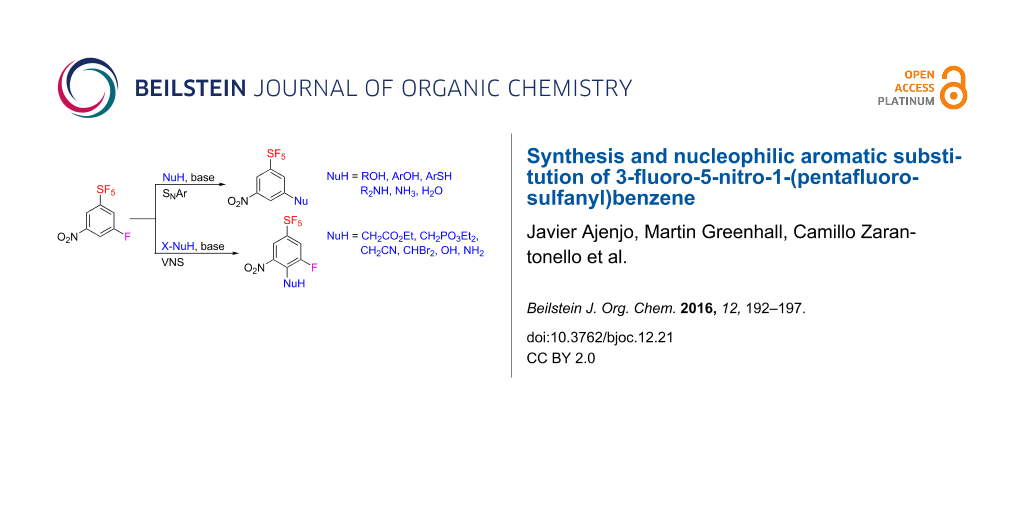
Abstract
3-Fluoro-5-nitro-1-(pentafluorosulfanyl)benzene was prepared by three different ways: as a byproduct of direct fluorination of 1,2-bis(3-nitrophenyl)disulfane, by direct fluorination of 4-nitro-1-(pentafluorosulfanyl)benzene, and by fluorodenitration of 3,5-dinitro-1-(pentafluorosulfanyl)benzene. The title compound was subjected to a nucleophilic aromatic substitution of the fluorine atom with oxygen, sulfur and nitrogen nucleophiles affording novel (pentafluorosulfanyl)benzenes with 3,5-disubstitution pattern. Vicarious nucleophilic substitution of the title compound with carbon, oxygen, and nitrogen nucleophiles provided 3-fluoro-5-nitro-1-(pentafluorosulfanyl)benzenes substituted in position four.

Graphical Abstract
Introduction
Organic compounds with a pentafluorosulfanyl (SF5) group are promising candidates in the development of new agrochemicals, pharmaceuticals and advanced materials [1-3]. This is due to an unusual combination of properties such as high stability [4], lipophilicity [5] and strong electron-withdrawing character [6] similar but more extreme than the trifluoromethyl group. Currently, the limiting factor for a more widespread use of SF5 compounds is the low accessibility of basic building blocks and the lack of understanding their chemical behavior. For aromatic SF5 compounds, two main synthetic approaches exist. The first one is a direct fluorination of nitro-substituted diaryl disulfides leading to 3- or 4-nitro-1-(pentafluorosulfanyl)benzenes [7-10]. This reaction is conducted in tens of kilogram scale in industry. The second method is known as Umemoto’s synthesis and starts with aromatic thiols or diaryl disulfides which are converted to arylsulfur chlorotetrafluorides [11,12]. Fluorination in the second step affords arylsulfur pentafluorides [11,13]. Umemoto’s synthesis does not require handling the elemental fluorine, gives better yields, and displays a wider substrate scope than the direct fluorination method. However, nitro(pentafluorosulfanyl)benzenes are widely-available primary industrial products and starting materials to other SF5-benzenes which were prepared by reduction followed by condensation or diazotization chemistry [8,14-16], SEAr [17], SNAr [18-24], or metal-catalyzed cross-coupling reactions [25-27]. Synthetic methods to novel SF5-containing building blocks are sought after and drive the development of applications of these compounds. In this work, we explore SNAr chemistry of 3-fluoro-5-nitro-1-(pentafluorosulfanyl)benzene which was initially obtained as a minor byproduct of direct fluorination of 1,2-bis(3-nitrophenyl)disulfane but can be also prepared by other methods, and show that substitution for aromatic fluorine or hydrogen atoms provide a range of novel structurally diverse SF5-benzenes.
