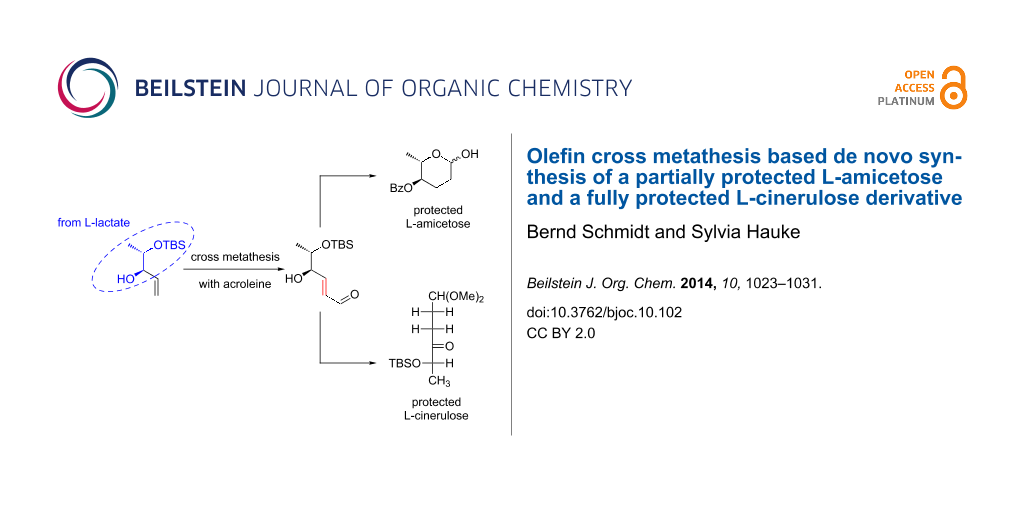
Abstract
Cross metathesis of a lactate derived allylic alcohol and acrolein is the entry point to a de novo synthesis of 4-benzoate protected L-amicetose and a cinerulose derivative protected at C5 and C1.

Graphical Abstract
Introduction
Many drugs and bioactive natural products are glycoconjugates, which contain an aglycon part linked through glycosidic bonds to one or more oligosaccharide side chains [1]. While it was assumed for quite some time that the carbohydrate side chain merely influences the pharmacokinetics, more recent investigations led to the conclusion that the oligosaccharide moiety contributes essentially to the mechanisms of action, e.g., through molecular recognition of a preferred binding site [2-5], thereby ensuring the selectivity of a chemotherapeutic agent. Particularly common are side chains composed of deoxygenated sugars [6]. For example, the kigamicins are bacterial secondary metabolites isolated from Amicolatopsis sp. [7,8] and display both antibiotic and cytotoxic activity [9]. They have a polycyclic xanthone aglycone in common, which is glycosylated at the C14–OH group. In Figure 1 the structure of kigamicin B, which carries a D-amicetose disaccharide unit, is shown as a representative example. Another antitumor antibiotic is aclacinomycin A, which has been used clinically under the name aclarubicin. It is an anthracycline [10] bearing a trisaccharide side chain consisting of L-rhodosamin, 2,6-didesoxy-L-lyxose, and L-cinerulose attached to the aglycon aclavinon [11,12]. It was found to be a potent antineoplastic agent with particular activity against different forms of leukemia [13] (Figure 1).
![[1860-5397-10-102-1]](/bjoc/content/figures/1860-5397-10-102-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of kigamicin B and aclacinomycin A as representative examples for antineoplastic glycoconjugates.
Figure 1: Structures of kigamicin B and aclacinomycin A as representative examples for antineoplastic glycoco...
Desoxy-sugars are sometimes scarcely available from natural sources and therefore the chemical syntheses of the monosaccharides [6,14] and their assembly to oligosaccharides [15-17] has attracted constant attention for many years. For example, D-amicetose has been obtained from other carbohydrates through ethanethiolysis and subsequent Ni-catalyzed desulfurization [18,19], by reduction of the corresponding aldonolactone [20], or by conversion of a protected mannopyranoside into the 2,3-unsaturated enopyranoside and subsequent hydrogenation and reductive 6-deoxygenation [21,22]. Successful de novo approaches [14] to both enantiomers of amicetose include an Achmatowicz rearrangement–hydrogenation sequence, starting from enantiomerically pure 2-(1-hydroxyethyl)furan [23-25], a sequence comprising perdeuteration of an alkynoate derived from L-threonine (giving a 2,2,3,3-tetradeuterated amicetose) [26], enantioselective hydroboration of hetero Diels–Alder adducts [27], stereoconservative diazotation of L-glutamic acid [28,29], diastereoselective addition of methylmagnesium bromide to enantiopure 2-benzyloxyhex-5-enal [30], oxidative degradation of aromatics and subsequent lactonization [31], and a two-carbon homologation of an enantiopure C4-building block with a metallated sulfone [32]. A combinatorial biosynthetic approach to D- and L-amicetose has also recently been established by combining different sugar biosynthesis genes [5].
Comparatively few transition metal mediated or -catalyzed reactions have hitherto been used for the synthesis of 2,3,6-tridesoxy sugars such as amicetose. An example is the W-promoted cycloisomerization of lactaldehyde derived alkynols, which yields the glycal of amicetose or its epimer rhodinose, respectively [33]. An approach to amicetose (and a few other 6-desoxy sugars) involves the formation of an enantiopure β,γ-unsaturated δ-valerolactone via ring closing metathesis (RCM). The RCM product is subsequently converted into L-amicetose in four steps [34]. Unfortunately, the steps determining the configuration at C4 proceed in both cases with virtually no diastereoselectivity, although the resulting epimers were conveniently separated by chromatography. We have recently established two different metathesis based routes to L-amicetose [35] or L-amicetal [36], respectively, which both use enantiomerically pure L-ethyl lactate (1) as the starting material (Scheme 1). The synthetic routes rely on the highly diastereoselective two-step conversion of ethyl lactate to allylic alcohol 2 [36,37] and further to the RCM precursor 3, which then undergoes RCM-isomerization to L-amicetal 4 [36]. Alternatively, we have used 2 as a substrate for a cross metathesis reaction with methyl acrylate under isomerization-free conditions to furnish enoate 5, which underwent cyclization to the lactone 6 after hydrogenation. Reduction of 6 with DIBAL-H eventually yields a protected L-amicetose 7 [35]. To avoid the last step of the sequence, we envisaged a cross metathesis with acroleine, rather than methyl acrylate, to give enal 8 which should undergo spontaneous lactol formation after hydrogenation and desilylation. However, compared to cross metathesis reactions with methyl acrylate literature precedence for the use of acroleins as CM partner is significantly smaller [38-44].
