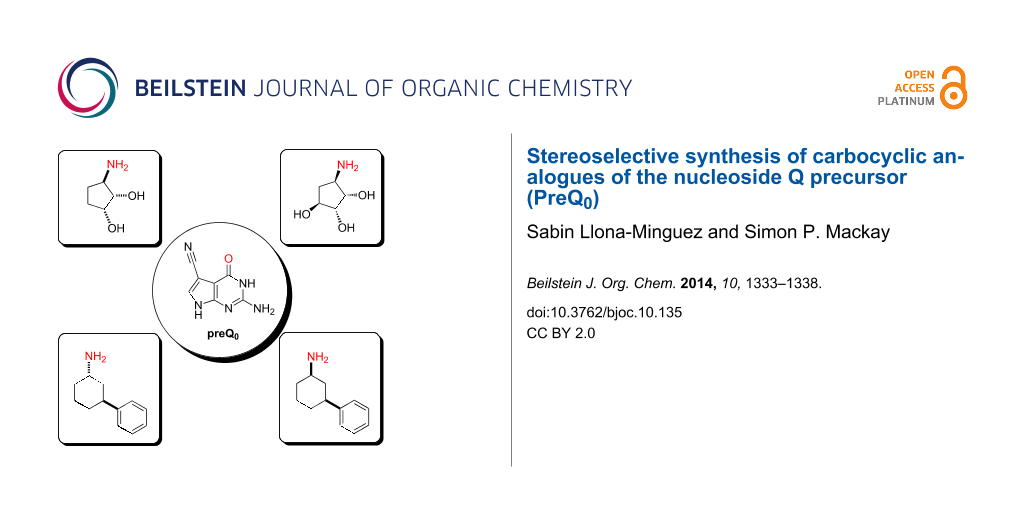
Abstract
A convergent and stereoselective synthesis of chiral cyclopentyl- and cyclohexylamine derivatives of nucleoside Q precursor (PreQ0) has been accomplished. This synthetic route allows for an efficient preparation of 4-substituted analogues with interesting three-dimensional character, including chiral cyclopentane-1,2-diol and -1,2,3-triol derivatives. This unusual substitution pattern provides a useful starting point for the discovery of novel bioactive molecules.

Graphical Abstract
Introduction
7-Deazapurine (pyrrolo[2,3-d]pyrimidine) nucleosides are commonly found in nature playing a variety of roles such as building blocks of nucleic acids and tRNA, metabolites or antimetabolites [1]. Deazapurine ribonucleosides also show interesting pharmacological profiles including antibacterial, antiviral and anticancer properties [2-4]. Nucleoside Q precursor (PreQ0) 1 is a common precursor in the biosynthesis of queuosine (Q, 2) and archaeosine (G+, 3), two hyper-modified nucleosides present in the tRNA of prokaryote/eukaryote and euryarchaeota, respectively [5,6]. In turn, the biosynthesis of PreQ0 originates from guanosine 5’-triphosphate (GTP, 4) [7] (Figure 1) and involves four steps via a tetrahydropterine intermediate.
![[1860-5397-10-135-1]](/bjoc/content/figures/1860-5397-10-135-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biosynthetic pathway leading to nucleosides queuosine and archaeosine.
Figure 1: Biosynthetic pathway leading to nucleosides queuosine and archaeosine.
The pyrrolo[2,3-d]pyrimidine core is a privileged scaffold for the development of kinase inhibitors; an inspection of the medicinal chemistry literature reveals >200 publications in the field. Additionally, PreQ0 meets all the criteria dictated by the “2-0” rule of kinase-likeness proposed by Aronov et al. [8]. It is likely that compounds derived from PreQ0 display kinase activity.
7-Deazapurine nucleoside chemistry has been the subject of extensive study [1] and several syntheses of the PreQ0 base or ribonucleoside [9-16] and queuosine [17] have been reported in the literature. Despite this long-lasting interest, examples of purine-based nucleosides containing a sugar or carbosugar motif at the 4-position of the heterocyclic core (systematic numbering) are scarce in the chemical literature and the methods available generally lack experimental information, making them unsatisfactory [18-25]. Inspired by the cyclopentane-1,2,3-triol motif present in noraristeromycin 5 (Figure 2), an IκB kinase inhibitor with antiviral and anti-inflammatory activity [26,27], we decided to investigate a synthetic route that would allow for the incorporation of carbocyclic systems with interesting three-dimensional character at the 4-position of PreQ0 as part of our fragment-based kinase inhibitor library generation programme.