Results and Discussion
Direct fluorination of 1,2-bis(3-nitrophenyl)disulfane on kilogram scale led to the main product 3-nitro-1-(pentafluorosulfanyl)benzene (1) which was isolated in 39% yield [8] and a byproduct 3-fluoro-5-nitro-1-(pentafluorosulfanyl)benzene (2) in 6% GC yield (2–3% isolated yield, Scheme 1). Compound 2 was isolated from the mixture by distillation; redistillation afforded 2 in 99% purity (detected by GC). However, for investigations of SNAr of 2, a more efficient process for its synthesis was required. With higher excess of fluorine, the 2:1 ratio increased. Therefore, the synthesis of 2 by the direct fluorination of 1 was investigated (Scheme 2 and Figure 1). The reaction progress was monitored by GC analysis. In acetonitrile, the reaction required more than 16 equivalents of F2 to reach maximal conversion of 2 (around 60%). With higher amounts of fluorine gas the conversion of 2 sharply decreased and the amounts of (poly)fluorinated aromatic byproducts 3 and 4 increased (Figure 1, left). The amount of non-volatile material (tar) in the final product mixture (after 20 equiv of F2 added) was 28% by weight as determined by Kugelrohr distillation. When the direct fluorination was performed in anhydrous HF a markedly different result was obtained. To reach maximal conversion of 2 (around 60%), only a 3.6-fold excess of F2 was required (Figure 1, right). However, in this case, higher amounts of byproducts, particularly difluoroderivatives 4 were observed. The amount of tar after the addition of 4.2 equivalents of F2 was only 5%. Purification of compound 2 from 3 and 4 by distillation or column chromatography was not successful. Therefore, for preparative experiments we decided to run the fluorination in MeCN to about 40% conversion and isolated 2 from unreacted 1 by flash chromatography (Scheme 3).
![[1860-5397-12-21-i1]](/bjoc/content/inline/1860-5397-12-21-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Direct fluorination of 1,2-bis(3-nitrophenyl)disulfane.
Scheme 1: Direct fluorination of 1,2-bis(3-nitrophenyl)disulfane.
![[1860-5397-12-21-i2]](/bjoc/content/inline/1860-5397-12-21-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Direct fluorination of 3-nitro-1-(pentafluorosulfanyl)benzene (1).
Scheme 2: Direct fluorination of 3-nitro-1-(pentafluorosulfanyl)benzene (1).
![[1860-5397-12-21-1]](/bjoc/content/figures/1860-5397-12-21-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Conversion vs added fluorine equivalents for the fluorination of 1 in MeCN (left) and anhydrous HF (right).
Figure 1: Conversion vs added fluorine equivalents for the fluorination of 1 in MeCN (left) and anhydrous HF ...
![[1860-5397-12-21-i3]](/bjoc/content/inline/1860-5397-12-21-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparative fluorination of 3-nitro-1-(pentafluorosulfanyl)benzene (1).
Scheme 3: Preparative fluorination of 3-nitro-1-(pentafluorosulfanyl)benzene (1).
Another method for the synthesis of 2 is the fluorodenitration of known 3,5-dinitro-1-(pentafluorosulfanyl)benzene (5) [10,28]. The reaction with TBAF hydrate resulted in clean substitution of only one nitro group for the fluorine atom (Scheme 4).
![[1860-5397-12-21-i4]](/bjoc/content/inline/1860-5397-12-21-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 2 by fluorodenitration of 5.
Scheme 4: Synthesis of 2 by fluorodenitration of 5.
Nucleophilic aromatic substitution of fluorine leading to compounds 3 was investigated (Table 1). With low-boiling alcohols (MeOH, EtOH), the reactions were heated under reflux using the alcohol as a solvent and excess of potassium hydroxide (Table 1, entries 1 and 2). For alcohols with higher boiling points, the reactions were performed with sodium hydride (Table 1, entries 3 and 4). For phenol, thiophenol and dialkylamines, heating with potassium carbonate in DMF gave good results (Table 1, entries 5–9). Finally, the reaction with potassium hydroxide was sluggish even under high temperature (Table 1, entry 10) and for amination, heating with aqueous ammonia solution in DMSO in a pressure vessel was required to form aniline 3k (Table 1, entry 11).