![[1860-5397-10-102-i1]](/bjoc/content/inline/1860-5397-10-102-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: RCM-isomerization approach to L-amicetal 4 and alternative CM approaches to L-amicetose.
Scheme 1: RCM-isomerization approach to L-amicetal 4 and alternative CM approaches to L-amicetose.
In this contribution we report a de novo synthesis of a protected L-amicetose, using a cross metathesis reaction of allyl alcohol 2 with acrolein, and the elaboration of the cross metathesis product 8 to a fully protected acyclic cinerulose derivative. We are aware of only one previous de novo synthesis of DL-cinerulose, which used an Achmatowicz rearrangement of 2-(1-hydroxyethyl)furan [45].
Results and Discussion
The three second generation catalysts A [46], B [47,48] and C [49,50] were initially tested for the cross metathesis of 2 and acrolein (Table 1). We started with the most common catalyst, the second generation Grubbs’ catalyst. In order to suppress undesired double bond isomerization reactions [51,52] we tried to avoid high reaction temperatures, which may cause catalyst decomposition to isomerization active species. This phenomenon has often been observed for second generation catalysts [53]. For these reasons, phenol was used as a rate accelerating additive. Phenols are believed to coordinate to the Ru and stabilize the catalytically active 14-electron species, resulting in a retarded catalyst decomposition [54,55]. In the presence of 5 mol % of A and 0.5 equiv of phenol, however, the yield of cross metathesis product 8 was very low at ambient temperature (Table 1, entry 1). Heating the mixture to reflux in dichloromethane resulted in a comparably low yield (Table 1, entry 2), whereas a significant improvement could be observed in toluene at 80 °C (Table 1, entry 3). We thought that the yield could not be further increased with catalyst A and therefore decided to test the phosphine free catalyst B. This and related catalysts [56], comprising a hemilabile ortho-isopropoxy substituted alkylidene ligand, are particularly suited for cross metathesis reactions [57,58]. Indeed, even at ambient temperature and without any rate accelerating additive, a good yield of 86% was obtained (Table 1, entry 4), which could be improved to quantitative by increasing the reaction temperature to 40 °C (Table 1, entry 5). Reducing the catalyst loading to 2.5 mol % is possible, but at the expense of isolated yield of product (Table 1, entry 6). We have previously used the Umicore M51 catalyst (C) [49] for cross metathesis reactions and discovered that high selectivities and rates of conversion can be accomplished even with comparatively low catalyst loadings [59,60]. For the cross metathesis of 2 and acroleine, C gave significantly better yields than A, but was found to be inferior to B (Table 1, entries 7–9).
Table 1: Catalyst screening for CM of allyl alcohol 2 and acrolein.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-102-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | catalyst | cat. loading | additive (equiv) | solvent | T/°C | yield of 8 |
|---|---|---|---|---|---|---|
| 1a | A | 5 mol % | phenol (0.5) | CH2Cl2 | 25 °C | 25% |
| 2a | A | 5 mol % | phenol (0.5) | CH2Cl2 | 40 °C | 20% |
| 3 | A | 5 mol % | phenol (0.5) | toluene | 80 °C | 68% |
| 4 | B | 5 mol % | – | CH2Cl2 | 25 °C | 86% |
| 5 | B | 5 mol % | – | CH2Cl2 | 40 °C | 99% |
| 6 | B | 2.5 mol % | – | CH2Cl2 | 40 °C | 76% |
| 7 | C | 5 mol % | – | CH2Cl2 | 40 °C | 65% |
| 8 | C | 5 mol % | – | toluene | 80 °C | 80% |
| 9 | C | 2.5 mol % | – | toluene | 80 °C | 65% |
aAllylic alcohol 2 was recovered in 21% yield (entry 1) and in 12% yield (entry 2), respectively.
In the following step we decided to protect the hydroxy group at C4 first to avoid any complications arising from the formation of a furanose after hydrogenation of the C–C double bond. Benzoyl was chosen as a protecting group, because of its UV-activity, its orthogonality to the TBS group at C5–OH and its stability under hydrogenation conditions. Interestingly, attempted benzoylation in pyridine resulted exclusively in the formation of furan 10 (Table 2, entry 1). We reasoned that pyridine initiates an E/Z-isomerization of the enal through nucleophilic attack at the β-position, followed by lactol formation, benzoylation of the lactol and finally elimination of benzoic acid. Replacing pyridine as a solvent by dichloromethane and using NEt(iPr)2 as an HCl-scavenger led indeed to a suppression of the formation of furan 10, but the desired benzoate 9 was obtained in yields lower than 20%, along with ca. 80% of recovered starting material 8 (Table 2, entry 2). Benzoate 9 was eventually obtained in high yields from benzoic acid using Steglich’s esterification [61] (Table 2, entry 3).
Table 2: C4–OH protection as benzoate.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-102-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | reagents and conditions | product | yield |
|---|---|---|---|
| 1 | benzoyl chloride (1.5 equiv), pyridine (0.4 M), 65 °C, 12 h | 10 | 47% |
| 2a | benzoyl chloride (2.6 equiv), NEt(iPr)2 (3.0 equiv), CH2Cl2, 40 °C, 12 h | 9 | <20% |
| 3 |
benzoic acid (1.8 equiv), DCC (1.8 equiv), DMAP
(0.1 equiv), CH2Cl2, 20 °C, 12 h |
9 | 82% |
aStarting material 8 was recovered in ca. 80% yield.
With enal 9 in hands, selective hydrogenation of the C–C double bond had to be accomplished in the next step. Lipshutz’ modification [62] of Stryker’s reagent [63], which we had previously used successfully for the conjugate reduction of a related enoate [35], failed completely in this case and resulted only in the isolation of unreacted starting material. For these reasons we resumed to a hydrogenation catalyzed by Pd/C, in spite of the well-known capricious nature of these transformations [64].