![[1860-5397-10-135-2]](/bjoc/content/figures/1860-5397-10-135-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Chemical structure of noraristeromycin.
Figure 2: Chemical structure of noraristeromycin.
Results and Discussion
Our retrosynthetic approach introduces the diversity point at a late stage and takes advantage of the heterocyclic lactam present in PreQ0 after activation and subsequent nucleophilic aromatic substitution. This convergent synthesis allowed us to prepare diverse chiral amine building blocks and react them with a common halo-purine intermediate to obtain the desired final products. The pyrrolo[2,3-d]pyrimidine core of PreQ0 was furnished following a method described by Klepper et al. [13] (Figure 3). The two step process started with the formylation of chloroacetonitrile with methyl formate. The resulting volatile and unstable chloroaldehyde 6 was used without further purification. Cyclocondensation of 6 with 2,4-diamino-4-hydroxypyrimidine afforded 1 regiospecifically with no detectable formation of the undesired 6-substituted-furo[2,3-d]pyrimidine 7. Direct chlorination of 1 in a moderate scale (1 g) using POCl3 proved to be very low yielding [28]. It remains unclear if this was due to the poor solubility of PreQ0 or to the presence of unprotected amino functionalities. In order to overcome this issue, the exocyclic amine was protected [14] (Figure 3). The resulting pivalamide 8 proved to be more soluble than 1 and the subsequent halogenation step was accomplished in the presence of a phase transfer catalyst, affording the desired chloro-intermediate 9 in fair yield. In our hands, nucleophilic aromatic substitution on 9 using amines of diverse nature usually proceeds smoothly and allows for a clean pivalamide deprotection [29]. For this reason we decided to couple the chiral amines of interest and remove protecting groups in a one-pot procedure.
![[1860-5397-10-135-3]](/bjoc/content/figures/1860-5397-10-135-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Synthesis of PreQ0 and chloro-intermediate 9. Reagents and conditions: (a) Methyl formate, NaOMe, PhMe, 3 h, 0 °C; (b) 2,6-diaminopyrimidin-4(3H)-one, NaOAc, H2O, 17 h, 100 °C, 60% (over two steps); (c) PivCl, pyridine, 2 h, 85 °C, 64%; (d) POCl3, DMA, BnEt3NCl, MeCN, 1 h, 90 °C, 35%.
Figure 3: Synthesis of PreQ0 and chloro-intermediate 9. Reagents and conditions: (a) Methyl formate, NaOMe, P...
First we investigated a more synthetically accessible (1RS,2SR,3RS)-3-aminocyclopentane-1,2-diol core. Our previous experience in coupling diols and triols at high temperatures with chloro-intermediate 9 showed that more than one unprotected alcohol functionality leads to complex reaction mixtures and very low yields of isolated products [29], hence we protected all hydroxy groups as esters. We chose the benzoate protecting group to generate UV–visible intermediates and because its ease of cleavage under basic conditions would converge with the final pivalamide deprotection step. We adapted this protecting group strategy to Bond’s synthetic route since it was the most concise and diastereospecific available [30] (Figure 4). The process started with a Wohl–Ziegler allylic bromination of cyclopentene. The volatile and unstable allylic halide 10 was immediately reacted with excess N,N-dibenzylamine and the resulting allylic amine 11 was obtained in good yield over two steps. Next, we introduced the two hydroxy groups trans- to the amine moiety using an Upjohn dihydroxylation. Freshly-prepared aqueous OsO4 stock solutions were required to obtain good yields in this step. The reaction proceeded smoothly and the 1H NMR spectra of the crude reaction mixture showed a 96:4 ratio of cis- to trans-isomers. After column chromatography the isolated diol 12 showed a diastereomeric purity of >99% by 1H NMR. The dibenzoate 13 was obtained in good yield following standard acylation conditions [31]. Final removal of the two benzyl groups was accomplished in excellent yield using catalytic hydrogenation [30], using EtOAc as a co-solvent to improve the substrate solubility. Amine 14 was coupled with 9 and the pivalamide and benzoate groups were cleaved in the one-pot procedure previously described to afford 15, the (1RS,2SR,3RS)-3-aminocyclopentane-1,2-diol derivative of PreQ0.
