang, J.; Pan, C. J. Phys. Chem. C 2012, 116, 7515–7519. doi:10.1021/jp301256h |
| 9. | Lo, S.-C.; Lin, C.-F.; Wu, C.-H.; Hsieh, P.-H. J. Hazard. Mater. 2004, 114, 183–190. doi:10.1016/j.jhazmat.2004.08.007 |
| 10. | Shen, Q.; Arae, D.; Toyoda, T. J. Photochem. Photobiol., A: Chem. 2004, 164, 75–80. doi:10.1016/j.jphotochem.2003.12.027 |
| 11. | Liu, H.; Gao, L. J. Am. Ceram. Soc. 2005, 88, 1020–1022. doi:10.1111/j.1551-2916.2005.00196.x |
| 12. | Ho, W.; Yu, J. C. J. Mol. Catal. A: Chem. 2006, 247, 268–274. doi:10.1016/j.molcata.2005.11.057 |
| 13. | Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2006, 128, 2385–2393. doi:10.1021/ja056494n |
| 14. | Lee, J.-C.; Kim, T. G.; Choi, H.-J.; Sung, Y.-M. Cryst. Growth Des. 2007, 7, 2588–2593. doi:10.1021/cg070588m |
| 15. | Robel, I.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2007, 129, 4136–4137. doi:10.1021/ja070099a |
| 16. | Shen, X.-C.; Zhang, Z.-L.; Zhou, B.; Peng, J.; Xie, M.; Zhang, M.; Pang, D.-W. Environ. Sci. Technol. 2008, 42, 5049–5054. doi:10.1021/es800668g |
| 17. | Kongkanand, A.; Tvrdy, K.; Takechi, K.; Kuno, M.; Kamat, P. V. J. Am. Chem. Soc. 2008, 130, 4007–4015. doi:10.1021/ja0782706 |
| 18. | López-Luke, T.; Wolcott, A.; Xu, L.-p.; Chen, S.; Wen, Z.; Li, J.; De La Rosa, E.; Zhang, J. Z. J. Phys. Chem. C 2008, 112, 1282–1292. doi:10.1021/jp077345p |
| 19. | Lee, H.; Wang, M.; Chen, P.; Gamelin, D. R.; Zakeeruddin, S. M.; Grätzel, M.; Nazeeruddin, M. K. Nano Lett. 2009, 9, 4221–4227. doi:10.1021/nl902438d |
| 20. | Zhang, H.; Quan, X.; Chen, S.; Yu, H.; Ma, N. Chem. Mater. 2009, 21, 3090–3095. doi:10.1021/cm900100k |
| 21. | Hensel, J.; Wang, G.; Li, Y.; Zhang, J. Z. Nano Lett. 2010, 10, 478–483. doi:10.1021/nl903217w |
| 22. | Yang, L.; Luo, S.; Liu, R.; Cai, Q.; Xiao, Y.; Liu, S.; Su, F.; Wen, L. J. Phys. Chem. C 2010, 114, 4783–4789. doi:10.1021/jp910489h |
| 23. | Wang, C.; Thompson, R. L.; Baltrus, J.; Matranga, C. J. Phys. Chem. Lett. 2010, 1, 48–53. doi:10.1021/jz9000032 |
| 24. | Chi, C.-F.; Liau, S.-Y.; Lee, Y.-L. Nanotechnology 2010, 21, 025202. doi:10.1088/0957-4484/21/2/025202 |
| 25. | Xie, Y.; Ali, G.; Yoo, S. H.; Cho, S. O. ACS Appl. Mater. Interfaces 2010, 2, 2910–2914. doi:10.1021/am100605a |
| 26. | Lim, C.-S.; Chen, M.-L.; Oh, W.-C. Bull. Korean Chem. Soc. 2011, 32, 1657–1661. doi:10.5012/bkcs.2011.32.5.1657 |
| 27. | Liu, L.; Wang, G.; Li, Y.; Li, Y.; Zhang, J. Z. Nano Res. 2011, 4, 249–258. doi:10.1007/s12274-010-0076-7 |
| 28. | Cheng, S.; Fu, W.; Yang, H.; Zhang, L.; Ma, J.; Zhao, H.; Sun, M.; Yang, L. J. Phys. Chem. C 2012, 116, 2615–2621. doi:10.1021/jp209258r |
| 29. | Meng, Z.-D.; Zhu, L.; Ye, S.; Sun, Q.; Ullah, K.; Cho, K.-Y.; Oh, W.-C. Nanoscale Res. Lett. 2013, 8, 189. doi:10.1186/1556-276X-8-189 |
