Abstract
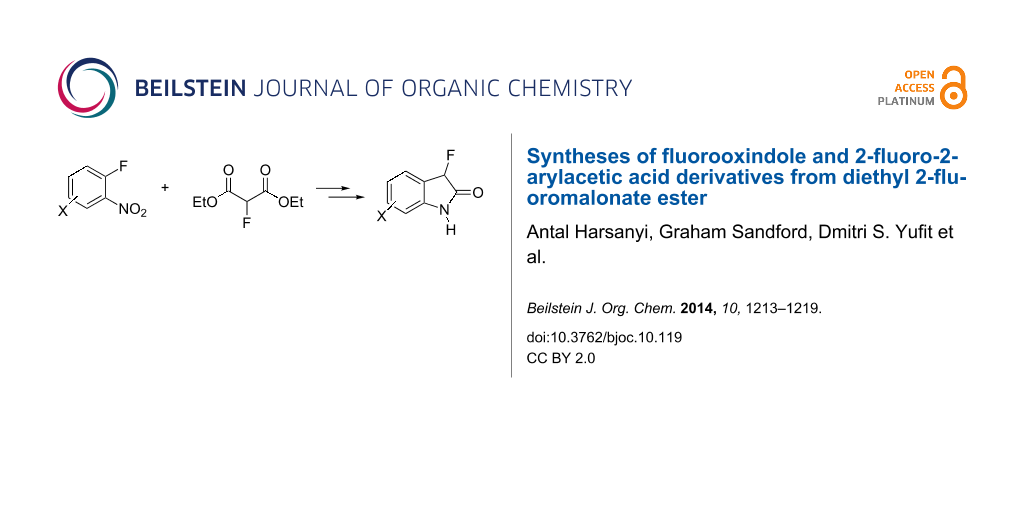
Diethyl 2-fluoromalonate ester is utilised as a building block for the synthesis of 2-fluoro-2-arylacetic acid and fluorooxindole derivatives by a strategy involving nucleophilic aromatic substitution reactions with ortho-fluoronitrobenzene substrates followed by decarboxylation, esterification and reductive cyclisation processes.

Graphical Abstract
Introduction
Since 1954, when Fried and Sabo observed that the incorporation of a fluorine atom into a corticosteroid derivative led to valuable enhanced biological activity [1], a growing number of commercially significant life science products, which owe their activity to the presence of fluorine atoms within their structures, have developed. Fluorine incorporation can lead, for example, to enhanced bioavailability, metabolic stability and lipophilicity of the organic system and these properties are exploited in a number of commercially valuable drugs including Ciprofloxacin, Lipitor and Voriconazole [2-6].
Given the very small number of fluorinated systems available from nature [7-9], in essence all organic molecules bearing carbon–fluorine bonds are ‘man-made’. Syntheses rely either on the construction of carbon–fluorine bonds using a fluorinating agent (‘late-stage’ fluorination) or the application of polyfunctional fluorine-containing small molecule building blocks (‘early stage’ fluorination) which may be employed in further transformations involving all the reactions and techniques available to synthetic organic chemists [10-13]. Of course, the success of an ‘early stage’ fluorination approach depends on the availability of a range of appropriately functionalised, fluorinated building blocks and the establishment of corresponding reactivity profiles [14]. However, it does not necessarily follow that reactions for which regio- and stereoselectivity profiles are well established for hydrocarbon systems will be similar to those for corresponding selectively fluorinated systems and, indeed, this is often not the case [15].
The use of 1,3-diketone, 1,3-ketoester and 1,3-diester derivatives in retrosynthetic planning is widespread in general organic chemistry and numerous terpenes, heterocycles and steroids originate from such simple yet synthetically versatile substrates [16-19]. In contrast, despite the availability of synthetic procedures for the preparation of various 2-fluoro-1,3-dicarbonyl systems [20-27], there is, surprisingly, only a relatively limited number of publications that report the use of such potentially useful fluorinated building blocks for the synthesis of more structurally sophisticated selectively fluorinated systems. For example, 2-fluoromalonate esters have been used for the preparation of various α-fluorocarboxylic acids [28-32], heterocycles, such as fluoropyrimidine [33] and quinolone [34] derivatives, alkylated [35] and Michael addition [36-40] products, providing an indication of the potential uses and opportunities available for the synthesis of fluoro-organic products from fluoromalonate precursors.
As part of a wider research programme aimed at developing routes for the synthesis of selectively fluorinated molecules using elemental fluorine for the key construction of the carbon–fluorine bond by complementary direct selective direct fluorination [41-44], continuous flow [45-49] and building block [50] strategies, in this paper, we describe nucleophilic aromatic substitution reactions of carbanions derived from diethyl 2-fluoromalonate ester as the first stage in the synthesis of fluoroacetic acid and fluoroxindole systems. While related palladium catalysed coupling processes between aryl bromides and diethyl 2-fluoromalonate have been described [51], reactions involving nucleophilic aromatic substitution between fluoromalonate systems [52] and appropriate aryl substrates have not been reported previously. Recently, various routes to fluorooxindoles have been discussed involving enantioselective fluorination of appropriate oxindole substrates by electrophilic fluorinating agents [53-62] or DAST [63] providing an indication of the importance of fluorooxindoles for medicinal chemistry applications.
