Abstract
The “one-pot” synthesis of several homochiral macrocycles has been achieved by using π-electron-rich, electron-deficient or extended aromatic dicarboxylic acids in combination with an axially-chiral dibenzylic alcohol, derived from enantiomerically-pure BINOL. Two series of cyclic adducts with average molecular D2 and D3 molecular symmetries, respectively, have been isolated in pure forms. Their yields and selectivities deviate substantially from statistical distributions. NMR and CD spectroscopic methods are efficient and functional in order to highlight the variability of shapes of the covalent macrocyclic frameworks. The larger D3 cyclic adducts exhibit recognition properties towards C60 in toluene solutions (up to log Ka = 3.2) with variable stoichiometries and variable intensities of the charge-tranfer band upon complexation.

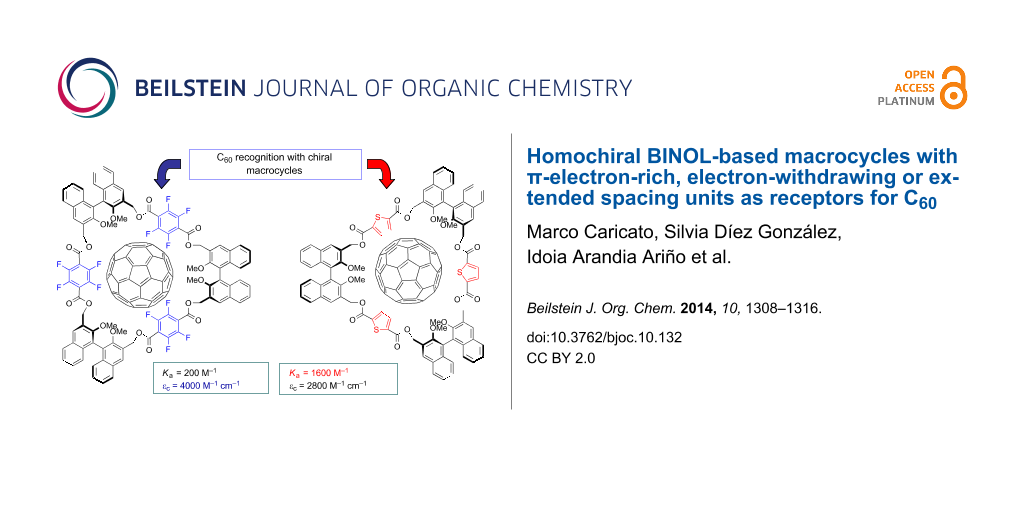
Graphical Abstract
Introduction
Shape persistent macrocycles are carbon-based nanomaterials and are more and more in demand. They enrich the molecular toolkit available to a variety of disciplines, e.g., supramolecular chemistry and materials science [1-4]. Shape persistency properties are traditionally sought after for enhancing the recognition toward suitable guests [5-10], and for the self-assembly, of stable organic nanotubes from the macrocyclic structures as molecular building blocks [11]. Cyclic peptides [12-14], phenylacetylene macrocycles [15], amide-containing macrocycles [16], and urea-based structures [17] have all been exploited to develop the nanotube concept.
Efficient supramolecular receptors for C60 and higher fullerenes have been already reported in the literature and research in this area is still very active [18,19]. Jasti and co-workers have demonstrated how cyclo-p-phenylenes of suitable size are able to form very stable complexes with C60 [20]. Aida and co-workers have reported on π-electron rich, porphyrin-based cyclic structures, which are able to selectively recognize C60 [21]. In subsequent work, similar porphyrin-based systems have been made chiral by substituting one of the nitrogen atoms, and the enantioselective complexation of chiral higher fullerenes (C84) has been demonstrated [22]. Complexes of C60 and C70 with large, calix-type macrocycles formed by π-electron deficient pyridine aromatic rings bridged by a nitrogen heteroatom, have been reported and characterized in terms of thermodynamic stability by using fluorescence measurements [23]. Martin, Perez et al. have reported on a series of extended TTF units able to form strong complexes with C60 and C70 in organic solutions [24,25].
We have recently reported an efficient protocol for the preparation of several chiral macrocycles incorporating BINOL (1,1′-bi-2-naphthol) units, through the formation of bridging ester functionalities, which ensure chemical inertness for the purposes of supramolecular sensing, recognition, or self-assembly. We have shown their application in the chiroptical sensing of organic or ionic species [26-39]. We have previously reported how some of our systems are able to sense C60 in toluene solutions, and how the recognition behavior is shape selective [28]. In the case of extended [2 + 2] macrocycles, induced CD activity in the characteristic UV–vis absorption bands of C60 showed how the chirality of the macrocyclic units could be transferred to the overall supramolecular ensemble [30].
In this paper, we extend on our previous studies by describing the synthesis of optically-active D2 and D3 macrocycles, whose spacing units are systematically changed in terms of their electronic nature, and we report on the recognition behavior towards C60.
Results and Discussion
Design, synthesis and spectroscopic characterization
In our design approach, rigid, aromatic dicarboxylic acid spacers are combined with a BINOL derivative with masked phenol functionalities in the 2,2’-positions and benzylic alcohol functionalities in the 3,3’-positions (diol 1 in Scheme 1). The incorporation of sp3 methylene carbon atoms in the cyclic structure adds a certain degree of conformational freedom to the covalent structure. This feature balances the distortion of the planarity which is inevitably introduced by the binaphthyl units. Enantiopure (R)-1 [40] was used in all cases, in order to achieve homochiral macrocycles. As previously reported in the case of terephthalic acid, both the [2 + 2] and the [3 + 3] macrocycles (compounds 3 and 4 in Scheme 1) were obtained.
![[1860-5397-10-132-i1]](/bjoc/content/inline/1860-5397-10-132-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of macrocycles 3 and 4.
Scheme 1: Synthesis of macrocycles 3 and 4.