| 30. | Lee, S.; Lee, K.; Kim, W. D.; Lee, S.; Shin, D. J.; Lee, D. C. J. Phys. Chem. C 2014, 118, 23627–23634. doi:10.1021/jp508315m |
| 31. | Wang, P.; Li, D.; Chen, J.; Zhang, X.; Xian, J.; Yang, X.; Zheng, X.; Li, X.; Shao, Y. Appl. Catal., B: Environ. 2014, 160–161, 217–226. doi:10.1016/j.apcatb.2014.05.032 |
| 32. | Wang, H.; Zhu, W.; Chong, B.; Qin, K. Int. J. Hydrogen Energy 2014, 39, 90–99. doi:10.1016/j.ijhydene.2013.10.048 |
| 33. | Zhang, M.; Xu, Y.; Lv, J.; Yang, L.; Jiang, X.; He, G.; Song, X.; Sun, Z. Nanoscale Res. Lett. 2014, 9, 636. doi:10.1186/1556-276X-9-636 |
| 34. | Lv, J.; Wang, H.; Gao, H.; Xu, G.; Wang, D.; Chen, Z.; Zhang, X.; Zheng, Z.; Wu, Y. Surf. Coat. Technol. 2015, 261, 356–363. doi:10.1016/j.surfcoat.2014.10.066 |
| 35. | Zhou, R.; Niu, H.; Zhang, Q.; Uchaker, E.; Guo, Z.; Wan, L.; Miao, S.; Xu, J.; Cao, G. J. Mater. Chem. A 2015, 3, 12539–12549. doi:10.1039/C5TA01461A |
| 36. | Wang, P.; Li, X.; Fang, J.; Li, D.; Chen, J.; Zhang, X.; Shao, Y.; He, Y. Appl. Catal., B: Environ. 2016, 181, 838–847. doi:10.1016/j.apcatb.2015.08.046 |
| 37. | Zhang, G.; Zhou, Y.; Fan, X.; Zou, J.; Dong, W.; Xu, X. Int. J. Hydrogen Energy 2017, 42, 19877–19884. doi:10.1016/j.ijhydene.2017.06.153 |
| 47. | Mastai, Y.; Polsky, R.; Koltypin, Yu.; Gedanken, A.; Hodes, G. J. Am. Chem. Soc. 1999, 121, 10047–10052. doi:10.1021/ja9908772 |
| 3. | Kumar, S. G.; Den, L. G. J. Phys. Chem. A 2011, 115, 13211–13241. doi:10.1021/jp204364a |
| 4. | Huang, Q.; Tian, S.; Zeng, D.; Wang, X.; Song, W.; Li, Y.; Xiao, W.; Xie, C. ACS Catal. 2013, 3, 1477–1485. doi:10.1021/cs400080w |
| 5. | Chen, H.; Xie, Y.; Sun, X.; Lv, M.; Wu, F.; Zhang, L.; Li, L.; Xu, X. Dalton Trans. 2015, 44, 13030–13039. doi:10.1039/C5DT01757B |
| 6. | Wang, H.; Zhang, L.; Chen, Z.; Hu, J.; Li, S.; Wang, Z.; Liu, J.; Wang, X. Chem. Soc. Rev. 2014, 43, 5234–5244. doi:10.1039/C4CS00126E |
| 7. | Labiadh, H.; Ben Chaabane, T.; Balan, L.; Becheik, N.; Corbel, S.; Medjahdi, G.; Schneider, R. Appl. Catal., B: Environ. 2014, 144, 29–35. doi:10.1016/j.apcatb.2013.07.004 |
| 8. | Donat, F.; Corbel, S.; Alem, H.; Pontvianne, S.; Balan, L.; Medjahdi, G.; Schneider, R. Beilstein J. Nanotechnol. 2017, 8, 1080–1093. doi:10.3762/bjnano.8.110 |
| 44. | Mills, A.; Davies, R. H.; Worksky, D. Chem. Soc. Rev. 1993, 22, 417–423. doi:10.1039/cs9932200417 |
| 3. | Kumar, S. G.; Den, L. G. J. Phys. Chem. A 2011, 115, 13211–13241. doi:10.1021/jp204364a |
| 4. | Huang, Q.; Tian, S.; Zeng, D.; Wang, X.; Song, W.; Li, Y.; Xiao, W.; Xie, C. ACS Catal. 2013, 3, 1477–1485. doi:10.1021/cs400080w |
| 5. | Chen, H.; Xie, Y.; Sun, X.; Lv, M.; Wu, F.; Zhang, L.; Li, L.; Xu, X. Dalton Trans. 2015, 44, 13030–13039. doi:10.1039/C5DT01757B |