Results and Discussion
Reactions of carbanions generated by the addition of sodium hydride to a solution of diethyl 2-fluoromalonate (1) in DMF with ortho-fluoronitrobenzene (2a) led to the efficient displacement of fluorine by a nucleophilic aromatic substitution process to provide diester 3 in good yield (Scheme 1). Displacement of fluorine from ortho-fluoronitrobenzene was quantitative as measured by 19F NMR spectroscopy of the crude reaction mixture and the structure of isolated diester 3 was confirmed by X-ray crystallography (Figure 1).
![[1860-5397-10-119-i1]](/bjoc/content/inline/1860-5397-10-119-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: SNAr reaction of 2-fluoronitrobenzene (2a) with diethyl 2-fluoromalonate (1).
Scheme 1: SNAr reaction of 2-fluoronitrobenzene (2a) with diethyl 2-fluoromalonate (1).
![[1860-5397-10-119-1]](/bjoc/content/figures/1860-5397-10-119-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In initial experiments, decarboxylation of 3 by reaction with potassium hydroxide gave good yields of the corresponding 2-fluoro-2-arylacetic acid 4a. However, in subsequent experiments, we found that further purification of the diester 3 after the initial SNAr step was not necessary and decarboxylation of crude diester 3 gave 4a very efficiently. Consequently, in all analogous experiments (Table 1), crude product diesters of type 3 were isolated and used without further purification, allowing the ready synthesis of a range of arylfluoroacetic acid derivatives 4a–f (Table 1). Structures 4a–f were confirmed by NMR techniques and, in particular, a doublet located at −190 ppm (2JHF = 50 Hz) in the 19F NMR spectra assigned to the CFH resonances and the corresponding doublets observed at ~6 ppm in the 1H NMR spectra, are diagnostic for the structures proposed.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-119-i4.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-119-i5.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-119-i6.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-119-i7.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-119-i8.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-119-i9.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-119-i10.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-119-i11.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-119-i12.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-119-i13.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-119-i14.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-119-i15.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-119-i16.svg?max-width=637&scale=1.0)
A nitro group ortho to a fluorine atom on the aryl ring is necessary under the present conditions to achieve full conversion of the starting fluoroarene. In related experiments, we found that a para-trifluoromethyl group is not sufficiently activating for reaction to occur whilst para-fluoronitrobenzene gave a complex mixture of unidentified products, most probably derived from competing benzyne formation.
This efficient methodology complements reported processes for the synthesis of various biologically active 2-fluoro-2-phenylacetic acids [64] which may be prepared using electrophilic fluorination of enolate esters [64-66], deoxofluorination [67-69] nucleophilic [70] and electrochemical fluorination [71,72] strategies.
Attempts to prepare 2-fluoro-2-(2,4-dinitrophenyl)acetic acid by an analogous process led to the isolation of a benzyl fluoride derivative 5, after evaporation of toluene and purification by column chromatography in 61% yield. The two consecutive decarboxylation reactions reflect the greater stability of the benzylic carbanion formed on loss of carbon dioxide from this system (Scheme 2).
![[1860-5397-10-119-i2]](/bjoc/content/inline/1860-5397-10-119-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of benzyl fluoride derivative 5.
Scheme 2: Synthesis of benzyl fluoride derivative 5.
With the series of 2-fluorophenylacetic acids in hand, we attempted the reduction of the nitro group in 4a using sodium dithionite, adapting reaction conditions similar to those described in the literature for the synthesis of biologically active system MaxiPost [63]. However, very low isolated yields of the cyclised product were obtained, presumably because of the high solubility of the amino acid intermediate in the aqueous reaction mixture and the well-established difficulty of direct amide bond formation processes. Consequently, before carrying out the nitro group reduction and amide forming cyclisation reactions, the acids 4a–e were transformed to the corresponding methyl esters 6a–e by stirring a mixture of the acid in hydrochloric acid and methanol (Table 2). The structure of 6a was confirmed unambiguously by X-ray crystallography (Figure 2) and all other methyl esters 6b–e were characterised by comparison with appropriate NMR data obtained for 6a.
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-119-i17.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-119-i18.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-119-i19.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-119-i20.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-119-i21.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-119-i22.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-119-i23.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-119-i24.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-119-i25.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-119-i26.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-119-i27.svg?max-width=637&scale=1.0)
![[1860-5397-10-119-2]](/bjoc/content/figures/1860-5397-10-119-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of methyl ester 6a.
Figure 2: Molecular structure of methyl ester 6a.
However, corresponding attempted esterification of the salt 4f with HCl in methanol gave 2-fluoromethyl-3-nitropyridine (7) in 68% yield (Scheme 3) after purification of the crude material by column chromatography and the structure was confirmed by X-ray analysis (Figure 3). In this case competing decarboxylation, rather than esterification, reflects the greater stabilisation of the carbanion system formed upon decarboxylation for this system.
![[1860-5397-10-119-i3]](/bjoc/content/inline/1860-5397-10-119-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of pyridyl fluoride 7.
Scheme 3: Synthesis of pyridyl fluoride 7.