The compounds have average molecular D2 and D3 point group symmetries, respectively. Our optimized esterification reaction protocol is carried out at intermediate dilution levels (each reagent 20–25 mM) in CH2Cl2. Several dicarboxylic acids with varying electronic structures and steric demands were tested. In the case of 9,10-anthracenedicarboxylic acid (2e), no cyclic product could be obtained, and only oligomeric, baseline materials were detected. Isolated yields, after column chromatography, are reported in Table 1. The yields and selectivities of isolated products are unusual, considering that the [3 + 3] macrocycles are sometimes formed with similar synthetic efficiency as the [2 + 2] macrocycles (entries 2 and 3 in Table 1). It is very likely that those conformational preferences dominate in this context.
Table 1: Yields of isolated cyclized products.a
| Entry | Diacid precursor | Macrocyle 3 | Macrocycle 4 |
|---|---|---|---|
| 1b | 2a | 18 | 9 |
| 2 | 2b | 6 | 4 |
| 3 | 2c | 8 | 5 |
| 4 | 2d | 18 | 4 |
| 5 | 2e | 0 | 0 |
aIsolated yield after colum chromatography. For conditions, see experimental. bData taken from ref. [28].
The macrocycles were correctly identified by NMR spectroscopy and mass spectrometry (see Supporting Information File 1). The room temperature 1H NMR spectra for all cyclic compounds display only one set of signals for each group of symmetry-related proton resonances, showing that all dynamic processes are fast on the NMR timescale at this temperature (Figure 1 and Figure 2).
![[1860-5397-10-132-1]](/bjoc/content/figures/1860-5397-10-132-1.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectra of macrocycles 3a–d, with key proton resonances for the spacing units and key benzylic and BINOL-based units highlighted with different colors.
Figure 1: 1H NMR spectra of macrocycles 3a–d, with key proton resonances for the spacing units and key benzyl...
![[1860-5397-10-132-2]](/bjoc/content/figures/1860-5397-10-132-2.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectroscopy of macrocycles 4a–d, with proton resonances for the spacing units and key benzylic and BINOL-based units highlighted with color.
Figure 2: 1H NMR spectroscopy of macrocycles 4a–d, with proton resonances for the spacing units and key benzy...
In the case of macrocycles 3, significant differences in the chemical shift of the proton resonances of the methoxy groups (Table 2, from 3.27 ppm to 3.48 ppm) and of the BINOL H-4,4‘ proton resonances (from 8.02 ppm to 8.19 ppm) could be detected. These significant differences tend to cancel out in the case of the larger macrocycles 4, pointing to a more flexible nature of the latter class of macrocycles. In the case of all the D3 macrocycles 4, the CH2 benzylic proton resonances appear as collapsed AB systems at room temperature (Figure 2). They demonstrate a peculiar arrangement for the two diastereotopic methylene protons, substantially different from that of the more rigid D2 symmetrical analogues 3, in which the methylene proton resonances appear as well-defined AB systems. Small amounts (5–10%) of impurities in macrocycles 4 were difficult to remove by flash column chromatography. These byproducts were tentatively characterized as higher oligomers from their NMR pattern.
Table 2: Selected chemical shifts for compounds in CDCl3 (25 °C).a
| Entry | Compound | Binol-H4,4'b | Benzylic CH2 | OCH3 |
|---|---|---|---|---|
| 1c | 3a | 8.19 | 5.63d | 3.48 |
| 2 | 3b | 8.10 | 5.60d | 3.27 |
| 4 | 3c | 8.02 | 5.72d | 3.34 |
| 5 | 3d | 8.15 | 5.67d | 3.44 |
| 6c | 4a | 8.15 | 5.68e | 3.35 |
| 7 | 4b | 8.10 | 5.63e | 3.31 |
| 8 | 4c | 8.13 | 5.79e | 3.35 |
| 9 | 4d | 8.11 | 5.71e | 3.29 |
aAll spectra recorded at 5–10 mM sample concentration. bResonances related to the singlet corresponding to the proton in the 4,4'-positions of the BINOL skeleton. cData taken from ref. [28]. dMultiplicity of the 1H NMR signal: Quartet, AB system. eMultiplicity of the 1H NMR signal: collapsed AB system.
The UV–vis absorption spectra in EtOH of a selection of macrocyclic compounds (3b, 3d, 4b and 4d with π-electron rich and π-electron deficient spacing units, Figure S1, Supporting Information File 1) show the major absorption band centered around 230 nm, typical of the binaphthyl chromophore [41]. Circular dichroism spectroscopy of the same macrocycles show the exciton couplet typical of binaphthyl moieties (Figure 3), corresponding to the maximum absorption band in the UV–vis spectra.
![[1860-5397-10-132-3]](/bjoc/content/figures/1860-5397-10-132-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CD spectra of macrocycles 3b, 3d, 4b, 4d in EtOH (0.5–12 × 10−6 M).
Figure 3: CD spectra of macrocycles 3b, 3d, 4b, 4d in EtOH (0.5–12 × 10−6 M).
Induced CD activity associated with other absorption bands in the UV–vis spectra (particularly intense in the case of 3b and 4b, Figure S1, Supporting Information File 1) could not be detected. The intensity of the low energy component of the couplet is significantly different in the case of the smallest macrocycles (Δε values of −113 for 3d and −326 for 3b). For substituted 2,2'-binaphthol derivatives, it has been reported that the low energy component values are related to variations of the dihedral angle between the naphthyl units due to the steric hindrance of the substituents in the 2,2'-positions [41]. Since compounds 3 possess the same substituent (OMe) in the 2,2'-positions, the variability of these values and thus the variation of the dihedral angle of the binaphthyl units, must depend on the buttressing effects of the neighbouring 3,3’-benzylic ester positions. They, in turn, can derive from the differing macrocyclic structural flexibility or from an equally rigid conformation altering the coupling of the benzylic protons. These data corroborate the NMR data in highlighting that the influence of the differing geometrical shapes of the spacing units on the macrocyclic conformation is strong in the case of the more rigid [2 + 2] macrocycles 3, but it tends to cancel out in the case of the more flexible [3 + 3] macrocycles 4.