With a commercial sample of Pd on charcoal (10 wt %) a plethora of products was detected, four of which could be isolated and identified as the hydrogenated acetal 11, the methyl ether 13, and the two desilylated products 14 and 15 (Table 3, entry 1). Literature precedence exists for the Pd/C-induced dealkoxylation of acetals [65] and for desilylation reactions of silyl ethers [66]. With in situ generated Pd/C (obtained from Pd(OAc)2 and charcoal according to Felpin’s protocol) [67] an improved selectivity was observed as the cleavage of the silyl groups could be suppressed. However, reductive dealkoxylation of the acetal remained a problem and the combined yield of the desired products 11 and 12 was still unsatisfactory (Table 3, entry 2). This situation changed when we used Pd(OH)2 on charcoal (10 wt %) as hydrogenation catalyst, which resulted in the exclusive formation of acetal 11 and aldehyde 12 in 83% combined yield (Table 3, entry 3).
Table 3: Hydrogenation of benzoyl protected enal 9.
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-102-i8.svg?max-width=637&scale=1.0)
|
||||||
| entry | reagents and conditions | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|
| 1 | Pd/C (10 wt %; 1.6 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 31% | – | 16% | 16% | 8% |
| 2 | Pd(OAc)2 (1 mol %), activated charcoal (9 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 28% | 9% | trace | – | – |
| 3a | Pd(OH)2/C (10 wt %; 1.2 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 51% | 32% | – | – | – |
aProducts 11 and 12 were obtained as an inseparable mixture, yields are estimated from 1H NMR spectrum.
For the deprotection of 11 and 12 a desilylation initiated by TBAF, followed by acidic hydrolysis was investigated first. With isolated acetal 11, these conditions induced a scrambling of the benzoate and led to a mixture of the desired pyranose 16 and the furanose 17. Facile migration of carboxylates upon TBAF-mediated desilylation of a vicinal alcohol has previously been observed by us in a different context [68]. We assume that this process starts with a nucleophilic attack of the alkoxylate 18 at the ester carbon, giving a five membered intermediate 19 (Scheme 2).
![[1860-5397-10-102-i2]](/bjoc/content/inline/1860-5397-10-102-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Two step desilylation–acetal hydrolysis.
Scheme 2: Two step desilylation–acetal hydrolysis.
The benzoate scrambling was completely suppressed by avoiding highly nucleophilic alkoxide intermediates, which was achieved with acidic deprotection conditions. Thus, treating the mixture of 11 and 12 with trifluoroacetic acid in dichloromethane at ambient temperature resulted in desilylation and acetal cleavage, giving 4-benzoyl protected L-amicetose 16 as an anomeric mixture in 65% yield (Scheme 3).
![[1860-5397-10-102-i3]](/bjoc/content/inline/1860-5397-10-102-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deprotection of 11 and 12 to L-amicetose derivative 16.
Scheme 3: Deprotection of 11 and 12 to L-amicetose derivative 16.
With a view towards L-cinerulose, formally the C4-oxidation product of L-amicetose, we started from cross metathesis product 8 which was first oxidized using Dess–Martin periodinane [69]. The α,β-unsaturated γ-keto aldehyde 21 was obtained in a very high yield of 90%, and subsequently subjected to hydrogenation conditions using a commercial sample of Pd/C (10 wt %, “batch 1”). We were pleased to find that the expected cinerulose derivative 22 could be isolated in analytically pure form in 85% yield (Scheme 4).
![[1860-5397-10-102-i4]](/bjoc/content/inline/1860-5397-10-102-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of a cinerulose-TBS ether 22.
Scheme 4: Synthesis of a cinerulose-TBS ether 22.
To our great dismay, we found that this result could not be reproduced with a different batch of commercial Pd/C (10 wt %, “batch 2”), purchased from the same supplier. As can be seen from Table 4, entry 1, the second batch of Pd/C catalyzes under otherwise identical conditions a chemoselective acetalization of the aldehyde, leading to 23 which was isolated in 52% yield. A similar result was obtained with Pd(OH)2/C (Table 4, entry 2). In situ formed Pd on charcoal [67], prepared from 0.5 mol % of Pd(OAc)2 as a precursor, gave an inseparable mixture of starting material 21 and acetal 23 in a 1.7:1 ratio (Table 4, entry 3). By increasing the amount of catalyst precursor to 1 mol %, conversion to acetal 23 was complete, and this product could be isolated in 82% yield (Table 4, entry 4).
Table 4: Chemoselective acetalization of oxidation product 21.
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-102-i9.svg?max-width=637&scale=1.0)
|
||
| entry | reagents and conditions | yield of 23 |
|---|---|---|
| 1 | Pd/C (“batch 2”, 10 wt %; 2.2 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 52% |
| 2 | Pd(OH)2/C (10 wt %; 1,7 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 43% |
| 3 | Pd(OAc)2 (0.5 mol %), activated charcoal (9 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 34%a |
| 4 | Pd(OAc)2 (1.0 mol %), activated charcoal (9 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 82% |
aEstimated from 1H NMR spectrum (21 and 23 were obtained as an inseparable mixture in ca. 90% combined yield).
Although the Pd(II)-catalyzed acetalization of aldehydes, including enals, is known [70], we are not aware of examples where Pd-catalysts immobilized on charcoal have been used for this transformation. Most likely the acetalization is catalyzed by residual Pd2+ via Lewis-acidic carbonyl activation. It is known that the reduction to Pd(0) is almost never complete, even if hydrogen is present [71,72].
At this stage we decided to reinvestigate the hydrogenation step, starting from acetal 23, using fresh samples of catalyst which have not been used previously for the acetalization (Table 5). With in situ prepared Pd/C (Table 5, entry 1) a major amount of starting material 23 was recovered and minor quantities of hydrogenated products 24 and 26, as well as desilylated starting material 25 were detected. With commercial Pd/C (10 wt %, “batch 2”) (Table 5, entry 2) the starting material was fully consumed and only hydrogenated products 24 and 26 could be detected in a 1:2 ratio, but unfortunately the isolated yields were only mediocre. As for the hydrogenation of benzoate 9 (Table 3), best results were reproducibly obtained with Pd(OH)2 on charcoal (Table 5, entry 3), leading to an isolated yield of 84% of a double protected cinerulose derivative 24.