![[1860-5397-10-119-3]](/bjoc/content/figures/1860-5397-10-119-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Reductive cyclization of methyl esters 6a–e using sodium dithionite provided fluorooxindoles 8a–e in acceptable yield after isolation by column chromatography (Table 3). In the 1H NMR spectrum, the characteristic CHF doublet located at 5.7 ppm (2JHF = 51 Hz) for the fluorooxindole systems 8 are 0.9 ppm upfield from the corresponding CHF resonances of the arylfluoroacetic esters 6a–e and, additionally, a broad NH singlet was detected at 9.0 ppm. The chemical shift of the doublet (−194.8 ppm) in the 19F NMR spectrum of fluorooxindoles 8a–e is also observed 10 ppm upfield from the fluorine resonance of the starting esters 6a–e.
![[Graphic 25]](/bjoc/content/inline/1860-5397-10-119-i28.svg?max-width=637&scale=1.0)
![[Graphic 26]](/bjoc/content/inline/1860-5397-10-119-i29.svg?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-10-119-i30.svg?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-10-119-i31.svg?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-10-119-i32.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-10-119-i33.svg?max-width=637&scale=1.0)
![[Graphic 31]](/bjoc/content/inline/1860-5397-10-119-i34.svg?max-width=637&scale=1.0)
![[Graphic 32]](/bjoc/content/inline/1860-5397-10-119-i35.svg?max-width=637&scale=1.0)
![[Graphic 33]](/bjoc/content/inline/1860-5397-10-119-i36.svg?max-width=637&scale=1.0)
![[Graphic 34]](/bjoc/content/inline/1860-5397-10-119-i37.svg?max-width=637&scale=1.0)
![[Graphic 35]](/bjoc/content/inline/1860-5397-10-119-i38.svg?max-width=637&scale=1.0)
Conclusion
Diethyl 2-fluoromalonate ester can be used as a highly effective fluorinated building block for the synthesis of various polyfunctional 2-fluoroacetic acid and 3-fluorooxindole systems. Fluorooxindoles are relatively rare fluorinated heterocyclic systems, even though several derivatives have useful biological activity, and current literature syntheses only involve fluorination of appropriate hydroxy and oxindole substrates. The strategy described here provides complementary building block syntheses from readily available fluorinated starting materials, further demonstrating the viability of using fluorinated dicarbonyl systems for the synthesis of more structurally sophisticated fluorinated derivatives.
References
-
Fried, J.; Sabo, E. F. J. Am. Chem. Soc. 1954, 76, 1455–1456. doi:10.1021/ja01634a101
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diedrich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Oxford, 2009.
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Isanbor, C.; O'Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011
Return to citation in text: [1] -
Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007
Return to citation in text: [1] -
Gribble, G. W. Chem. Soc. Rev. 1999, 28, 335–346. doi:10.1039/a900201d
Return to citation in text: [1] -
Hall, R. J. New Phytol. 1972, 71, 855–871. doi:10.1111/j.1469-8137.1972.tb01965.x
Return to citation in text: [1] -
O'Hagan, D.; Schaffrath, C.; Cobb, S. L.; Hamilton, J. T. G.; Murphy, C. D. Nature 2002, 416, 279. doi:10.1038/416279a
Return to citation in text: [1] -
Baasner, B.; Hagemann, H.; Tatlow, J. C. Houben-Weyl Organofluorine Compounds; Thieme: Stuttgart, 2000; Vol. E10a.
Return to citation in text: [1] -
Chambers, R. D. Fluorine in Organic Chemistry; Blackwell: Oxford, 2004.
Return to citation in text: [1] -
Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, 2006. doi:10.1002/9780470988589
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Percy, J. M. Top. Curr. Chem. 1997, 199, 131–195. doi:10.1007/3-540-69197-9_4
Return to citation in text: [1] -
Seebach, D. Angew. Chem., Int. Ed. Engl. 1990, 29, 1320–1367. doi:10.1002/anie.199013201
Return to citation in text: [1] -
Warren, S. Designing Organic Syntheses: A Progrmmed Introduction to the Synthon Approach; John Wiley and Sons: New York, 1978.
Return to citation in text: [1] -
Warren, S.; Wyatt, P. Organic Synthesis: The Disconnection Approach, 2nd ed.; John Wiley and Sons: Chichester, 2008.
Return to citation in text: [1] -
Wyatt, P.; Warren, S. Organic Synthesis: Strategy and Control, 2nd ed.; John Wiley and Sons: Chichester, 2007.
Return to citation in text: [1] -
Corey, E. J.; Cheng, X.-M. The logic of chemical synthesis; John Wiley and Sons: New York, 1995.
Return to citation in text: [1] -
Chambers, R. D.; Hutchinson, J.; Thomson, J. J. Fluorine Chem. 1996, 78, 165–166. doi:10.1016/0022-1139(96)03422-7
Return to citation in text: [1] -
Chambers, R. D.; Fox, M. A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Chem. Eng. Technol. 2005, 28, 344–352. doi:10.1002/ceat.200407123
Return to citation in text: [1] -
Chambers, R. D.; Hutchinson, J. J. Fluorine Chem. 1998, 92, 45–52. doi:10.1016/S0022-1139(98)00254-1
Return to citation in text: [1] -
Müh, T.; Fiedler, P.; Weintritt, H.; Westerkamp, W.; Reinecke, A. Method for producing alpha fluoromalonic acid dialkyl esters. W.O. Patent WO2002016304, Feb 28, 2002.