Table 1: SNAr reactions of 2.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-21-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entry | NuH (equiv) | Base (equiv) | Solvent | Temp. (°C) | Time (h) | 3, Yield (%)a |
|---|---|---|---|---|---|---|
| 1 | MeOH (excess) | KOH (5) | MeOH | 80 | 0.5 | 3a, 85 |
| 2 | EtOH (excess) | KOH (3) | EtOH | 80 | 0.6 | 3b, 83 |
| 3 | iPrOH (excess) | NaH (3) | iPrOH | rt | 6 | 3c, 72 |
| 4 | HC≡C-CH2OH (1.5) | NaH (3) | THF | rt | 2 | 3d, 70 |
| 5 | PhOH (1.5) | K2CO3 (3) | DMF | 80 | 3 | 3e, 67 |
| 6 | PhSH (1.5) | K2CO3 (3) | DMF | 90 | 3 | 3f, 46 |
| 7 | Morpholine (3) | K2CO3 (3) | DMF | 85 | 7 | 3g, 63 |
| 8 | Piperidine (3) | K2CO3 (3) | DMF | 85 | 3 | 3h, 51 |
| 9 | Pyrrolidine (3) | K2CO3 (3) | DMF | 85 | 2 | 3i, 67 |
| 10 | ’OH’ | KOH (5) | DMSOb | 135 | 6 | 3j, 33 |
| 11 | ’NH2’ | NH4OHc (2.5) | DMSO | 135 | 5 | 3k, 44 |
aIsolated yield. bDMSO/H2O (2:1, v/v). c28% aqueous ammonia solution.
It is well known that ipso attack of nitroaromatics by nucleophiles (SNAr) is only a secondary process. Under kinetic conditions the aromatic system is initially attacked by a nucleophile in ortho or para position to the nitro group, which was exploited in oxidative nucleophilic substitution for hydrogen reactions (ONSH) with organolithium or magnesium species or in vicarious nucleophilic substitution reactions (VNS) with carbon, oxygen or nitrogen nucleophiles [29]. VNS is a very powerful process for selective alkylation, amination and hydroxylation of nitroaromatics and was reported to proceed efficiently on 1 and its para-isomer [19-21]. In general, the reactions are characterized by short reaction times, low temperatures and an equimolar amount of the nucleophile. The results of VNS reactions with compound 2 are shown in Table 2. With carbon nucleophiles the reactions proceeded in good yields except for diethyl chloromethylphosphonate. A very short reaction time was needed in the reaction with bromoform to avoid decomposition of the tribromomethyl anion to dibromocarbene (Table 2, entry 4). Direct hydroxylation with cumene hydroperoxide required the use of liquid ammonia as a co-solvent (Table 2, entry 5). For direct amination 1,1,1-trimethylhydrazinium iodide was used, which upon deprotonation with strong base provided the nitrogen nucleophile containing the leaving group (Me3N) (Table 2, entry 6). High regioselectivities were observed in all cases except for the cyanomethylation reaction (Table 2, entry 3). In general, regioselectivities in VNS reactions were higher than for analogous reactions of 1 which can be explained by the presence of additional steric hindrance of the fluorine substituent in 2. It is important to note that under the VNS conditions and in the presence of strong nucleophiles and base (t-BuOK), fluorine atoms of 2 and 4 remained intact.
Table 2: VNS reactions of 2.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-21-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | X-NuH (equiv) | Solvent | Temp. (°C) | Time (min) | 4, Yield (%)a | 4:4’b | |
|---|---|---|---|---|---|---|---|
| 1 | Cl-CH2CO2Et (1) | DMF | −30 | 10 | 4a, 71 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-21-i7.svg?max-width=637&scale=1.0)
|
97:3 |
| 2 | Cl-CH2PO3Et2 (1) | DMF | −60 | 10 | 4b, 31 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-21-i8.svg?max-width=637&scale=1.0)
|
97:3c |
| 3 | PhO-CH2CN (1) | DMF | −30 | 10 | 4c, 50 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-21-i9.svg?max-width=637&scale=1.0)
|
87:13 |
| 4 | Br-CHBr2 (1.1) | DMF/THFd | −70 | 2 | 4d, 81 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-21-i10.svg?max-width=637&scale=1.0)
|
>98:2c |
| 5 | PhC(CH3)2O-OH (1) | NH3/THFe | −50 | 15 | 4e, 60 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-21-i11.svg?max-width=637&scale=1.0)
|
96:4 |
| 6f | I− Me3N+-NH2 (1.8) | DMSO | rt | 5 | 4f, 85 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-21-i12.svg?max-width=637&scale=1.0)
|
>98:2 |
aIsolated yield of the major isomer 4. bDetermined by GC–MS of the crude reaction mixture. cDetermined by 19F NMR of the crude reaction mixture. dDMF/THF (7:2, v/v). eNH3/THF (4:1, v/v). fUsing t-BuOK (4 equiv).