![[1860-5397-10-135-4]](/bjoc/content/figures/1860-5397-10-135-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Synthesis of 15, a (1RS,2SR,3RS)-3-aminocyclopentane-1,2-diol derivative of PreQ0. Reagents and conditions: (a) NBS, (PhCO2)2, CCl4, 1 h, 90 °C; (b) NH(Bn)2, CCl4, 12 h, rt, 70% (over two steps); (c) OsO4, NMO, acetone/H2O, 4 h, rt, 72%, 96% ds; (d) BzCl, pyr, 24 h, 0 °C to rt, 84%; (e) H2 (1 atm), Pd(OH)2, EtOH/EtOAc, 16 h, rt, 98%; (f) 9, Et3N, n-BuOH, 16 h, 130 °C; (g) KOH, n-BuOH/EtOH, 16 h, 80 °C, 42%.
Figure 4: Synthesis of 15, a (1RS,2SR,3RS)-3-aminocyclopentane-1,2-diol derivative of PreQ0. Reagents and con...
Adapting a protocol developed by Springthorpe et al. [32], we then investigated a route to prepare the enantiopure (1S,2R,3S,4R)-4-aminocyclopentane-1,2,3-triol analogue of PreQ0 16 (Figure 5). The first step is a Tsuji–Trost allylation of sodium di-tert-butyliminodicarboxylate. The reaction proceeded with an overall retention of configuration as expected and the 1H NMR spectra of the crude reaction mixture only showed the desired diastereomer 17. Several known catalytic systems were tested [29]: Pd(PPh3)4/PPh3 in THF/DMF [33], Pd2(dba)3/diphos in THF/DMF [34], Pd2(dba)3/dppf in THF [35]. The first set of conditions proved to be the most successful, although addition of DMF was required to improve the solubility of the reactants. It is worth noting that this reaction proved to be extremely sensitive to the presence of moisture and oxygen. The bulky nature of the nucleophile used aided in the diastereoselectivity of the following syn-dihydroxylation. Using the Upjohn conditions previously described we obtained the desired triol 18 in good yield and excellent diastereoselectivity (>99% by 1H NMR after column chromatography) [30]. Tri-benzoate 19 was subsequently obtained in good yield using the standard benzoylation conditions [31]. Final removal of the two BOC protecting groups using 4 M HCl in 1,4-dioxane yielded amine 20 as the hydrochloride salt. Amine 20 was coupled with chloro-intermediate 9 and the remaining four protecting groups were cleaved in a one-pot procedure under basic conditions, generating the desired triol 16.
![[1860-5397-10-135-5]](/bjoc/content/figures/1860-5397-10-135-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Synthesis of 16, a (1S,2R,3S,4R)-4-aminocyclopentane-1,2,3-triol derivative of PreQ0. Reagents and conditions: (a) Pd(PPh3)4, PPh3, NaH, NH(Boc)2, THF/DMF, 1 day, 50 °C, 42%; (b) OsO4, NMO, acetone/H2O, 1 day, rt, 83%; (c) BzCl, pyr, 17 h, 0 °C to rt, 74%; (d) 4 M HCl in 1,4-dioxane, 16 h, 0 °C to rt, 76%; (e) 9, Et3N, n-BuOH, 16 h, 130 °C; (f) KOH, n-BuOH/EtOH, 16 h, 80 °C, 33%.
Figure 5: Synthesis of 16, a (1S,2R,3S,4R)-4-aminocyclopentane-1,2,3-triol derivative of PreQ0. Reagents and ...