Return to citation in text: [1] -
Günther, A.; Weintritt, H.; Böhm, S. Method for producing a-fluoromalonic acid dialkyl esters. W.O. Patent WO2005019154, March 3, 2005.
Return to citation in text: [1] -
Braun, M.; Brosch, C. Production of fluorine compounds. W.O. Patent WO2002060838, Aug 8, 2002.
Return to citation in text: [1] -
Ishikawa, N.; Takaoka, A. Chem. Lett. 1981, 10, 107–110. doi:10.1246/cl.1981.107
Return to citation in text: [1] -
Ishikawa, N.; Takaoka, A.; Ibrahim, M. K. J. Fluorine Chem. 1984, 25, 203–212. doi:10.1016/S0022-1139(00)80949-5
Return to citation in text: [1] -
Miller, T. A.; Sloman, D. L.; Stanton, M. G.; Wilson, K. J.; Witter, D. J. Fluorinated arylamide derivatives. W.O. Patent WO2007087129, Aug 2, 2007.
Return to citation in text: [1] -
Close, J.; Heidebrecht, R. W.; Kattar, S.; Miller, T. A.; Sloman, D.; Stanton, M. G.; Tempest, P.; Witter, D. J. Histone deacetylase inhibitors with aryl-pyrazolyl motifs. W.O. Patent WO2007055941, May 18, 2007.
Return to citation in text: [1] -
Hubbs, J. L.; Mampreian, D. M.; Methot, J. L.; Miller, T. A.; Otte, K. M.; Siliphaivanh, P.; Sloman, D. L.; Stanton, M. G.; Wilson, K. J.; Witter, D. J. Benzothiophene derivatives. W.O. Patent WO2006115845, Nov 2, 2006.
Return to citation in text: [1] -
Bressi, J. C.; Chu, S.; Erickson, P.; Komandla, M.; Kwok, L.; Lawson, J. D.; Stafford, J. A.; Wallace, M. B.; Zhang, Z.; Das, J. cMET inhibitors. W.O. Patent WO2010019899, Feb 18, 2010.
Return to citation in text: [1] -
Leonardi, A.; Riva, C.; Tavecchia, P.; Sironi, G. Aldosterone receptor antagonists. W.O. Patent WO2007025780, March 8, 2007.
Return to citation in text: [1] -
Bergmann, E. D.; Cohen, S.; Shahak, I. J. Chem. Soc. 1959, 3286–3289. doi:10.1039/jr9590003286
Return to citation in text: [1] -
Brickner, S. J.; Chen, J. M.; Li, Z. B.; Marfat, A.; Mitton-Fry, M. J.; Plotkin, M. A.; Reilly, U. D.; Subramanyam, C.; Zhang, Z.; Robinson, S. Substituted heterocyclic derivatives and their pharmaceutical use and compositions. U.S. Patent US20080280879, Nov 13, 2008.
Return to citation in text: [1] -
Polla, M. O.; Tottie, L.; Nordén, C.; Linschoten, M.; Müsil, D.; Trumpp-Kallmeyer, S.; Aukrust, I. R.; Ringom, R.; Holm, K. H.; Neset, S. M.; Sandberg, M.; Thurmond, J.; Yu, P.; Hategan, G.; Anderson, H. Bioorg. Med. Chem. 2004, 12, 1151–1175. doi:10.1016/j.bmc.2003.12.039
Return to citation in text: [1] -
Kim, D.-Y.; Kim, S.-M.; Koh, K.-O.; Mang, J.-Y.; Lee, K.-S. Bull. Korean Chem. Soc. 2003, 24, 1425–1426. doi:10.5012/bkcs.2003.24.10.1425
Return to citation in text: [1] -
Cho, M.-J.; Cho, M.-G.; Huh, S.-C.; Kim, S.-M.; Lee, K.-s.; Koh, K.-O.; Mang, J.-Y.; Kim, D.-Y. Bull. Korean Chem. Soc. 2006, 27, 857–862. doi:10.5012/bkcs.2006.27.6.857
Return to citation in text: [1] -
Kang, S.-H.; Kim, D.-Y. Bull. Korean Chem. Soc. 2009, 30, 1439–1440. doi:10.5012/bkcs.2009.30.7.1439
Return to citation in text: [1] -
Kwon, B. K.; Kim, S. M.; Kim, D. Y. J. Fluorine Chem. 2009, 130, 759–761. doi:10.1016/j.jfluchem.2009.06.002
Return to citation in text: [1] -
Li, H.; Zu, L.; Xie, H.; Wang, W. Synthesis 2009, 1525–1530. doi:10.1055/s-0028-1088124
Return to citation in text: [1] -
Sandford, G. J. Fluorine Chem. 2007, 128, 90–104. doi:10.1016/j.jfluchem.2006.10.019
Return to citation in text: [1] -
Chambers, R. D.; Nakano, T.; Parsons, M.; Sandford, G.; Batsanov, A. S.; Howard, J. A. K. J. Fluorine Chem. 2008, 129, 811–816. doi:10.1016/j.jfluchem.2008.04.010
Return to citation in text: [1] -
Chambers, R. D.; Sandford, G.; Trmcic, J.; Okazoe, T. Org. Process Res. Dev. 2008, 12, 339–344. doi:10.1021/op700194r