Conclusion
In conclusion, 3-fluoro-5-nitro-1-(pentafluorosulfanyl)benzene was prepared by direct fluorination and fluorodenitration pathways. It underwent nucleophilic aromatic substitutions of the fluorine atom with oxygen, sulfur and nitrogen nucleophiles affording novel 3-substituted-5-nitro-1-(pentafluorosulfanyl)benzenes. Regioselective vicarious nucleophilic substitution with carbon, oxygen, and nitrogen nucleophiles afforded 4-substituted-3-fluoro-5-nitro-1-(pentafluorosulfanyl)benzenes.
Supporting Information
Synthesis and characterization of all products, copies of 1H, 13C, and 19F NMR spectra of newly synthesized products.
| Supporting Information File 1: Experimental part and copies of NMR spectra. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u
Return to citation in text: [1] -
Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030
Return to citation in text: [1] -
Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. doi:10.2533/000942904777678172
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072–3076. doi:10.1021/ja00875a007
Return to citation in text: [1] -
Bowden, R. D.; Greenhall, M. P.; Moilliet, J. S.; Thomson, J. The preparation of fluorinated organic compounds. WO Patent WO9705106 A1, Feb 13, 1997.
Chem. Abstr. 1997, 126, 199340.
Return to citation in text: [1] -
Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8
Return to citation in text: [1] [2] [3] -
Chambers, R. D.; Spink, R. C. H. Chem. Commun. 1999, 883–884. doi:10.1039/A901473J
Return to citation in text: [1] -
Sipyagin, A. M.; Bateman, C. P.; Matsev, A. V.; Waterfeld, A.; Jilek, R. E.; Key, C. D.; Szulczewski, G. J.; Thrasher, J. S. J. Fluorine Chem. 2014, 167, 203–210. doi:10.1016/j.jfluchem.2014.07.031
Return to citation in text: [1] [2] -
Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53
Return to citation in text: [1] [2] -
Kanishchev, O. S.; Dolbier, W. R., Jr. Angew. Chem., Int. Ed. 2015, 54, 280–284. doi:10.1002/anie.201409990
Return to citation in text: [1] -
Lummer, K.; Ponomarenko, M. V.; Röschenthaler, G.-V.; Bremer, M.; Beier, P. J. Fluorine Chem. 2014, 157, 79–83. doi:10.1016/j.jfluchem.2013.10.009
Return to citation in text: [1] -
Iakobson, G.; Du, J.; Slawin, A. M. Z.; Beier, P. Beilstein J. Org. Chem. 2015, 11, 1494–1502. doi:10.3762/bjoc.11.162
Return to citation in text: [1] -
Okazaki, T.; Laali, K. K.; Bunge, S. D.; Adas, S. K. Eur. J. Org. Chem. 2014, 1630–1644. doi:10.1002/ejoc.201301538
Return to citation in text: [1] -
Okazaki, T.; Laali, K. K.; Reddy, A. S. J. Fluorine Chem. 2014, 165, 91–95. doi:10.1016/j.jfluchem.2014.06.021
Return to citation in text: [1] -
Frischmuth, A.; Unsinn, A.; Groll, K.; Stadtmüller, H.; Knochel, P. Chem. – Eur. J. 2012, 18, 10234–10238. doi:10.1002/chem.201201485
Return to citation in text: [1] -
Beier, P.; Pastýříková, T.; Vida, N.; Iakobson, G. Org. Lett. 2011, 13, 1466–1469. doi:10.1021/ol2001478
Return to citation in text: [1] -
Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p
Return to citation in text: [1] [2] -
Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011
Return to citation in text: [1] [2] -
Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127
Return to citation in text: [1] [2] -
Vida, N.; Beier, P. J. Fluorine Chem. 2012, 143, 130–134. doi:10.1016/j.jfluchem.2012.04.001
Return to citation in text: [1] -
Beier, P.; Pastýříková, T. Beilstein J. Org. Chem. 2013, 9, 411–416. doi:10.3762/bjoc.9.43
Return to citation in text: [1] -
Sipyagin, A. M.; Bateman, C. P.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2001, 112, 287–295. doi:10.1016/s0022-1139(01)00514-0
Return to citation in text: [1] -
Joliton, A.; Carreira, E. M. Org. Lett. 2013, 15, 5147–5149. doi:10.1021/ol4025666
Return to citation in text: [1] -
Wang, C.; Yu, Y.-B.; Fan, S.; Zhang, X. Org. Lett. 2013, 15, 5004–5007. doi:10.1021/ol4023326
Return to citation in text: [1] -
Joliton, A.; Carreira, E. M. Synlett 2015, 26, 737–740. doi:10.1055/s-0034-1379981
Return to citation in text: [1] -
Umemoto, T.; Chika, J. Processes for Preparing 1,3-Dinitro-5-(Pentafluorosulfanyl)Benzene and its Intermediates. U.S. Patent US2011301382 A1, Dec 8, 2011.