Complexation studies
Titrations of a solution of C60 with increasing amounts of macrocycles 3b–d in toluene solutions resulted in no detectable changes in the UV–vis spectra, similarly to what was previously described for the [2 + 2] adduct 3a. In the case of macrocyles 4b and 4d, instead, an enhancement of the absorption band above 400 nm could be readily detected (Figure 4). This behavior is similar to previously reported cases in terms of band shape, involving both cyclic π-electron rich and π-electron deficient substrates [23,25].
![[1860-5397-10-132-4]](/bjoc/content/figures/1860-5397-10-132-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis titration of C60 (1.8 × 10−4 M) in toluene with increasing amounts of macrocycle 4b (top) and 4d (bottom). Inset: titration profiles at 405 nm, and relative best fitting curves obtained with the Hill equation (top) or a 1:1 binding equation (bottom).
Figure 4: UV–vis titration of C60 (1.8 × 10−4 M) in toluene with increasing amounts of macrocycle 4b (top) an...
The calculated thermodynamic binding constants are reported in Table 3. In the case of macrocycle 4d, a 1:1 binding isotherm could be efficiently employed to fit the titration data, strongly indicating that a 1:1 binding behavior between the host and the C60 guest is predominant in solution. The insertion of the π-electron deficient tetrafluoroterephthalic moieties lowers the affinity with the electron acceptor C60 guest, when compared with the reference host 4a (Table 3, entry 1 vs entry 4). This is reasonable as the C60 core is an acceptor of electron density, so that π-electron neutral or rich substrates are more suitable for complexation.
Table 3: Thermodynamic binding constants of the complexation of C60 with macrocycles 4 in toluene (25 °C).a
| Entry | Compound | kab | Hill coefficientc |
|---|---|---|---|
| 1c | 4a | 1100 ± 100 | 1 |
| 2 | 4b | 1600 ± 200 | 1.5 |
| 3 | 4c | No binding | – |
| 4 | 4d | 200 ± 40 | 1 |
aC60 concentration constant at 1.8 × 10−4 M in all cases. The ka values are the average of two independent tirations. bIn M−1. cData taken from ref. [28].
In the case of 4b, an acceptable fitting with the 1:1 binding equation could not be achieved, indicating multiple binding stoichiometries in solution. Data treatment with the Hill equation [42] gave an average binding constant of 1600 M−1, thus confirming a higher affinity for the π-electron rich thiophene-derived spacer units. The Hill coefficient [43,44] suggests the presence of concomitant 1:1 and 1:2 C60:macrocycle complexes in solution.
Even more interestingly, the titration profiles in the insets of Figure 4 display marked differences for the calculated molar absorbivity values of the 4b@C60 and 4d@C60 complexes at saturation (2800 and 4000 M−1 cm−1, respectively). In the case of 4c, no variation in the UV–vis spectra was detected. It is likely that in this case the steric demand of the naphthalene spacing units does not allow for the optimal positioning of the C60 and the formation of a 4c@C60 complex of measurable stability.
Conclusion
We have reported on the synthesis and characterization of novel homochiral macrocycles, built upon resolved 1,1’-binaphthyl scaffolds, which incorporate either π-electron rich, π-electron deficient or π-extended spacing units. The cyclic adducts are obtained in an acceptable yield in a one-pot synthetic procedure, and easily purified by flash column chromatography. NMR and CD spectroscopy give an insight into the conformational properties of these cyclic structures and indicate a more rigid structure for the [2 + 2] adducts, whereas the [3 + 3] adducts are more flexible. The latter are also capable of binding C60 in toluene solutions. Both the thermodynamic strengths and the optical absorptivity coefficients of the complexes in solution give insights into the role of nonspecific host–guest interactions (such as π–π stacking) for the overall stabilization of the complexes. We are currently designing binaphthyl-based hosts for the enantioselective recognition and separation of higher fullerenes and chiral nanotubes. These binaphthyl-based hosts may also be utilized as chiroptical sensors for chiral carbon-based nanomaterials in functional nanochemical environments.
Experimental
General experimental. All commercially available compounds were purchased from commercial sources and used as received. Compounds (R)-1 [40], 3a [28], 4a [28] and p-toluenesulfonic acid 4-dimethylaminopyridinium salt (PTSA-DMAP) [45] were prepared according to literature procedures. THF (Na, benzophenone) and CH2Cl2 (CaH2) were dried before use. Analytical thin-layer chromatography was performed on silica gel, chromophore loaded, and with commercially available plates. Flash chromatography was carried out by using silica gel (pore size 60 Å, 230–400 mesh). 1H and 13C NMR spectra were recorded from solutions in CDCl3 on 200 or 300 MHz spectrometer with the solvent residual proton signal or tetramethylsilane as a standard. The UV–vis spectroscopic studies were recorded by means of commercially-available spectrophotometers. Mass spectra were recorded by using an electrospray ionization instrument. Optical rotations were measured on a polarimeter with a sodium lamp (λ = 589 nm) and are reported as follows: [α]Drt (c = g (100 mL solvent)−1). CD spectroscopy was performed with a spectropolarimeter; spectra were recorded at 25 °C at a scanning speed of 50 nm min−1 and were background corrected. The reported spectra are the instrument-averaged results of four consecutive scans.
General procedure for the preparation of macrocycles 3 and 4. In a manner similar to the procedure described in [28], a solution of DICD (diisopropylcarbodiimide, 3 equivalents vs diol and dicarboxylic acid) in a minimum amount of dry CH2Cl2 is added to a solution of the appropriate dicarboxylic acid (20–25 mM), (R)-1 (20–25 mM), PTSA-DMAP (2 equivalents) in dry CH2Cl2 under stirring and N2. The solution is stirred overnight, and then H2O (10 mL) is added. The aqueous phase is extracted with CH2Cl2 (3×), the organic phase is washed with H2O (3×) and dried (MgSO4). The products are then purified by flash chromatography.