To extend into hydrophobic chemical space around our PreQ0 analogues, we prepared two novel derivatives containing the unusual 3-arylcyclohexylamine chiral motif present in 21 and 22. Zhou et al. had reported an asymmetric synthesis leading to the cis-3-arylcyclohexanamines with reasonable diastereoselectivity [36], but since initially we did not require an enantioselective synthesis and the Zhou method employed rather expensive reagents, we investigated a simpler and cheaper route to access both cis- and trans-isomers. We envisioned a stereoselective synthesis that would potentially allow for the introduction of diverse aryl groups at the 3-position of the cyclohexane ring using commercially available arylboronic acids as building blocks, and Pd catalysis to form the new C–C bond, followed by a highly diastereoselective ketone-to-amine conversion. Others have reported on similar preparations of 3-phenylcyclohexanamines, although with poor diastereomeric control [37,38]. 1-Cyclohex-2-enone provided the two required synthetic handles: a sp2 carbon for Pd chemistry and a ketone for further derivatization into an amine group (Figure 6). The synthesis of cis- and trans-3-arylcyclohexylamines 23 and 24 started with a Pd(II)-catalyzed Miyaura 1,4-conjugate addition of phenylboronic acid to cyclohexenone [39]. The resulting ketone 25 was reduced to the axial [40,41] and equatorial [40] alcohols 26 and 27 with excellent diastereoselectivity thanks to steric control of the hydride source. After column chromatography both alcohols showed a diastereomeric purity of >99% by 1H NMR. Mitsunobu reaction on the secondary alcohols using DEAD or DIAD did not provide the desired azides [42,43] nor did a one-pot Appel reaction/nucleophilic substitution/Staudinger reaction protocol involving a double inversion of configuration [44]. Mesylation of 26 and 27 lead to intermediates 28 and 29 [45], which were subsequently reacted with sodium azide inverting the stereochemistry as required [46]. A final transfer hydrogenation of 30 and 31 yielded the desired amines rapidly and with excellent yields [47]. Amines 23 and 24 were reacted with chloro-intermediate 9 and the pivalamide groups were cleaved under basic hydrolysis conditions to yield 21 and 22.
![[1860-5397-10-135-6]](/bjoc/content/figures/1860-5397-10-135-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Synthesis of 21 and 22, 3-arylcyclohexylamine derivatives of PreQ0. Reagents and conditions: (a) PhB(OH)2, Pd(OAc)2, bpy, H2O/THF/AcOH, 3 days, 60 °C, 98%; (b) K-Selectride, THF, 2 h, −90 °C; then KOH (aq), H2O2, 30 min, rt, 80%, 99% ds; (c) LiAlH4, Et2O, 1,5 h, 0 °C to rt; then NaOH (aq), 30 min, rt, 76%, 97% ds (d) MsCl, Et3N, THF, 16 h, rt, (trans- 67%, cis- 64%); (d) NaN3, DMF, 2 days, 80 °C, (trans- 56%, cis- 67%); (f) HCO2NH4, Pd/C, MeOH, 1.5 h, reflux, (trans- 92%, cis- 94%); (g) 9, Et3N, n-BuOH, 16 h, 130 °C; (h) KOH, n-BuOH/EtOH, 16 h, 80 °C, (trans- 38%, cis- 50%).
Figure 6: Synthesis of 21 and 22, 3-arylcyclohexylamine derivatives of PreQ0. Reagents and conditions: (a) Ph...
Conclusion
In conclusion, a concise and stereoselective synthesis of novel cyclopentyl and cyclohexyl analogues of PreQ0 has been developed to expand our fragment-based kinase library. This synthetic protocol involves asymmetric syntheses of hydroxy-protected (1RS,2SR,3RS)-3-aminocyclopentane-1,2-diol and (1S,2R,3S,4R)-4-aminocyclopentane-1,2,3-triol or cis- and trans-3-arylcyclohexylamines, which are in turn reacted with a conveniently PreQ0-derived halo-intermediate and subsequently deprotected in a one-pot fashion. Pharmacological assessment of these novel PreQ0 derivatives is currently underway in a variety of kinase-inhibitory studies and will be reported in due course.
Supporting Information
| Supporting Information File 1: General methods, experimental procedures and copies of 1H/13C NMR spectra and HPLC UV traces of final compounds 15, 16, 21 and 22. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
The authors would like to thank Stuart Lang for insightful comments and manuscript reviewing. S. Llona-Minguez and S. P. Mackay have received funding from the University of Strathclyde studentship scheme and Cancer Research UK to pursue inhibitory IkB kinase inhibitors and a patent application is in preparation.
References
-
Seela, F.; Peng, X. Curr. Top. Med. Chem. 2006, 6, 867–892. doi:10.2174/156802606777303649
Return to citation in text: [1] [2] -
Suhadolnik, R. J. Pyrrolopyrimidine Nucleosides. "Nucleoside Antibiotics"; Wiley-Interscience: New York, 1970.