Table 5: Hydrogenation of acetal 23.
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-102-i10.svg?max-width=637&scale=1.0)
|
||||
| entry | reagents and conditions | 24 | 25 | 26 |
|---|---|---|---|---|
| 1a | Pd(OAc)2 (1.0 mol %), activated charcoal (9 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 7% | 16% | 7% |
| 2 | Pd/C (10 wt %; 2.2 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 19% | n.d. | 38% |
| 3 | Pd(OH)2/C (10 wt %; 1.0 mol %), H2 (1 bar), methanol, 20 °C, 12 h | 84% | n.d. | n.d. |
aStarting material 23 (49%) was recovered.
All attempts to isolate cinerulose were hampered by the high volatility of the product. To accomplish complete deprotection of the precursor, compound 24 was first treated with TBAF trihydrate in THF, followed by treatment with diluted aqueous HCl. Although only small amounts of material were obtained via this procedure, 1H NMR spectroscopical analysis was possible. The data suggest that cinerulose exists in CDCl3 as a 2:1 mixture of acyclic aldose 27 and lactol 28 (Scheme 5).
![[1860-5397-10-102-i5]](/bjoc/content/inline/1860-5397-10-102-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we showed that an allylic alcohol, available diastereoselectively from enantiomerically pure L-lactate in few steps, is a useful starting material for a metathesis based synthesis of protected L-amicetose and L-cinerulose. It undergoes a clean and high yielding cross metathesis reaction with acrolein, however, the most commonly used second generation Grubbs’ catalyst is not the ideal initiator for this particular transformation. Significantly better results were obtained with two different phosphine free catalysts comprising a hemilabile alkoxy substituted benzylidene ligand, even at only moderately elevated temperatures. The acrolein cross metathesis product can be converted into the 4-benzoate of L-amicetose via benzoylation, Pd-catalyzed hydrogenation and global deprotection, whereas a cinerulose derivative protected at C1 as a dimethyl acetal and at C5–OH as a TBS ether becomes available via Dess–Martin oxidation, Pd-catalyzed acetalization and Pd-catalyzed hydrogenation. Notably, our results underline once again that apparently trivial olefin hydrogenation reactions catalyzed by commercial “Pd/C” can be capricious and very difficult to reproduce, unless the specific properties of the catalyst used and the conditions for its preparation are documented by the supplier. Unfortunately, this is very often not the case and the catalytic activities of apparently identical catalysts which are often designated just as “10 wt % Pd on carbon” may vary dramatically.
References
-
La Ferla, B.; Airoldi, C.; Zona, C.; Orsato, A.; Cardona, F.; Merlo, S.; Sironi, E.; D'Orazio, G.; Nicotra, F. Nat. Prod. Rep. 2011, 28, 630–648. doi:10.1039/c0np00055h
Return to citation in text: [1] -
Gräfe, U. Biochemie der Antibiotika; Spektrum Verlag: Heidelberg, Germany, 1992.
Return to citation in text: [1] -
Hoffmeister, D.; Dräger, G.; Ichinose, K.; Rohr, J.; Bechthold, A. J. Am. Chem. Soc. 2003, 125, 4678–4679. doi:10.1021/ja029645k
Return to citation in text: [1] -
Hoffmeister, D.; Weber, M.; Dräger, G.; Ichinose, K.; Dürr, C.; Bechthold, A. ChemBioChem 2004, 5, 369–371. doi:10.1002/cbic.200300793
Return to citation in text: [1] -
Pérez, M.; Lombó, F.; Zhu, L.; Gibson, M.; Braña, A. F.; Rohr, J.; Salas, J. A.; Méndez, C. Chem. Commun. 2005, 1604–1606. doi:10.1039/b417815g
Return to citation in text: [1] [2] -
Kirschning, A.; Bechthold, A. F.-W.; Rohr, J. Top. Curr. Chem. 1997, 188, 1–84. doi:10.1007/BFb0119234
Return to citation in text: [1] [2] -
Kunimoto, S.; Someno, T.; Yamazaki, Y.; Lu, J.; Esumi, H.; Naganawa, H. J. Antibiot. 2003, 56, 1012–1017. doi:10.7164/antibiotics.56.1012
Return to citation in text: [1] -
Someno, T.; Kunimoto, S.; Nakamura, H.; Naganawa, H.; Ikeda, D. J. Antibiot. 2005, 58, 56–60. doi:10.1038/ja.2005.6
Return to citation in text: [1] -
Lu, J.; Kunimoto, S.; Yamazaki, Y.; Kaminishi, M.; Esumi, H. Cancer Sci. 2004, 95, 547–552. doi:10.1111/j.1349-7006.2004.tb03247.x
Return to citation in text: [1] -
Arcamone, F.; Cassinelli, G. Curr. Med. Chem. 1998, 5, 391–419.
Return to citation in text: [1] -
Oki, T.; Matsuzawa, Y.; Yoshimoto, A.; Numata, K.; Kitamura, I.; Hori, S.; Takamatsu, A.; Umezawa, H.; Ishizuka, M.; Naganawa, H.; Suda, H.; Hamada, M.; Takeuchi, T. J. Antibiot. 1975, 28, 830–834. doi:10.7164/antibiotics.28.830
Return to citation in text: [1] -
Komiyama, T.; Oki, T.; Inui, T.; Takeuchi, T.; Umezawa, H. Gann 1979, 70, 395–401.