Macrocycles (R,R)-3b and (R,R,R)-4b. From 2,5-thiophenedicarboxylic acid (115 mg, 0.67 mmol, 1 equiv), DICD (312 µL, 2.01 mmol, 3 equiv), (R)-1 (250 mg, 0.67 mmol, 1 equiv), PTSA-DMAP (414 mg, 1.337 mmol, 2 equiv). Purified by flash column chromatography (hexanes/EtOAc 8:2 to 7:3) to yield 3b (21 mg, 6%) and 4b (13 mg, 4%) as white solids. (R,R)-3b. [α]D25 +54 (c 0.0015, CH2Cl2); ESIMS m/z (%): 1043 ([M + Na]+, 100); 1H NMR (CDCl3, 200 MHz, 25 °C) δ 8.10 (s, 4H, binaphthyl), 7.89 (d, 4H, binaphthyl), 7.81 (s, 4H, thiophene), 7.38 (t, 4H, binaphthyl), 7.27 (m, 4H, binaphthyl), 7.16 (d, 4H, binaphthyl), 5.60 (dd, 8H, CH2), 3.27 (s, 12H, -OCH3); 13C NMR (CDCl3, 75 MHz, 25 °C) δ 161.3 (Cq), 155.9 (Cq), 138.2 (Cq), 134.6 (Cq), 133.5 (CH), 132.9 (CH), 130.0 (Cq), 128.3 (CH), 128.2 (Cq), 127.1 (CH), 125.5 (CH), 125.0 (CH), 124.2 (Cq), 64.2 (CH2), 61.4 (CH3). (R,R,R)-4b. [α]D25 +43 (c 0.0015, CH2Cl2); ESIMS m/z (%): 1553 ([M + Na]+, 100); 1H NMR (CDCl3, 200 MHz, 25 °C) δ 8.10 (s, 6H, binaphthyl), 7.95–7.80 (m, 12H, binaphthyl + thiophene), 7.35 (t, 6H, binaphthyl), 7.5–7.1 (m, 12H, binaphthyl), 5.63 (s, 12H, CH2), 3.31 (s, 18H, OCH3); 13C NMR (CDCl3, 75 MHz, 25 °C) δ 161.2 (Cq), 154.9 (Cq), 138.7 (Cq), 134.3 (Cq), 133.3 (CH), 130.8 (CH), 130.1 (Cq), 128.5 (Cq), 128.1 (CH), 126.9 (CH), 125.5 (CH), 125.1 (CH), 124.4 (Cq), 63.6 (CH2), 61.3 (CH3).
Macrocycles (R,R)-3c and (R,R,R)-4c. From 1,4-naphthalenedicarboxylic acid (144 mg, 0.67 mmol, 1 equiv), DICD (312 µL, 2.0 mmol, 3 equiv), (R)-1 (250 mg, 0.67 mmol, 1 equiv), PTSA-DMAP (414 mg, 1.34 mmol, 2 equiv). Purified by flash column chromatography (hexanes/EtOAc 7:3) to yield 3c (30 mg, 8%) and 4b (17 mg, 5%) as white solids. (R,R)-3c. [α]D25 +97 (c 0.0015, CH2Cl2); ESIMS m/z (%): 1131 ([M + Na]+, 100); 1H NMR (CDCl3, 200 MHz, 25 °C) δ 8.80 (m, 4H, naphthalene), 8.22 (s, 4H, naphthalene), 8.02 (s, 4H, binaphthyl), 7.94 (d, 4H, binaphthyl), 7.53 (m, 4H, naphthalene), 7.43 (t, 4H, binaphthyl), 7.27 (m, 4H, binaphthyl), 7.16 (d, 4H, binaphthyl), 5.72 (q, 8H, CH2), 3.34 (s, 12H, OCH3); 13C NMR (CDCl3, 75 MHz, 25 °C) δ 167.0 (Cq), 155.8 (Cq), 134.8 (Cq), 133.1 (CH), 131.9 (Cq), 131.2 (Cq), 130.1 (Cq), 128.6 (Cq), 128.3 (CH), 127.9 (Cq), 127.7 (CH), 127.6 (CH), 127.1 (CH), 125.8 (CH), 125.5 (CH), 125.1 (CH), 124.4 (Cq), 64.1 (CH2), 61.3 (CH3). (R,R,R)-4c. [α]D25 +80 (c 0.0015, CH2Cl2). ESIMS m/z (%): 1687 ([M + Na]+, 100); 1H NMR (CDCl3, 200 MHz, 25 °C) δ 8.90 (m, 6H, naphthalene), 8.19 (s, 6H, naphthalene), 8.13 (s, 6H, binaphthyl); 7.90 (d, 6H, binaphthyl), 7.61 (m, 6H, naphthalene), 7.38 (t, 6H, binaphthyl), 7.20 (m, 12H, binaphthyl), 5.79 (s, 12H, CH2), 3.35 (s, 18H, OCH3); 13C NMR (CDCl3, 75 MHz, 25 °C) δ 166.8 (Cq), 155.1 (Cq), 134.4 (Cq), 131.7 (Cq), 131.4 (Cq), 131.0 (CH), 130.2 (Cq), 128.9 (Cq), 128.2 (CH), 127.9 (CH), 127.7 (CH), 126.9 (CH), 125.9 (CH), 125.5 (CH), 125.1 (CH), 124.3 (Cq), 63.6 (CH2), 61.2 (CH3).