Return to citation in text: [1] -
Chambers, R. D.; Nakano, T.; Okazoe, T.; Sandford, G. J. Fluorine Chem. 2009, 130, 792–798. doi:10.1016/j.jfluchem.2009.06.003
Return to citation in text: [1] -
Chambers, R. D.; Fox, M. A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Lab Chip 2005, 5, 191–198. doi:10.1039/b416400h
Return to citation in text: [1] -
Chambers, R. D.; Fox, M. A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Chem. Eng. Technol. 2005, 28, 344–352. doi:10.1002/ceat.200407123
Return to citation in text: [1] -
Chambers, R. D.; Fox, M. A.; Sandford, G. Lab Chip 2005, 5, 1132–1139. doi:10.1039/b504675k
Return to citation in text: [1] -
Breen, J. R.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Fray, J.; Patel, B. Beilstein J. Org. Chem. 2011, 7, 1048–1054. doi:10.3762/bjoc.7.120
Return to citation in text: [1] -
McPake, C. B.; Sandford, G. Org. Process Res. Dev. 2012, 16, 844–851. doi:10.1021/op200331s
Return to citation in text: [1] -
Hutchinson, J.; Sandford, G.; Vaughan, J. F. S. Tetrahedron 1998, 54, 2867–2876. doi:10.1016/S0040-4020(98)83023-8
Return to citation in text: [1] -
Beare, N. A.; Hartwig, J. F. J. Org. Chem. 2002, 67, 541–555. doi:10.1021/jo016226h
Return to citation in text: [1] -
Harsanyi, A. M.Sc. Thesis, Synthesis of 3-fluoro-oxindoles and phenyl fluoroacetic acid derivatives; Durham University, 2013, available from http://etheses.dur.ac.uk/6357/.
Return to citation in text: [1] -
Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t
Return to citation in text: [1] -
Shibata, N.; Ishimaru, T.; Suzuki, E.; Kirk, K. L. J. Org. Chem. 2003, 68, 2494–2497. doi:10.1021/jo026792s
Return to citation in text: [1] -
Zoute, L.; Audouard, C.; Plaquevent, J.-C.; Cahard, D. Org. Biomol. Chem. 2003, 1, 1833–1834. doi:10.1039/b303113f
Return to citation in text: [1] -
Shibata, N.; Kohno, J.; Takai, K.; Ishimaru, T.; Nakamura, S.; Toru, T.; Kanemasa, S. Angew. Chem., Int. Ed. 2005, 44, 4204–4207. doi:10.1002/anie.200501041
Return to citation in text: [1] -
Hamashima, Y.; Suzuki, N.; Takano, H.; Shimura, Y.; Sodeoka, M. J. Am. Chem. Soc. 2005, 127, 10164–10165. doi:10.1021/ja0513077
Return to citation in text: [1] -
Deng, Q.-H.; Wadepohl, H.; Gade, L. H. Chem.–Eur. J. 2011, 17, 14922–14928. doi:10.1002/chem.201102375
Return to citation in text: [1] -
Li, J.; Cai, Y.; Chen, W.; Liu, X.; Lin, L.; Feng, X. J. Org. Chem. 2012, 77, 9148–9155. doi:10.1021/jo301705t
Return to citation in text: [1] -
Wu, L.; Falivene, L.; Drinkel, E.; Grant, S.; Linden, A.; Cavallo, L.; Dorta, R. Angew. Chem., Int. Ed. 2012, 51, 2870–2873. doi:10.1002/anie.201200206
Return to citation in text: [1] -
Ishimaru, T.; Shibata, N.; Horikawa, T.; Yasuda, N.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2008, 47, 4157–4161. doi:10.1002/anie.200800717
Return to citation in text: [1] -
Dou, X.; Lu, Y. Org. Biomol. Chem. 2013, 11, 5217–5221. doi:10.1039/c3ob41267a
Return to citation in text: [1] -
Hewawasam, P.; Gribkoff, V. K.; Pendri, Y.; Dworetzky, S. I.; Meanwell, N. A.; Martinez, E.; Boissard, C. G.; Post-Munson, D. J.; Trojnacki, J. T.; Yeleswaram, K.; Pajor, L. M.; Knipe, J.; Gao, Q.; Perrone, R.; Starret, J. E. Bioorg. Med. Chem. Lett. 2002, 12, 1023–1026. doi:10.1016/S0960-894X(02)00101-4
Return to citation in text: [1] [2] -
Schiefer, I. T.; Abdul-Hay, S.; Wang, H.; Vanni, M.; Qin, Z.; Thatcher, G. R. J. J. Med. Chem. 2011, 54, 2293–2306. doi:10.1021/jm101450p
Return to citation in text: [1] [2] -
Zhang, F.; Song, J. Z. Tetrahedron Lett. 2006, 47, 7641–7644. doi:10.1016/j.tetlet.2006.08.057
Return to citation in text: [1] -