Return to citation in text: [1] -
Lee, Y.-L.; Chen, C.-W.; Liu, F.-H.; Huang, Y.-W.; Huang, H.-M. PLoS One 2013, 8, e61939. doi:10.1371/journal.pone.0061939
Return to citation in text: [1] -
Kirschning, A.; Jesberger, M.; Schöning, K.-U. Synthesis 2001, 507–540. doi:10.1055/s-2001-12342
Return to citation in text: [1] [2] -
Thiem, J.; Klaffke, W. Top. Curr. Chem. 1990, 154, 285–332. doi:10.1007/BFb0111564
Return to citation in text: [1] -
Seeberger, P. H.; Danishefsky, S. J. Acc. Chem. Res. 1998, 31, 685–695. doi:10.1021/ar9600648
Return to citation in text: [1] -
Seeberger, P. H.; Haase, W.-C. Chem. Rev. 2000, 100, 4349–4394. doi:10.1021/cr9903104
Return to citation in text: [1] -
Lajšić, S.; Miljković, D.; Ćetković, G. Carbohydr. Res. 1992, 233, 261–264. doi:10.1016/S0008-6215(00)90940-6
Return to citation in text: [1] -
Bethell, G. S.; Ferrier, R. J. J. Chem. Soc., Perkin Trans. 1 1973, 1400–1405. doi:10.1039/p19730001400
Return to citation in text: [1] -
Pedersen, C.; Jensen, H. S. Acta Chem. Scand. 1994, 48, 222–227. doi:10.3891/acta.chem.scand.48-0222
Return to citation in text: [1] -
Spohr, U.; Paszkiewicz-Hnatiw, E.; Morishima, N.; Lemieux, R. U. Can. J. Chem. 1992, 70, 254–271. doi:10.1139/v92-036
Return to citation in text: [1] -
Kjølberg, O.; Neumann, K. Acta Chem. Scand. 1992, 46, 877–882. doi:10.3891/acta.chem.scand.46-0877
Return to citation in text: [1] -
Wang, H.-Y. L.; Wu, B.; Zhang, Q.; Kang, S.-W.; Rojanasakul, Y.; O'Doherty, G. A. ACS Med. Chem. Lett. 2011, 2, 259–263. doi:10.1021/ml100291n
Return to citation in text: [1] -
Wang, H.-Y. L.; Xin, W.; Zhou, M.; Stueckle, T. A.; Rojanasakul, Y.; O'Doherty, G. A. ACS Med. Chem. Lett. 2011, 2, 73–78. doi:10.1021/ml100219d
Return to citation in text: [1] -
Zhu, L.; Talukdar, A.; Zhang, G.; Kedenburg, J. P.; Wang, P. G. Synlett 2005, 1547–1550. doi:10.1055/s-2005-869846
Return to citation in text: [1] -
Kirschning, A.; Hary, U.; Ries, M. Tetrahedron 1995, 51, 2297–2304. doi:10.1016/0040-4020(94)01081-A
Return to citation in text: [1] -
Noecker, L. A.; Martino, J. A.; Foley, P. J.; Rush, D. M.; Giuliano, R. M.; Villani, F. J., Jr. Tetrahedron: Asymmetry 1998, 9, 203–212. doi:10.1016/S0957-4166(97)00631-9
Return to citation in text: [1] -
Berti, G.; Caroti, P.; Catelani, G.; Monti, L. Carbohydr. Res. 1983, 124, 35–42. doi:10.1016/0008-6215(83)88353-0
Return to citation in text: [1] -
Marco, J. L. Synth. Commun. 1989, 19, 485–490. doi:10.1080/00397918908050690
Return to citation in text: [1] -
Itoh, T.; Yoshinaka, A.; Sato, T.; Fujisawa, T. Chem. Lett. 1985, 14, 1679–1680. doi:10.1246/cl.1985.1679
Return to citation in text: [1] -
Fuganti, C.; Grasselli, P. J. Chem. Soc., Chem. Commun. 1978, 299–300. doi:10.1039/c39780000299
Return to citation in text: [1] -
Aidhen, I. S.; Satyamurthi, N. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2008, 47, 1851–1857.
Return to citation in text: [1] -
McDonald, F. E.; Zhu, H. Y. H. J. Am. Chem. Soc. 1998, 120, 4246–4247. doi:10.1021/ja980196r
Return to citation in text: [1] -
Zhu, L.; Kedenburg, J. P.; Xian, M.; Wang, P. G. Tetrahedron Lett. 2005, 46, 811–813. doi:10.1016/j.tetlet.2004.12.014
Return to citation in text: [1] -
Schmidt, B.; Hauke, S. Org. Biomol. Chem. 2013, 11, 4194–4206. doi:10.1039/c3ob40167g
Return to citation in text: [1] [2] [3] -
Schmidt, B.; Biernat, A. Chem.–Eur. J. 2008, 14, 6135–6141. doi:10.1002/chem.200800567
Return to citation in text: [1] [2] [3] -
Ley, S. V.; Armstrong, A.; Díez-Martín, D.; Ford, M. J.; Grice, P.; Knight, J. G.; Kolb, H. C.; Madin, A.; Marby, C. A.; Mukherjee, S.; Shaw, A. N.; Slawin, A. M. Z.; Vile, S.; White, A. D.; Williams, D. J.; Woods, M. J. Chem. Soc., Perkin Trans. 1 1991, 667–692. doi:10.1039/p19910000667
Return to citation in text: [1] -
Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 3783–3784. doi:10.1021/ja9939744
Return to citation in text: [1] -
Cossy, J.; BouzBouz, S.; Hoveyda, A. H. J. Organomet. Chem. 2001, 624, 327–332. doi:10.1016/S0022-328X(00)00932-3
Return to citation in text: [1] -
Dinh, M.-T.; BouzBouz, S.; Peglion, J.-L.; Cossy, J. Synlett 2005, 2851–2853. doi:10.1055/s-2005-918943
Return to citation in text: [1] -
Chandra Rao, D.; Kumar Reddy, D.; Shekhar, V.; Venkateswarlu, Y. Tetrahedron Lett. 2013, 54, 828–829. doi:10.1016/j.tetlet.2012.11.061
Return to citation in text: [1] -
Miao, X.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H. ChemSusChem 2009, 2, 542–545. doi:10.1002/cssc.200900028
Return to citation in text: [1] -
Mohapatra, D. K.; Maity, S.; Rao, T. S.; Yadav, J. S.; Sridhar, B. Eur. J. Org. Chem. 2013, 2859–2863. doi:10.1002/ejoc.201300053
Return to citation in text: [1] -
Paul, T.; Andrade, R. B. Tetrahedron Lett. 2007, 48, 5367–5370. doi:10.1016/j.tetlet.2007.06.031
Return to citation in text: [1] -
Achmatowicz, O.; Szechner, B. B. Bull. Acad. Pol. Sci., Ser. Sci. Chim. 1971, 19, 309–311.