Macrocycles (R,R)-3d and (R,R,R)-4d. From tetrafluoroterephthalic acid (162 mg, 0.68 mmol, 1 equiv), DICD (317 µL, 2.04 mmol, 3 equiv), (R)-1 (255 mg, 0.68 mmol, 1 equiv), PTSA-DMAP (423 mg, 1.36 mmol, 2 equiv). Purified by column chromatography (hexanes/EtOAc 8:2) to yield 3d (71 mg, 18%), 4d (10 mg, 4%) and a noticeable quantity of a higher cyclic adduct (8 mg), as white solids. (R,R)-3d. [α]D25 +8.3 (c 0.005, CH2Cl2); ESIMS m/z (%): 1176 ([M + Na]+, 100); 1H NMR (CDCl3, 200 MHz, 25 °C) δ 8.15 (s, 4H, binaphthyl), 7.93 (d, 4H, binaphthyl), 7.43 (t, 4H, binaphthyl), 7.27 (s, 4H, binaphthyl), 7.11 (d, 4H, binaphthyl), 5.67 (dd, 8H, CH2), 3.44 (s, 12H, -OCH3); 13C NMR (CDCl3, 75 MHz) δ 158.7 (Cq), 155.4 (Cq), 144.5 (Cq, dm, J = 255 Hz), 134.9 (Cq), 133.4 (CH), 130.0 (Cq), 128.3 (CH), 127.4 (CH), 127.3 (Cq), 125.4 (CH), 125.4 (Cq), 125.2 (CH), 118.0 (Cq, m), 65.4 (CH2), 61.1 (CH3). (R,R,R)-4d. [α]D25 +4.8 (c 0.005, CH2Cl2); ESIMS m/z (%): 1752 ([M + Na]+, 100); 1H NMR (CDCl3, 200 MHz, 25°C) δ 8.11 (s, 6H, binaphthyl), 7.92 (d, 6H, binaphthyl), 7.43 (t, 6H, binaphthyl), 7.27 (m, 6H, binaphthyl), 7.17 (d, 6H, binaphthyl), 5.71 (s, 12H, CH2), 3.29 (s, 18H, -OCH3); 13C NMR (CDCl3, 75 MHz) δ 158.7 (Cq), 154.9 (Cq), 144.3 (Cq, J = 263 Hz), 134.6 (Cq), 131.5 (CH), 130.1 (Cq), 128.3 (CH), 127.6 (CH), 127.2 (Cq), 125.5 (CH), 125.2 (CH), 124.1 (Cq), 117.6 (Cq, m), 64.9 (CH2), 61.0 (CH3).
UV–vis titrations. As described in [28], the titration experiments were conducted as follows: to a stock solution of C60 (solution A) in toluene were added several aliquots of the host (solution B) in toluene. Solution B is formed by the ligand at a higher concentration dissolved in solution A, so that the guest always remains at the same, constant concentration. In the case of a 1:1 binding isotherm (Figure 4, bottom), by employing a nonlinear fitting curve program, the plot of A against the macrocycle concentration x was fitted by Equation 1, thus affording the value of the association constant Ka and of the molar absorptivity of the complex εc:
![[1860-5397-10-132-i2]](/bjoc/content/inline/1860-5397-10-132-i2.svg?max-width=590&scale=1.18182)
where A is the measured absorbance, x is the total concentration of titrant added, εc is the molar absorptivity of the complex, εs is the molar absorbivity of the substrate at the desired wavelength, which could be directly determined, C is the total concentration of the titrate (which is a constant quantity), and Ka is the association constant for the 1:1 complex [28].
The data for titrations of 4b with C60 (Figure 4, top) were fitted to a general form of the Hill equation:
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-132-i4.svg?max-width=637&scale=1.18182)
which can be conveniently rewritten in:
![[1860-5397-10-132-i3]](/bjoc/content/inline/1860-5397-10-132-i3.svg?max-width=590&scale=1.18182)
Equation 2 could be fitted employing a nonlinear fitting program according to the general equation: f(x) = a·xb/(cb + xb), obtaining values of a = ΔAbsmax, b = n (the Hill coefficient), c = 1/Ka.
Supporting Information
| Supporting Information File 1: UV spectra for selected macrocycles, additional NMR and MS spectra for all newly synthesized macrocyles. | ||
| Format: PDF | Size: 937.2 KB | Download |
References
-
Grave, C.; Schlüter, A. D. Eur. J. Org. Chem. 2002, 3075–3098. doi:10.1002/1099-0690(200209)2002:18<3075::AID-EJOC3075>3.0.CO;2-3
Return to citation in text: [1] -
Yamaguchi, Y.; Yoshida, Z.-i. Chem.–Eur. J. 2003, 9, 5430–5440. doi:10.1002/chem.200305099
Return to citation in text: [1] -
Höger, S. Chem.–Eur. J. 2004, 10, 1320–1329. doi:10.1002/chem.200305496
Return to citation in text: [1] -
Zhang, W.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 45, 4416–4439. doi:10.1002/anie.200503988
Return to citation in text: [1] -
Hua, Y.; Ramabhadran, R. O.; Karty, J. A.; Raghavachari, K.; Flood, A. H. Chem. Commun. 2011, 47, 5979–5981. doi:10.1039/c1cc10428d
Return to citation in text: [1] -
Pasini, D. Molecules 2013, 18, 9512–9530. doi:10.3390/molecules18089512
Return to citation in text: [1] -
Droz, A. S.; Diederich, F. J. Chem. Soc., Perkin Trans. 1 2000, 4224–4226. doi:10.1039/b007706m
Return to citation in text: [1] -
Droz, A. S.; Neidlein, U.; Anderson, S.; Seiler, P.; Diederich, F. Helv. Chim. Acta 2001, 84, 2243–2289. doi:10.1002/1522-2675(20010815)84:8<2243::AID-HLCA2243>3.0.CO;2-G
Return to citation in text: [1] -
Yus, M. Arene-catalyzed lithiation. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; John Wiley and Sons: Chichester, U.K., 2004; pp 647–748.
Return to citation in text: [1] -
Campbell, K.; Tykwinski, R. R. Chiral Carbon-rich Macrocycles and Cyclophanes. In Carbon-Rich Compounds: From Molecules to Materials; Haley, M. M.; Tykwinski, R. R., Eds.; Wiley-VCH: Weinheim, 2006; pp 229–294.