Return to citation in text: [1] -
Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, 2013.
Return to citation in text: [1]
| 1. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 2. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 3. | Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. doi:10.2533/000942904777678172 |
| 7. |
Bowden, R. D.; Greenhall, M. P.; Moilliet, J. S.; Thomson, J. The preparation of fluorinated organic compounds. WO Patent WO9705106 A1, Feb 13, 1997.
Chem. Abstr. 1997, 126, 199340. |
| 8. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8 |
| 9. | Chambers, R. D.; Spink, R. C. H. Chem. Commun. 1999, 883–884. doi:10.1039/A901473J |
| 10. | Sipyagin, A. M.; Bateman, C. P.; Matsev, A. V.; Waterfeld, A.; Jilek, R. E.; Key, C. D.; Szulczewski, G. J.; Thrasher, J. S. J. Fluorine Chem. 2014, 167, 203–210. doi:10.1016/j.jfluchem.2014.07.031 |
| 19. | Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p |
| 20. | Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011 |
| 21. | Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127 |
| 5. | Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003 |
| 10. | Sipyagin, A. M.; Bateman, C. P.; Matsev, A. V.; Waterfeld, A.; Jilek, R. E.; Key, C. D.; Szulczewski, G. J.; Thrasher, J. S. J. Fluorine Chem. 2014, 167, 203–210. doi:10.1016/j.jfluchem.2014.07.031 |
| 28. | Umemoto, T.; Chika, J. Processes for Preparing 1,3-Dinitro-5-(Pentafluorosulfanyl)Benzene and its Intermediates. U.S. Patent US2011301382 A1, Dec 8, 2011. |
| 29. | Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, 2013. |
| 17. | Frischmuth, A.; Unsinn, A.; Groll, K.; Stadtmüller, H.; Knochel, P. Chem. – Eur. J. 2012, 18, 10234–10238. doi:10.1002/chem.201201485 |
| 25. | Joliton, A.; Carreira, E. M. Org. Lett. 2013, 15, 5147–5149. doi:10.1021/ol4025666 |
| 26. | Wang, C.; Yu, Y.-B.; Fan, S.; Zhang, X. Org. Lett. 2013, 15, 5004–5007. doi:10.1021/ol4023326 |
| 27. | Joliton, A.; Carreira, E. M. Synlett 2015, 26, 737–740. doi:10.1055/s-0034-1379981 |
| 8. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8 |
| 14. | Iakobson, G.; Du, J.; Slawin, A. M. Z.; Beier, P. Beilstein J. Org. Chem. 2015, 11, 1494–1502. doi:10.3762/bjoc.11.162 |
| 15. | Okazaki, T.; Laali, K. K.; Bunge, S. D.; Adas, S. K. Eur. J. Org. Chem. 2014, 1630–1644. doi:10.1002/ejoc.201301538 |
| 16. | Okazaki, T.; Laali, K. K.; Reddy, A. S. J. Fluorine Chem. 2014, 165, 91–95. doi:10.1016/j.jfluchem.2014.06.021 |
| 8. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8 |
| 11. | Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53 |
| 13. | Lummer, K.; Ponomarenko, M. V.; Röschenthaler, G.-V.; Bremer, M.; Beier, P. J. Fluorine Chem. 2014, 157, 79–83. doi:10.1016/j.jfluchem.2013.10.009 |
| 11. | Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53 |
| 12. | Kanishchev, O. S.; Dolbier, W. R., Jr. Angew. Chem., Int. Ed. 2015, 54, 280–284. doi:10.1002/anie.201409990 |
| 18. | Beier, P.; Pastýříková, T.; Vida, N.; Iakobson, G. Org. Lett. 2011, 13, 1466–1469. doi:10.1021/ol2001478 |
| 19. | Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p |
| 20. | Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011 |
| 21. | Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127 |
| 22. | Vida, N.; Beier, P. J. Fluorine Chem. 2012, 143, 130–134. doi:10.1016/j.jfluchem.2012.04.001 |
| 23. | Beier, P.; Pastýříková, T. Beilstein J. Org. Chem. 2013, 9, 411–416. doi:10.3762/bjoc.9.43 |
| 24. | Sipyagin, A. M.; Bateman, C. P.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2001, 112, 287–295. doi:10.1016/s0022-1139(01)00514-0 |
© 2016 Ajenjo et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)