Return to citation in text: [1] -
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q
Return to citation in text: [1] -
Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g
Return to citation in text: [1] -
Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett. 2000, 41, 9973–9976. doi:10.1016/S0040-4039(00)01808-6
Return to citation in text: [1] -
Arlt, D.; Bieniek, M.; Karch, R. Novel Metathesis Catalysts. WO Pantent Applcation WO2008/034552 A1, March 27, 2008.
Return to citation in text: [1] [2] -
Mutlu, H.; Montero de Espinosa, L.; Türünç, O.; Meier, M. A. R. Beilstein J. Org. Chem. 2010, 6, 1149–1158. doi:10.3762/bjoc.6.131
Return to citation in text: [1] -
Schmidt, B. Eur. J. Org. Chem. 2004, 1865–1880. doi:10.1002/ejoc.200300714
Return to citation in text: [1] -
Schmidt, B. J. Mol. Catal. A 2006, 254, 53–57. doi:10.1016/j.molcata.2006.03.026
Return to citation in text: [1] -
Higman, C. S.; Plais, L.; Fogg, D. E. ChemCatChem 2013, 5, 3548–3551. doi:10.1002/cctc.201300886
Return to citation in text: [1] -
Forman, G. S.; McConnell, A. E.; Tooze, R. P.; van Rensburg, W. J.; Meyer, W. H.; Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W. Organometallics 2005, 24, 4528–4542. doi:10.1021/om0503848
Return to citation in text: [1] -
Forman, G. S.; Tooze, R. P. J. Organomet. Chem. 2005, 690, 5863–5866. doi:10.1016/j.jorganchem.2005.07.107
Return to citation in text: [1] -
Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f
Return to citation in text: [1] -
Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900–1923. doi:10.1002/anie.200200556
Return to citation in text: [1] -
Connon, S. J.; Blechert, S. Top. Organomet. Chem. 2004, 11, 93–124.
Return to citation in text: [1] -
Schmidt, B.; Kunz, O.; Petersen, M. H. J. Org. Chem. 2012, 77, 10897–10906. doi:10.1021/jo302359h
Return to citation in text: [1] -
Schmidt, B.; Kunz, O. Beilstein J. Org. Chem. 2013, 9, 2544–2555. doi:10.3762/bjoc.9.289
Return to citation in text: [1] -
Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. doi:10.1002/anie.197805221
Return to citation in text: [1] -
Baker, B. A.; Bošković, Z. V.; Lipshutz, B. H. Org. Lett. 2008, 10, 289–292. doi:10.1021/ol702689v
Return to citation in text: [1] -
Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291–293. doi:10.1021/ja00209a048
Return to citation in text: [1] -
Young, J. G.; Hartung, W. H.; Daniels, H. H. J. Org. Chem. 1953, 18, 229–234. doi:10.1021/jo01130a015
Return to citation in text: [1] -
Haukaas, M. H.; O'Doherty, G. A. Org. Lett. 2002, 4, 1771–1774. doi:10.1021/ol025844x
Return to citation in text: [1] -
Kim, S.; Jacobo, S. M.; Chang, C.-T.; Bellone, S.; Powell, W. S.; Rokach, J. Tetrahedron Lett. 2004, 45, 1973–1976. doi:10.1016/j.tetlet.2003.12.145
Return to citation in text: [1] -
Felpin, F.-X.; Fouquet, E. Chem.–Eur. J. 2010, 16, 12440–12445. doi:10.1002/chem.201001377
Return to citation in text: [1] [2] -
Schmidt, B.; Kunz, O.; Biernat, A. J. Org. Chem. 2010, 75, 2389–2394. doi:10.1021/jo1002642
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c
Return to citation in text: [1] -
Gorla, F.; Venanzi, L. M. Helv. Chim. Acta 1990, 73, 690–697. doi:10.1002/hlca.19900730317
Return to citation in text: [1] -
Seki, M. Synthesis 2006, 2975–2992. doi:10.1055/s-2006-950197
Return to citation in text: [1] -
Felpin, F.-X.; Ayad, T.; Mitra, S. Eur. J. Org. Chem. 2006, 2679–2690. doi:10.1002/ejoc.200501004
Return to citation in text: [1]
| 36. | Schmidt, B.; Biernat, A. Chem.–Eur. J. 2008, 14, 6135–6141. doi:10.1002/chem.200800567 |
| 35. | Schmidt, B.; Hauke, S. Org. Biomol. Chem. 2013, 11, 4194–4206. doi:10.1039/c3ob40167g |
| 38. | Chatterjee, A. K.; Morgan, J. P.; Scholl, M.; Grubbs, R. H. J. Am. Chem. Soc. 2000, 122, 3783–3784. doi:10.1021/ja9939744 |
| 39. | Cossy, J.; BouzBouz, S.; Hoveyda, A. H. J. Organomet. Chem. 2001, 624, 327–332. doi:10.1016/S0022-328X(00)00932-3 |
| 40. | Dinh, M.-T.; BouzBouz, S.; Peglion, J.-L.; Cossy, J. Synlett 2005, 2851–2853. doi:10.1055/s-2005-918943 |
| 41. | Chandra Rao, D.; Kumar Reddy, D.; Shekhar, V.; Venkateswarlu, Y. Tetrahedron Lett. 2013, 54, 828–829. doi:10.1016/j.tetlet.2012.11.061 |
| 42. | Miao, X.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H. ChemSusChem 2009, 2, 542–545. doi:10.1002/cssc.200900028 |