Return to citation in text: [1] -
Pasini, D.; Ricci, M. Curr. Org. Synth. 2007, 4, 59–80. doi:10.2174/157017907779981606
Return to citation in text: [1] -
Ghadiri, M. R.; Granja, J. R.; Buehler, L. K. Nature 1994, 369, 301–304. doi:10.1038/369301a0
Return to citation in text: [1] -
Leclair, S.; Baillargeon, P.; Skouta, R.; Gauthier, D.; Zhao, Y.; Dory, Y. L. Angew. Chem., Int. Ed. 2004, 43, 349–353. doi:10.1002/anie.200352259
Return to citation in text: [1] -
Fischer, L.; Decossas, M.; Briand, J.-P.; Didierjean, C.; Guichard, G. Angew. Chem., Int. Ed. 2009, 48, 1625–1628. doi:10.1002/anie.200804019
Return to citation in text: [1] -
Sakamoto, J.; Schlüter, A. D. Eur. J. Org. Chem. 2007, 2700–2712. doi:10.1002/ejoc.200700118
Return to citation in text: [1] -
Gong, B. Acc. Chem. Res. 2008, 41, 1376–1386. doi:10.1021/ar700266f
Return to citation in text: [1] -
Xu, Y.; Smith, M. D.; Geer, M. F.; Pellechia, P. J.; Brown, J. C.; Wibowo, A. C.; Shimizu, L. S. J. Am. Chem. Soc. 2010, 132, 5334–5335. doi:10.1021/ja9107066
Return to citation in text: [1] -
Pérez, E. M.; Martin, N. Org. Biomol. Chem. 2012, 10, 3577–3583. doi:10.1039/c2ob07159b
Return to citation in text: [1] -
Canevet, D.; Pérez, E. M.; Martin, N. Angew. Chem., Int. Ed. 2011, 50, 9248–9259. doi:10.1002/anie.201101297
Return to citation in text: [1] -
Xia, J.; Bacon, J. W.; Jasti, R. Chem. Sci. 2012, 3, 3018–3021. doi:10.1039/c2sc20719b
Return to citation in text: [1] -
Tashiro, K.; Aida, T.; Zheng, J.-Y.; Kinbara, K.; Saigo, K.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 1999, 121, 9477–9478. doi:10.1021/ja992416m
Return to citation in text: [1] -
Shoji, Y.; Tashiro, K.; Aida, T. J. Am. Chem. Soc. 2006, 128, 10690–10691. doi:10.1021/ja063828f
Return to citation in text: [1] -
Liu, S.-Q.; Wang, D.-X.; Zheng, Q.-Y.; Wang, M.-X. Chem. Commun. 2007, 3856–3858. doi:10.1039/b705595a
Return to citation in text: [1] [2] -
Canevet, D.; Gallego, M.; Isla, H.; de Juan, A.; Pérez, E. M.; Martin, N. J. Am. Chem. Soc. 2011, 133, 3184–3190. doi:10.1021/ja111072j
Return to citation in text: [1] -
Cao, R., Jr.; Isla, H.; Cao, R.; Pérez, E. M.; Martin, N. Chem. Sci. 2011, 2, 1384–1388. doi:10.1039/C1SC00179E
Return to citation in text: [1] [2] -
Moletti, A.; Coluccini, C.; Pasini, D.; Taglietti, A. Dalton Trans. 2007, 1588–1592. doi:10.1039/b700059f
Return to citation in text: [1] -
Coluccini, C.; Castelluccio, A.; Pasini, D. J. Org. Chem. 2008, 73, 4237–4240. doi:10.1021/jo800315s
Return to citation in text: [1] -
Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Coluccini, C.; Mazzanti, A.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1807–1815. doi:10.1039/b924400j
Return to citation in text: [1] -
Caricato, M.; Coluccini, C.; Dondi, D.; Vander Griend, D. A.; Pasini, D. Org. Biomol. Chem. 2010, 8, 3272–3280. doi:10.1039/c004379f
Return to citation in text: [1] [2] -
Colombo, S.; Coluccini, C.; Caricato, M.; Gargiulli, C.; Gattuso, G.; Pasini, D. Tetrahedron 2010, 66, 4206–4211. doi:10.1016/j.tet.2010.03.102
Return to citation in text: [1] -
Caricato, M.; Olmo, A.; Gargiulli, C.; Gattuso, G.; Pasini, D. Tetrahedron 2012, 68, 7861–7866. doi:10.1016/j.tet.2012.07.038
Return to citation in text: [1] -
Boiocchi, M.; Bonizzoni, M.; Moletti, A.; Pasini, D.; Taglietti, A. New J. Chem. 2007, 31, 352–356. doi:10.1039/b616492g
Return to citation in text: [1] -
Bencini, A.; Coluccini, C.; Garau, A.; Giorgi, C.; Lippolis, V.; Messori, L.; Pasini, D.; Puccioni, S. Chem. Commun. 2012, 48, 10428–10430. doi:10.1039/c2cc35383k
Return to citation in text: [1] -
Caricato, M.; Leza, N. J.; Gargiulli, C.; Gattuso, G.; Dondi, D.; Pasini, D. Beilstein J. Org. Chem. 2012, 8, 967–976. doi:10.3762/bjoc.8.109
Return to citation in text: [1] -
Caricato, M.; Coluccini, C.; Vander Griend, D. A.; Forni, A.; Pasini, D. New J. Chem. 2013, 37, 2792–2799. doi:10.1039/C3NJ00466J
Return to citation in text: [1] -
Caricato, M.; Leza, N. J.; Roy, K.; Dondi, D.; Gattuso, G.; Shimizu, L. S.; Vander Griend, D. A.; Pasini, D. Eur. J. Org. Chem. 2013, 6078–6083. doi:10.1002/ejoc.201300884
Return to citation in text: [1] -