| 45. | Dzhagan, V. M.; Valakh, M. Ya.; Raevskaya, A. E.; Stroyuk, A. L.; Kuchmiy, S. Ya.; Zahn, D. R. T. Nanotechnology 2007, 18, 285701. doi:10.1088/0957-4484/18/28/285701 |
| 41. | Lee, S.; Flanagan, J. C.; Kang, J.; Kim, J.; Shim, M.; Park, B. Sci. Rep. 2015, 5, 17472. doi:10.1038/srep17472 |
| 43. | Li, Y.; Hu, Y.; Peng, S.; Lu, G.; Li, S. J. Phys. Chem. C 2009, 113, 9352–9358. doi:10.1021/jp901505j |
| 53. | Zhao, J.; Hidaka, H.; Takamura, A.; Pelizzetti, E.; Serpone, N. Langmuir 1993, 3, 1646–1650. doi:10.1021/la00031a008 |
| 40. | Esparza, D.; Zarazùa, I.; López-Luke, T.; Cerdán-Pasarán, A.; Sánchez-Solis, A.; Torres-Castro, A.; Mora-Sero, I.; De La Rosa, E. J. Phys. Chem. C 2015, 119, 13394–13403. doi:10.1021/acs.jpcc.5b01525 |
| 43. | Li, Y.; Hu, Y.; Peng, S.; Lu, G.; Li, S. J. Phys. Chem. C 2009, 113, 9352–9358. doi:10.1021/jp901505j |
| 9. | Lo, S.-C.; Lin, C.-F.; Wu, C.-H.; Hsieh, P.-H. J. Hazard. Mater. 2004, 114, 183–190. doi:10.1016/j.jhazmat.2004.08.007 |
| 11. | Liu, H.; Gao, L. J. Am. Ceram. Soc. 2005, 88, 1020–1022. doi:10.1111/j.1551-2916.2005.00196.x |
| 12. | Ho, W.; Yu, J. C. J. Mol. Catal. A: Chem. 2006, 247, 268–274. doi:10.1016/j.molcata.2005.11.057 |
| 22. | Yang, L.; Luo, S.; Liu, R.; Cai, Q.; Xiao, Y.; Liu, S.; Su, F.; Wen, L. J. Phys. Chem. C 2010, 114, 4783–4789. doi:10.1021/jp910489h |
| 26. | Lim, C.-S.; Chen, M.-L.; Oh, W.-C. Bull. Korean Chem. Soc. 2011, 32, 1657–1661. doi:10.5012/bkcs.2011.32.5.1657 |
| 31. | Wang, P.; Li, D.; Chen, J.; Zhang, X.; Xian, J.; Yang, X.; Zheng, X.; Li, X.; Shao, Y. Appl. Catal., B: Environ. 2014, 160–161, 217–226. doi:10.1016/j.apcatb.2014.05.032 |
| 33. | Zhang, M.; Xu, Y.; Lv, J.; Yang, L.; Jiang, X.; He, G.; Song, X.; Sun, Z. Nanoscale Res. Lett. 2014, 9, 636. doi:10.1186/1556-276X-9-636 |
| 34. | Lv, J.; Wang, H.; Gao, H.; Xu, G.; Wang, D.; Chen, Z.; Zhang, X.; Zheng, Z.; Wu, Y. Surf. Coat. Technol. 2015, 261, 356–363. doi:10.1016/j.surfcoat.2014.10.066 |
| 39. | Luo, J.; Ma, L.; He, T.; Ng, C. F.; Wang, S.; Sun, H.; Fan, H. J. J. Phys. Chem. C 2012, 116, 11956–11963. doi:10.1021/jp3031754 |
| 51. | Wagner, C. D.; Gale, L. H.; Raymond, R. H. Anal. Chem. 1979, 51, 466–482. doi:10.1021/ac50040a005 |
| 38. | Huynh, W. U.; Dittmer, J. J.; Alivisatos, A. P. Science 2002, 295, 2425–2457. doi:10.1126/science.1069156 |
| 42. | Hassan, Y.; Chuang, C.-H.; Kobayashi, Y.; Coombs, N.; Gorantla, S.; Botton, G. A.; Winnik, M. A.; Burda, C.; Scholes, G. D. J. Phys. Chem. C 2014, 118, 3347–3358. doi:10.1021/jp411830u |
| 52. | Fu, H.; Pan, C.; Yao, W.; Zhu, Y. J. Phys. Chem. B 2005, 109, 22432–22439. doi:10.1021/jp052995j |
© 2017 Laatar et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)