| 43. | Mohapatra, D. K.; Maity, S.; Rao, T. S.; Yadav, J. S.; Sridhar, B. Eur. J. Org. Chem. 2013, 2859–2863. doi:10.1002/ejoc.201300053 |
| 44. | Paul, T.; Andrade, R. B. Tetrahedron Lett. 2007, 48, 5367–5370. doi:10.1016/j.tetlet.2007.06.031 |
| 54. | Forman, G. S.; McConnell, A. E.; Tooze, R. P.; van Rensburg, W. J.; Meyer, W. H.; Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W. Organometallics 2005, 24, 4528–4542. doi:10.1021/om0503848 |
| 55. | Forman, G. S.; Tooze, R. P. J. Organomet. Chem. 2005, 690, 5863–5866. doi:10.1016/j.jorganchem.2005.07.107 |
| 56. | Samojłowicz, C.; Bieniek, M.; Grela, K. Chem. Rev. 2009, 109, 3708–3742. doi:10.1021/cr800524f |
| 51. | Schmidt, B. Eur. J. Org. Chem. 2004, 1865–1880. doi:10.1002/ejoc.200300714 |
| 52. | Schmidt, B. J. Mol. Catal. A 2006, 254, 53–57. doi:10.1016/j.molcata.2006.03.026 |
| 53. | Higman, C. S.; Plais, L.; Fogg, D. E. ChemCatChem 2013, 5, 3548–3551. doi:10.1002/cctc.201300886 |
| 47. | Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g |
| 48. | Gessler, S.; Randl, S.; Blechert, S. Tetrahedron Lett. 2000, 41, 9973–9976. doi:10.1016/S0040-4039(00)01808-6 |
| 49. | Arlt, D.; Bieniek, M.; Karch, R. Novel Metathesis Catalysts. WO Pantent Applcation WO2008/034552 A1, March 27, 2008. |
| 50. | Mutlu, H.; Montero de Espinosa, L.; Türünç, O.; Meier, M. A. R. Beilstein J. Org. Chem. 2010, 6, 1149–1158. doi:10.3762/bjoc.6.131 |
| 45. | Achmatowicz, O.; Szechner, B. B. Bull. Acad. Pol. Sci., Ser. Sci. Chim. 1971, 19, 309–311. |
| 46. | Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q |
| 57. | Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900–1923. doi:10.1002/anie.200200556 |
| 58. | Connon, S. J.; Blechert, S. Top. Organomet. Chem. 2004, 11, 93–124. |
| 49. | Arlt, D.; Bieniek, M.; Karch, R. Novel Metathesis Catalysts. WO Pantent Applcation WO2008/034552 A1, March 27, 2008. |
| 59. | Schmidt, B.; Kunz, O.; Petersen, M. H. J. Org. Chem. 2012, 77, 10897–10906. doi:10.1021/jo302359h |
| 60. | Schmidt, B.; Kunz, O. Beilstein J. Org. Chem. 2013, 9, 2544–2555. doi:10.3762/bjoc.9.289 |
| 66. | Kim, S.; Jacobo, S. M.; Chang, C.-T.; Bellone, S.; Powell, W. S.; Rokach, J. Tetrahedron Lett. 2004, 45, 1973–1976. doi:10.1016/j.tetlet.2003.12.145 |
| 67. | Felpin, F.-X.; Fouquet, E. Chem.–Eur. J. 2010, 16, 12440–12445. doi:10.1002/chem.201001377 |
| 64. | Young, J. G.; Hartung, W. H.; Daniels, H. H. J. Org. Chem. 1953, 18, 229–234. doi:10.1021/jo01130a015 |
| 65. | Haukaas, M. H.; O'Doherty, G. A. Org. Lett. 2002, 4, 1771–1774. doi:10.1021/ol025844x |
| 63. | Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291–293. doi:10.1021/ja00209a048 |
| 35. | Schmidt, B.; Hauke, S. Org. Biomol. Chem. 2013, 11, 4194–4206. doi:10.1039/c3ob40167g |
| 61. | Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. doi:10.1002/anie.197805221 |
| 62. | Baker, B. A.; Bošković, Z. V.; Lipshutz, B. H. Org. Lett. 2008, 10, 289–292. doi:10.1021/ol702689v |
| 69. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c |
| 67. | Felpin, F.-X.; Fouquet, E. Chem.–Eur. J. 2010, 16, 12440–12445. doi:10.1002/chem.201001377 |
| 68. | Schmidt, B.; Kunz, O.; Biernat, A. J. Org. Chem. 2010, 75, 2389–2394. doi:10.1021/jo1002642 |
| 1. | La Ferla, B.; Airoldi, C.; Zona, C.; Orsato, A.; Cardona, F.; Merlo, S.; Sironi, E.; D'Orazio, G.; Nicotra, F. Nat. Prod. Rep. 2011, 28, 630–648. doi:10.1039/c0np00055h |
| 9. | Lu, J.; Kunimoto, S.; Yamazaki, Y.; Kaminishi, M.; Esumi, H. Cancer Sci. 2004, 95, 547–552. doi:10.1111/j.1349-7006.2004.tb03247.x |
| 23. | Wang, H.-Y. L.; Wu, B.; Zhang, Q.; Kang, S.-W.; Rojanasakul, Y.; O'Doherty, G. A. ACS Med. Chem. Lett. 2011, 2, 259–263. doi:10.1021/ml100291n |
| 24. | Wang, H.-Y. L.; Xin, W.; Zhou, M.; Stueckle, T. A.; Rojanasakul, Y.; O'Doherty, G. A. ACS Med. Chem. Lett. 2011, 2, 73–78. doi:10.1021/ml100219d |
| 25. | Zhu, L.; Talukdar, A.; Zhang, G.; Kedenburg, J. P.; Wang, P. G. Synlett 2005, 1547–1550. doi:10.1055/s-2005-869846 |
| 7. | Kunimoto, S.; Someno, T.; Yamazaki, Y.; Lu, J.; Esumi, H.; Naganawa, H. J. Antibiot. 2003, 56, 1012–1017. doi:10.7164/antibiotics.56.1012 |
| 8. | Someno, T.; Kunimoto, S.; Nakamura, H.; Naganawa, H.; Ikeda, D. J. Antibiot. 2005, 58, 56–60. doi:10.1038/ja.2005.6 |
| 26. | Kirschning, A.; Hary, U.; Ries, M. Tetrahedron 1995, 51, 2297–2304. doi:10.1016/0040-4020(94)01081-A |