Caricato, M.; Sharma, A. K.; Coluccini, C.; Pasini, D. Nanoscale 2014. doi:10.1039/C4NR00801D
Return to citation in text: [1] -
Asakawa, M.; Ashton, P. R.; Boyd, S. E.; Brown, C. L.; Menzer, S.; Pasini, D.; Stoddart, J. F.; Tolley, M. S.; White, A. J. P.; Williams, D. J.; Wyatt, P. G. Chem.–Eur. J. 1997, 3, 463–481. doi:10.1002/chem.19970030319
Return to citation in text: [1] -
Stock, H. T.; Kellogg, R. M. J. Org. Chem. 1996, 61, 3093–3105. doi:10.1021/jo952107o
Return to citation in text: [1] [2] -
Rosini, C.; Superchi, S.; Peerlings, H. W. I.; Meijer, E. W. Eur. J. Org. Chem. 2000, 61–71. doi:10.1002/(SICI)1099-0690(200001)2000:1<61::AID-EJOC61>3.0.CO;2-I
Return to citation in text: [1] [2] -
Baudry, Y.; Bollot, G.; Gorteau, V.; Litvinchuk, S.; Mareda, J.; Nishihara, M.; Pasini, D.; Perret, F.; Ronan, D.; Sakai, N.; Shah, M. R.; Som, A.; Sordé, N.; Talukdar, P.; Tran, D.-H.; Matile, S. Adv. Funct. Mater. 2006, 16, 169–179. doi:10.1002/adfm.200500198
Return to citation in text: [1] -
Ercolani, G. J. Am. Chem. Soc. 2003, 125, 16097–16103. doi:10.1021/ja038396c
Return to citation in text: [1] -
Hamacek, J.; Piguet, C. J. Phys. Chem. B 2006, 110, 7783–7792. doi:10.1021/jp056932c
Return to citation in text: [1] -
Moore, S.; Stupp, S. I. Macromolecules 1990, 23, 65–70. doi:10.1021/ma00203a013
Return to citation in text: [1]
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 45. | Moore, S.; Stupp, S. I. Macromolecules 1990, 23, 65–70. doi:10.1021/ma00203a013 |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 1. | Grave, C.; Schlüter, A. D. Eur. J. Org. Chem. 2002, 3075–3098. doi:10.1002/1099-0690(200209)2002:18<3075::AID-EJOC3075>3.0.CO;2-3 |
| 2. | Yamaguchi, Y.; Yoshida, Z.-i. Chem.–Eur. J. 2003, 9, 5430–5440. doi:10.1002/chem.200305099 |
| 3. | Höger, S. Chem.–Eur. J. 2004, 10, 1320–1329. doi:10.1002/chem.200305496 |
| 4. | Zhang, W.; Moore, J. S. Angew. Chem., Int. Ed. 2006, 45, 4416–4439. doi:10.1002/anie.200503988 |
| 15. | Sakamoto, J.; Schlüter, A. D. Eur. J. Org. Chem. 2007, 2700–2712. doi:10.1002/ejoc.200700118 |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 12. | Ghadiri, M. R.; Granja, J. R.; Buehler, L. K. Nature 1994, 369, 301–304. doi:10.1038/369301a0 |
| 13. | Leclair, S.; Baillargeon, P.; Skouta, R.; Gauthier, D.; Zhao, Y.; Dory, Y. L. Angew. Chem., Int. Ed. 2004, 43, 349–353. doi:10.1002/anie.200352259 |
| 14. | Fischer, L.; Decossas, M.; Briand, J.-P.; Didierjean, C.; Guichard, G. Angew. Chem., Int. Ed. 2009, 48, 1625–1628. doi:10.1002/anie.200804019 |
| 30. | Caricato, M.; Coluccini, C.; Dondi, D.; Vander Griend, D. A.; Pasini, D. Org. Biomol. Chem. 2010, 8, 3272–3280. doi:10.1039/c004379f |
| 11. | Pasini, D.; Ricci, M. Curr. Org. Synth. 2007, 4, 59–80. doi:10.2174/157017907779981606 |
| 24. | Canevet, D.; Gallego, M.; Isla, H.; de Juan, A.; Pérez, E. M.; Martin, N. J. Am. Chem. Soc. 2011, 133, 3184–3190. doi:10.1021/ja111072j |
| 25. | Cao, R., Jr.; Isla, H.; Cao, R.; Pérez, E. M.; Martin, N. Chem. Sci. 2011, 2, 1384–1388. doi:10.1039/C1SC00179E |
| 5. | Hua, Y.; Ramabhadran, R. O.; Karty, J. A.; Raghavachari, K.; Flood, A. H. Chem. Commun. 2011, 47, 5979–5981. doi:10.1039/c1cc10428d |
| 6. | Pasini, D. Molecules 2013, 18, 9512–9530. doi:10.3390/molecules18089512 |
| 7. | Droz, A. S.; Diederich, F. J. Chem. Soc., Perkin Trans. 1 2000, 4224–4226. doi:10.1039/b007706m |
| 8. | Droz, A. S.; Neidlein, U.; Anderson, S.; Seiler, P.; Diederich, F. Helv. Chim. Acta 2001, 84, 2243–2289. doi:10.1002/1522-2675(20010815)84:8<2243::AID-HLCA2243>3.0.CO;2-G |
| 9. | Yus, M. Arene-catalyzed lithiation. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; John Wiley and Sons: Chichester, U.K., 2004; pp 647–748. |
| 10. | Campbell, K.; Tykwinski, R. R. Chiral Carbon-rich Macrocycles and Cyclophanes. In Carbon-Rich Compounds: From Molecules to Materials; Haley, M. M.; Tykwinski, R. R., Eds.; Wiley-VCH: Weinheim, 2006; pp 229–294. |
| 26. | Moletti, A.; Coluccini, C.; Pasini, D.; Taglietti, A. Dalton Trans. 2007, 1588–1592. doi:10.1039/b700059f |
| 27. | Coluccini, C.; Castelluccio, A.; Pasini, D. J. Org. Chem. 2008, 73, 4237–4240. doi:10.1021/jo800315s |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 29. | Coluccini, C.; Mazzanti, A.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1807–1815. doi:10.1039/b924400j |