Rozen, S.; Hagooly, A.; Harduf, R. J. Org. Chem. 2001, 66, 7464–7468. doi:10.1021/jo010677k
Return to citation in text: [1] -
Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1996, 60, 4730–4737. doi:10.1021/jo00120a014
Return to citation in text: [1] -
Cantrell, G. L.; Filler, R. J. Fluorine Chem. 1985, 27, 35–45. doi:10.1016/S0022-1139(00)80895-7
Return to citation in text: [1] -
Bresciani, S.; O’Hagan, D. Tetrahedron Lett. 2010, 51, 5795–5797. doi:10.1016/j.tetlet.2010.08.104
Return to citation in text: [1] -
Watanabe, S.; Fujita, T.; Sakamoto, M.; Endo, H.; Kitazume, T. J. Fluorine Chem. 1990, 47, 187–192. doi:10.1016/S0022-1139(00)82371-4
Return to citation in text: [1] -
Kim, K.-Y.; Kim, B. C.; Lee, H. B.; Shin, H. J. Org. Chem. 2008, 73, 8106–8108. doi:10.1021/jo8015659
Return to citation in text: [1] -
Yin, J.; Zarkowsky, D. S.; Thomas, D. W.; Zhao, M. M.; Huffman, M. A. Org. Lett. 2004, 6, 1465–1468. doi:10.1021/ol049672a
Return to citation in text: [1]
| 1. | Fried, J.; Sabo, E. F. J. Am. Chem. Soc. 1954, 76, 1455–1456. doi:10.1021/ja01634a101 |
| 14. | Percy, J. M. Top. Curr. Chem. 1997, 199, 131–195. doi:10.1007/3-540-69197-9_4 |
| 45. | Chambers, R. D.; Fox, M. A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Lab Chip 2005, 5, 191–198. doi:10.1039/b416400h |
| 46. | Chambers, R. D.; Fox, M. A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Chem. Eng. Technol. 2005, 28, 344–352. doi:10.1002/ceat.200407123 |
| 47. | Chambers, R. D.; Fox, M. A.; Sandford, G. Lab Chip 2005, 5, 1132–1139. doi:10.1039/b504675k |
| 48. | Breen, J. R.; Sandford, G.; Yufit, D. S.; Howard, J. A. K.; Fray, J.; Patel, B. Beilstein J. Org. Chem. 2011, 7, 1048–1054. doi:10.3762/bjoc.7.120 |
| 49. | McPake, C. B.; Sandford, G. Org. Process Res. Dev. 2012, 16, 844–851. doi:10.1021/op200331s |
| 10. | Baasner, B.; Hagemann, H.; Tatlow, J. C. Houben-Weyl Organofluorine Compounds; Thieme: Stuttgart, 2000; Vol. E10a. |
| 11. | Chambers, R. D. Fluorine in Organic Chemistry; Blackwell: Oxford, 2004. |
| 12. | Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, 2006. doi:10.1002/9780470988589 |
| 13. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 50. | Hutchinson, J.; Sandford, G.; Vaughan, J. F. S. Tetrahedron 1998, 54, 2867–2876. doi:10.1016/S0040-4020(98)83023-8 |
| 7. | Gribble, G. W. Chem. Soc. Rev. 1999, 28, 335–346. doi:10.1039/a900201d |
| 8. | Hall, R. J. New Phytol. 1972, 71, 855–871. doi:10.1111/j.1469-8137.1972.tb01965.x |
| 9. | O'Hagan, D.; Schaffrath, C.; Cobb, S. L.; Hamilton, J. T. G.; Murphy, C. D. Nature 2002, 416, 279. doi:10.1038/416279a |
| 36. | Kim, D.-Y.; Kim, S.-M.; Koh, K.-O.; Mang, J.-Y.; Lee, K.-S. Bull. Korean Chem. Soc. 2003, 24, 1425–1426. doi:10.5012/bkcs.2003.24.10.1425 |
| 37. | Cho, M.-J.; Cho, M.-G.; Huh, S.-C.; Kim, S.-M.; Lee, K.-s.; Koh, K.-O.; Mang, J.-Y.; Kim, D.-Y. Bull. Korean Chem. Soc. 2006, 27, 857–862. doi:10.5012/bkcs.2006.27.6.857 |
| 38. | Kang, S.-H.; Kim, D.-Y. Bull. Korean Chem. Soc. 2009, 30, 1439–1440. doi:10.5012/bkcs.2009.30.7.1439 |
| 39. | Kwon, B. K.; Kim, S. M.; Kim, D. Y. J. Fluorine Chem. 2009, 130, 759–761. doi:10.1016/j.jfluchem.2009.06.002 |
| 40. | Li, H.; Zu, L.; Xie, H.; Wang, W. Synthesis 2009, 1525–1530. doi:10.1055/s-0028-1088124 |
| 2. | Müller, K.; Faeh, C.; Diedrich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 3. | Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Oxford, 2009. |
| 4. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 5. | Isanbor, C.; O'Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011 |
| 6. | Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007 |