| 6. | Kirschning, A.; Bechthold, A. F.-W.; Rohr, J. Top. Curr. Chem. 1997, 188, 1–84. doi:10.1007/BFb0119234 |
| 21. | Spohr, U.; Paszkiewicz-Hnatiw, E.; Morishima, N.; Lemieux, R. U. Can. J. Chem. 1992, 70, 254–271. doi:10.1139/v92-036 |
| 22. | Kjølberg, O.; Neumann, K. Acta Chem. Scand. 1992, 46, 877–882. doi:10.3891/acta.chem.scand.46-0877 |
| 2. | Gräfe, U. Biochemie der Antibiotika; Spektrum Verlag: Heidelberg, Germany, 1992. |
| 3. | Hoffmeister, D.; Dräger, G.; Ichinose, K.; Rohr, J.; Bechthold, A. J. Am. Chem. Soc. 2003, 125, 4678–4679. doi:10.1021/ja029645k |
| 4. | Hoffmeister, D.; Weber, M.; Dräger, G.; Ichinose, K.; Dürr, C.; Bechthold, A. ChemBioChem 2004, 5, 369–371. doi:10.1002/cbic.200300793 |
| 5. | Pérez, M.; Lombó, F.; Zhu, L.; Gibson, M.; Braña, A. F.; Rohr, J.; Salas, J. A.; Méndez, C. Chem. Commun. 2005, 1604–1606. doi:10.1039/b417815g |
| 14. | Kirschning, A.; Jesberger, M.; Schöning, K.-U. Synthesis 2001, 507–540. doi:10.1055/s-2001-12342 |
| 6. | Kirschning, A.; Bechthold, A. F.-W.; Rohr, J. Top. Curr. Chem. 1997, 188, 1–84. doi:10.1007/BFb0119234 |
| 14. | Kirschning, A.; Jesberger, M.; Schöning, K.-U. Synthesis 2001, 507–540. doi:10.1055/s-2001-12342 |
| 18. | Lajšić, S.; Miljković, D.; Ćetković, G. Carbohydr. Res. 1992, 233, 261–264. doi:10.1016/S0008-6215(00)90940-6 |
| 19. | Bethell, G. S.; Ferrier, R. J. J. Chem. Soc., Perkin Trans. 1 1973, 1400–1405. doi:10.1039/p19730001400 |
| 13. | Lee, Y.-L.; Chen, C.-W.; Liu, F.-H.; Huang, Y.-W.; Huang, H.-M. PLoS One 2013, 8, e61939. doi:10.1371/journal.pone.0061939 |
| 20. | Pedersen, C.; Jensen, H. S. Acta Chem. Scand. 1994, 48, 222–227. doi:10.3891/acta.chem.scand.48-0222 |
| 11. | Oki, T.; Matsuzawa, Y.; Yoshimoto, A.; Numata, K.; Kitamura, I.; Hori, S.; Takamatsu, A.; Umezawa, H.; Ishizuka, M.; Naganawa, H.; Suda, H.; Hamada, M.; Takeuchi, T. J. Antibiot. 1975, 28, 830–834. doi:10.7164/antibiotics.28.830 |
| 12. | Komiyama, T.; Oki, T.; Inui, T.; Takeuchi, T.; Umezawa, H. Gann 1979, 70, 395–401. |
| 70. | Gorla, F.; Venanzi, L. M. Helv. Chim. Acta 1990, 73, 690–697. doi:10.1002/hlca.19900730317 |
| 15. | Thiem, J.; Klaffke, W. Top. Curr. Chem. 1990, 154, 285–332. doi:10.1007/BFb0111564 |
| 16. | Seeberger, P. H.; Danishefsky, S. J. Acc. Chem. Res. 1998, 31, 685–695. doi:10.1021/ar9600648 |
| 17. | Seeberger, P. H.; Haase, W.-C. Chem. Rev. 2000, 100, 4349–4394. doi:10.1021/cr9903104 |
| 71. | Seki, M. Synthesis 2006, 2975–2992. doi:10.1055/s-2006-950197 |
| 72. | Felpin, F.-X.; Ayad, T.; Mitra, S. Eur. J. Org. Chem. 2006, 2679–2690. doi:10.1002/ejoc.200501004 |
| 30. | Itoh, T.; Yoshinaka, A.; Sato, T.; Fujisawa, T. Chem. Lett. 1985, 14, 1679–1680. doi:10.1246/cl.1985.1679 |
| 27. | Noecker, L. A.; Martino, J. A.; Foley, P. J.; Rush, D. M.; Giuliano, R. M.; Villani, F. J., Jr. Tetrahedron: Asymmetry 1998, 9, 203–212. doi:10.1016/S0957-4166(97)00631-9 |
| 28. | Berti, G.; Caroti, P.; Catelani, G.; Monti, L. Carbohydr. Res. 1983, 124, 35–42. doi:10.1016/0008-6215(83)88353-0 |
| 29. | Marco, J. L. Synth. Commun. 1989, 19, 485–490. doi:10.1080/00397918908050690 |
| 36. | Schmidt, B.; Biernat, A. Chem.–Eur. J. 2008, 14, 6135–6141. doi:10.1002/chem.200800567 |
| 36. | Schmidt, B.; Biernat, A. Chem.–Eur. J. 2008, 14, 6135–6141. doi:10.1002/chem.200800567 |
| 37. | Ley, S. V.; Armstrong, A.; Díez-Martín, D.; Ford, M. J.; Grice, P.; Knight, J. G.; Kolb, H. C.; Madin, A.; Marby, C. A.; Mukherjee, S.; Shaw, A. N.; Slawin, A. M. Z.; Vile, S.; White, A. D.; Williams, D. J.; Woods, M. J. Chem. Soc., Perkin Trans. 1 1991, 667–692. doi:10.1039/p19910000667 |
| 34. | Zhu, L.; Kedenburg, J. P.; Xian, M.; Wang, P. G. Tetrahedron Lett. 2005, 46, 811–813. doi:10.1016/j.tetlet.2004.12.014 |
| 35. | Schmidt, B.; Hauke, S. Org. Biomol. Chem. 2013, 11, 4194–4206. doi:10.1039/c3ob40167g |
| 5. | Pérez, M.; Lombó, F.; Zhu, L.; Gibson, M.; Braña, A. F.; Rohr, J.; Salas, J. A.; Méndez, C. Chem. Commun. 2005, 1604–1606. doi:10.1039/b417815g |
| 33. | McDonald, F. E.; Zhu, H. Y. H. J. Am. Chem. Soc. 1998, 120, 4246–4247. doi:10.1021/ja980196r |
| 31. | Fuganti, C.; Grasselli, P. J. Chem. Soc., Chem. Commun. 1978, 299–300. doi:10.1039/c39780000299 |
| 32. | Aidhen, I. S.; Satyamurthi, N. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2008, 47, 1851–1857. |
© 2014 Schmidt and Hauke; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)