| 30. | Caricato, M.; Coluccini, C.; Dondi, D.; Vander Griend, D. A.; Pasini, D. Org. Biomol. Chem. 2010, 8, 3272–3280. doi:10.1039/c004379f |
| 31. | Colombo, S.; Coluccini, C.; Caricato, M.; Gargiulli, C.; Gattuso, G.; Pasini, D. Tetrahedron 2010, 66, 4206–4211. doi:10.1016/j.tet.2010.03.102 |
| 32. | Caricato, M.; Olmo, A.; Gargiulli, C.; Gattuso, G.; Pasini, D. Tetrahedron 2012, 68, 7861–7866. doi:10.1016/j.tet.2012.07.038 |
| 33. | Boiocchi, M.; Bonizzoni, M.; Moletti, A.; Pasini, D.; Taglietti, A. New J. Chem. 2007, 31, 352–356. doi:10.1039/b616492g |
| 34. | Bencini, A.; Coluccini, C.; Garau, A.; Giorgi, C.; Lippolis, V.; Messori, L.; Pasini, D.; Puccioni, S. Chem. Commun. 2012, 48, 10428–10430. doi:10.1039/c2cc35383k |
| 35. | Caricato, M.; Leza, N. J.; Gargiulli, C.; Gattuso, G.; Dondi, D.; Pasini, D. Beilstein J. Org. Chem. 2012, 8, 967–976. doi:10.3762/bjoc.8.109 |
| 36. | Caricato, M.; Coluccini, C.; Vander Griend, D. A.; Forni, A.; Pasini, D. New J. Chem. 2013, 37, 2792–2799. doi:10.1039/C3NJ00466J |
| 37. | Caricato, M.; Leza, N. J.; Roy, K.; Dondi, D.; Gattuso, G.; Shimizu, L. S.; Vander Griend, D. A.; Pasini, D. Eur. J. Org. Chem. 2013, 6078–6083. doi:10.1002/ejoc.201300884 |
| 38. | Caricato, M.; Sharma, A. K.; Coluccini, C.; Pasini, D. Nanoscale 2014. doi:10.1039/C4NR00801D |
| 39. | Asakawa, M.; Ashton, P. R.; Boyd, S. E.; Brown, C. L.; Menzer, S.; Pasini, D.; Stoddart, J. F.; Tolley, M. S.; White, A. J. P.; Williams, D. J.; Wyatt, P. G. Chem.–Eur. J. 1997, 3, 463–481. doi:10.1002/chem.19970030319 |
| 20. | Xia, J.; Bacon, J. W.; Jasti, R. Chem. Sci. 2012, 3, 3018–3021. doi:10.1039/c2sc20719b |
| 22. | Shoji, Y.; Tashiro, K.; Aida, T. J. Am. Chem. Soc. 2006, 128, 10690–10691. doi:10.1021/ja063828f |
| 18. | Pérez, E. M.; Martin, N. Org. Biomol. Chem. 2012, 10, 3577–3583. doi:10.1039/c2ob07159b |
| 19. | Canevet, D.; Pérez, E. M.; Martin, N. Angew. Chem., Int. Ed. 2011, 50, 9248–9259. doi:10.1002/anie.201101297 |
| 23. | Liu, S.-Q.; Wang, D.-X.; Zheng, Q.-Y.; Wang, M.-X. Chem. Commun. 2007, 3856–3858. doi:10.1039/b705595a |
| 17. | Xu, Y.; Smith, M. D.; Geer, M. F.; Pellechia, P. J.; Brown, J. C.; Wibowo, A. C.; Shimizu, L. S. J. Am. Chem. Soc. 2010, 132, 5334–5335. doi:10.1021/ja9107066 |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 21. | Tashiro, K.; Aida, T.; Zheng, J.-Y.; Kinbara, K.; Saigo, K.; Sakamoto, S.; Yamaguchi, K. J. Am. Chem. Soc. 1999, 121, 9477–9478. doi:10.1021/ja992416m |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 40. | Stock, H. T.; Kellogg, R. M. J. Org. Chem. 1996, 61, 3093–3105. doi:10.1021/jo952107o |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 40. | Stock, H. T.; Kellogg, R. M. J. Org. Chem. 1996, 61, 3093–3105. doi:10.1021/jo952107o |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 42. | Baudry, Y.; Bollot, G.; Gorteau, V.; Litvinchuk, S.; Mareda, J.; Nishihara, M.; Pasini, D.; Perret, F.; Ronan, D.; Sakai, N.; Shah, M. R.; Som, A.; Sordé, N.; Talukdar, P.; Tran, D.-H.; Matile, S. Adv. Funct. Mater. 2006, 16, 169–179. doi:10.1002/adfm.200500198 |
| 43. | Ercolani, G. J. Am. Chem. Soc. 2003, 125, 16097–16103. doi:10.1021/ja038396c |
| 44. | Hamacek, J.; Piguet, C. J. Phys. Chem. B 2006, 110, 7783–7792. doi:10.1021/jp056932c |
| 23. | Liu, S.-Q.; Wang, D.-X.; Zheng, Q.-Y.; Wang, M.-X. Chem. Commun. 2007, 3856–3858. doi:10.1039/b705595a |
| 25. | Cao, R., Jr.; Isla, H.; Cao, R.; Pérez, E. M.; Martin, N. Chem. Sci. 2011, 2, 1384–1388. doi:10.1039/C1SC00179E |
| 28. | Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Org. Biomol. Chem. 2010, 8, 1640–1649. doi:10.1039/b920867d |
| 41. | Rosini, C.; Superchi, S.; Peerlings, H. W. I.; Meijer, E. W. Eur. J. Org. Chem. 2000, 61–71. doi:10.1002/(SICI)1099-0690(200001)2000:1<61::AID-EJOC61>3.0.CO;2-I |
| 41. | Rosini, C.; Superchi, S.; Peerlings, H. W. I.; Meijer, E. W. Eur. J. Org. Chem. 2000, 61–71. doi:10.1002/(SICI)1099-0690(200001)2000:1<61::AID-EJOC61>3.0.CO;2-I |
© 2014 Caricato et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)