| 41. | Sandford, G. J. Fluorine Chem. 2007, 128, 90–104. doi:10.1016/j.jfluchem.2006.10.019 |
| 42. | Chambers, R. D.; Nakano, T.; Parsons, M.; Sandford, G.; Batsanov, A. S.; Howard, J. A. K. J. Fluorine Chem. 2008, 129, 811–816. doi:10.1016/j.jfluchem.2008.04.010 |
| 43. | Chambers, R. D.; Sandford, G.; Trmcic, J.; Okazoe, T. Org. Process Res. Dev. 2008, 12, 339–344. doi:10.1021/op700194r |
| 44. | Chambers, R. D.; Nakano, T.; Okazoe, T.; Sandford, G. J. Fluorine Chem. 2009, 130, 792–798. doi:10.1016/j.jfluchem.2009.06.003 |
| 28. | Miller, T. A.; Sloman, D. L.; Stanton, M. G.; Wilson, K. J.; Witter, D. J. Fluorinated arylamide derivatives. W.O. Patent WO2007087129, Aug 2, 2007. |
| 29. | Close, J.; Heidebrecht, R. W.; Kattar, S.; Miller, T. A.; Sloman, D.; Stanton, M. G.; Tempest, P.; Witter, D. J. Histone deacetylase inhibitors with aryl-pyrazolyl motifs. W.O. Patent WO2007055941, May 18, 2007. |
| 30. | Hubbs, J. L.; Mampreian, D. M.; Methot, J. L.; Miller, T. A.; Otte, K. M.; Siliphaivanh, P.; Sloman, D. L.; Stanton, M. G.; Wilson, K. J.; Witter, D. J. Benzothiophene derivatives. W.O. Patent WO2006115845, Nov 2, 2006. |
| 31. | Bressi, J. C.; Chu, S.; Erickson, P.; Komandla, M.; Kwok, L.; Lawson, J. D.; Stafford, J. A.; Wallace, M. B.; Zhang, Z.; Das, J. cMET inhibitors. W.O. Patent WO2010019899, Feb 18, 2010. |
| 32. | Leonardi, A.; Riva, C.; Tavecchia, P.; Sironi, G. Aldosterone receptor antagonists. W.O. Patent WO2007025780, March 8, 2007. |
| 34. | Brickner, S. J.; Chen, J. M.; Li, Z. B.; Marfat, A.; Mitton-Fry, M. J.; Plotkin, M. A.; Reilly, U. D.; Subramanyam, C.; Zhang, Z.; Robinson, S. Substituted heterocyclic derivatives and their pharmaceutical use and compositions. U.S. Patent US20080280879, Nov 13, 2008. |
| 20. | Chambers, R. D.; Hutchinson, J.; Thomson, J. J. Fluorine Chem. 1996, 78, 165–166. doi:10.1016/0022-1139(96)03422-7 |
| 21. | Chambers, R. D.; Fox, M. A.; Holling, D.; Nakano, T.; Okazoe, T.; Sandford, G. Chem. Eng. Technol. 2005, 28, 344–352. doi:10.1002/ceat.200407123 |
| 22. | Chambers, R. D.; Hutchinson, J. J. Fluorine Chem. 1998, 92, 45–52. doi:10.1016/S0022-1139(98)00254-1 |
| 23. | Müh, T.; Fiedler, P.; Weintritt, H.; Westerkamp, W.; Reinecke, A. Method for producing alpha fluoromalonic acid dialkyl esters. W.O. Patent WO2002016304, Feb 28, 2002. |
| 24. | Günther, A.; Weintritt, H.; Böhm, S. Method for producing a-fluoromalonic acid dialkyl esters. W.O. Patent WO2005019154, March 3, 2005. |
| 25. | Braun, M.; Brosch, C. Production of fluorine compounds. W.O. Patent WO2002060838, Aug 8, 2002. |
| 26. | Ishikawa, N.; Takaoka, A. Chem. Lett. 1981, 10, 107–110. doi:10.1246/cl.1981.107 |
| 27. | Ishikawa, N.; Takaoka, A.; Ibrahim, M. K. J. Fluorine Chem. 1984, 25, 203–212. doi:10.1016/S0022-1139(00)80949-5 |
| 35. | Polla, M. O.; Tottie, L.; Nordén, C.; Linschoten, M.; Müsil, D.; Trumpp-Kallmeyer, S.; Aukrust, I. R.; Ringom, R.; Holm, K. H.; Neset, S. M.; Sandberg, M.; Thurmond, J.; Yu, P.; Hategan, G.; Anderson, H. Bioorg. Med. Chem. 2004, 12, 1151–1175. doi:10.1016/j.bmc.2003.12.039 |
| 16. | Warren, S. Designing Organic Syntheses: A Progrmmed Introduction to the Synthon Approach; John Wiley and Sons: New York, 1978. |
| 17. | Warren, S.; Wyatt, P. Organic Synthesis: The Disconnection Approach, 2nd ed.; John Wiley and Sons: Chichester, 2008. |
| 18. | Wyatt, P.; Warren, S. Organic Synthesis: Strategy and Control, 2nd ed.; John Wiley and Sons: Chichester, 2007. |
| 19. | Corey, E. J.; Cheng, X.-M. The logic of chemical synthesis; John Wiley and Sons: New York, 1995. |
| 15. | Seebach, D. Angew. Chem., Int. Ed. Engl. 1990, 29, 1320–1367. doi:10.1002/anie.199013201 |
| 33. | Bergmann, E. D.; Cohen, S.; Shahak, I. J. Chem. Soc. 1959, 3286–3289. doi:10.1039/jr9590003286 |
| 53. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 54. | Shibata, N.; Ishimaru, T.; Suzuki, E.; Kirk, K. L. J. Org. Chem. 2003, 68, 2494–2497. doi:10.1021/jo026792s |
| 55. | Zoute, L.; Audouard, C.; Plaquevent, J.-C.; Cahard, D. Org. Biomol. Chem. 2003, 1, 1833–1834. doi:10.1039/b303113f |
| 56. | Shibata, N.; Kohno, J.; Takai, K.; Ishimaru, T.; Nakamura, S.; Toru, T.; Kanemasa, S. Angew. Chem., Int. Ed. 2005, 44, 4204–4207. doi:10.1002/anie.200501041 |
| 57. | Hamashima, Y.; Suzuki, N.; Takano, H.; Shimura, Y.; Sodeoka, M. J. Am. Chem. Soc. 2005, 127, 10164–10165. doi:10.1021/ja0513077 |
| 58. | Deng, Q.-H.; Wadepohl, H.; Gade, L. H. Chem.–Eur. J. 2011, 17, 14922–14928. doi:10.1002/chem.201102375 |
| 59. | Li, J.; Cai, Y.; Chen, W.; Liu, X.; Lin, L.; Feng, X. J. Org. Chem. 2012, 77, 9148–9155. doi:10.1021/jo301705t |
| 60. | Wu, L.; Falivene, L.; Drinkel, E.; Grant, S.; Linden, A.; Cavallo, L.; Dorta, R. Angew. Chem., Int. Ed. 2012, 51, 2870–2873. doi:10.1002/anie.201200206 |
| 61. | Ishimaru, T.; Shibata, N.; Horikawa, T.; Yasuda, N.; Nakamura, S.; Toru, T.; Shiro, M. Angew. Chem., Int. Ed. 2008, 47, 4157–4161. doi:10.1002/anie.200800717 |
| 62. | Dou, X.; Lu, Y. Org. Biomol. Chem. 2013, 11, 5217–5221. doi:10.1039/c3ob41267a |
| 51. | Beare, N. A.; Hartwig, J. F. J. Org. Chem. 2002, 67, 541–555. doi:10.1021/jo016226h |
| 52. | Harsanyi, A. M.Sc. Thesis, Synthesis of 3-fluoro-oxindoles and phenyl fluoroacetic acid derivatives; Durham University, 2013, available from http://etheses.dur.ac.uk/6357/. |
| 63. | Hewawasam, P.; Gribkoff, V. K.; Pendri, Y.; Dworetzky, S. I.; Meanwell, N. A.; Martinez, E.; Boissard, C. G.; Post-Munson, D. J.; Trojnacki, J. T.; Yeleswaram, K.; Pajor, L. M.; Knipe, J.; Gao, Q.; Perrone, R.; Starret, J. E. Bioorg. Med. Chem. Lett. 2002, 12, 1023–1026. doi:10.1016/S0960-894X(02)00101-4 |
| 70. | Watanabe, S.; Fujita, T.; Sakamoto, M.; Endo, H.; Kitazume, T. J. Fluorine Chem. 1990, 47, 187–192. doi:10.1016/S0022-1139(00)82371-4 |
| 71. | Kim, K.-Y.; Kim, B. C.; Lee, H. B.; Shin, H. J. Org. Chem. 2008, 73, 8106–8108. doi:10.1021/jo8015659 |
| 72. | Yin, J.; Zarkowsky, D. S.; Thomas, D. W.; Zhao, M. M.; Huffman, M. A. Org. Lett. 2004, 6, 1465–1468. doi:10.1021/ol049672a |
| 64. | Schiefer, I. T.; Abdul-Hay, S.; Wang, H.; Vanni, M.; Qin, Z.; Thatcher, G. R. J. J. Med. Chem. 2011, 54, 2293–2306. doi:10.1021/jm101450p |
| 65. | Zhang, F.; Song, J. Z. Tetrahedron Lett. 2006, 47, 7641–7644. doi:10.1016/j.tetlet.2006.08.057 |
| 66. | Rozen, S.; Hagooly, A.; Harduf, R. J. Org. Chem. 2001, 66, 7464–7468. doi:10.1021/jo010677k |
| 67. | Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1996, 60, 4730–4737. doi:10.1021/jo00120a014 |
| 68. | Cantrell, G. L.; Filler, R. J. Fluorine Chem. 1985, 27, 35–45. doi:10.1016/S0022-1139(00)80895-7 |
| 69. | Bresciani, S.; O’Hagan, D. Tetrahedron Lett. 2010, 51, 5795–5797. doi:10.1016/j.tetlet.2010.08.104 |
| 63. | Hewawasam, P.; Gribkoff, V. K.; Pendri, Y.; Dworetzky, S. I.; Meanwell, N. A.; Martinez, E.; Boissard, C. G.; Post-Munson, D. J.; Trojnacki, J. T.; Yeleswaram, K.; Pajor, L. M.; Knipe, J.; Gao, Q.; Perrone, R.; Starret, J. E. Bioorg. Med. Chem. Lett. 2002, 12, 1023–1026. doi:10.1016/S0960-894X(02)00101-4 |
| 64. | Schiefer, I. T.; Abdul-Hay, S.; Wang, H.; Vanni, M.; Qin, Z.; Thatcher, G. R. J. J. Med. Chem. 2011, 54, 2293–2306. doi:10.1021/jm101450p |
© 2014 Harsanyi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)