Return to citation in text: [1] -
Ritch, P. S.; Glazer, R. I. Pyrrolo[2,3-d]pyrimidine nucleosides. Developments in Cancer Chemotherapy; CRC Press: Florida, 1984.
Return to citation in text: [1] -
Martin, J. C. Nucleotide Analogues as Antiviral Agents; American Chemical Society: Washington, 1989. doi:10.1021/bk-1989-0401
Return to citation in text: [1] -
Morris, R. C.; Elliott, M. S. Mol. Genet. Metab. 2001, 74, 147–159. doi:10.1006/mgme.2001.3216
Return to citation in text: [1] -
Phillips, G.; Swairjo, M. A.; Gaston, K. W.; Bailly, M.; Limbach, P. A.; Iwata-Reuyl, D.; de Crécy-Lagard, V. ACS Chem. Biol. 2011, 7, 300–305. doi:10.1021/cb200361w
Return to citation in text: [1] -
McCarty, R. M.; Somogyi, A.; Lin, G.; Jacobsen, N. E.; Bandarian, V. Biochemistry 2009, 48, 3847–3852. doi:10.1021/bi900400e
Return to citation in text: [1] -
Aronov, A. M.; McClain, B.; Moody, C. S.; Murcko, M. A. J. Med. Chem. 2008, 51, 1214–1222. doi:10.1021/jm701021b
Return to citation in text: [1] -
Cheng, C. S.; Hinshaw, B. C.; Panzica, R. P.; Townsend, L. B. J. Am. Chem. Soc. 1976, 98, 7870–7872. doi:10.1021/ja00440a094
Return to citation in text: [1] -
Kondo, T.; Okamoto, K.; Ohgi, T.; Goto, T. Tetrahedron 1986, 42, 207–213. doi:10.1016/S0040-4020(01)87419-6
Return to citation in text: [1] -
Migawa, M. T.; Hinkley, J. M.; Hoops, G. C.; Townsend, L. B. Synth. Commun. 1996, 26, 3317–3322. doi:10.1080/00397919608004641
Return to citation in text: [1] -
Ramzaeva, N.; Becher, G.; Seela, F. Synthesis 1998, 1327–1330. doi:10.1055/s-1998-6088
Return to citation in text: [1] -
Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201
Return to citation in text: [1] [2] -
Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j
Return to citation in text: [1] [2] -
Brückl, T.; Thoma, I.; Wagner, A. J.; Knochel, P.; Carell, T. Eur. J. Org. Chem. 2010, 6517–6519. doi:10.1002/ejoc.201000987
Return to citation in text: [1] -
Ming, X.; Seela, F. Chem. Biodiversity 2010, 7, 2616–2621. doi:10.1002/cbdv.201000239
Return to citation in text: [1] -
Klepper, F.; Jahn, E.-M.; Hickmann, V.; Carell, T. Angew. Chem., Int. Ed. 2007, 46, 2325–2327. doi:10.1002/anie.200604579
Return to citation in text: [1] -
Arndt, D.; Graffi, A. Z. Chem. 1977, 17, 224–225.
Return to citation in text: [1] -
Anderson, B. G.; Bauta, W. E.; Cantrell, W. R., Jr.; Engles, T.; Lovett, D. P. Org. Process Res. Dev. 2008, 12, 1229–1237. doi:10.1021/op800182x
Return to citation in text: [1] -
Arndt, D.; Graffi, A. S-Glycosides of arabinofuranose. German Democratic Republic Patent DD107278A1, 1974.
Return to citation in text: [1] -
Claiborne, C. F.; Critchley, S.; Langston, S. P.; Olhava, E. J.; Peluso, S.; Weatherhead, G. S.; Vyskocil, S.; Visiers, I.; Mizutani, H.; Cullis, C. Preparation of carbocyclic purine nucleoside analogs as antitumor agents and inhibitors of E1 activating enzymes. WO Patent WO2008019124A1, Feb 14, 2008.
Return to citation in text: [1] -
Fujishima, T.; Uchida, K.; Yoshino, H. N6-(β-D-Ribofuranosyl)adenine. Japanese Patent JP50034040B, 1975.
Return to citation in text: [1] -
Lüipke, U.; Seela, F. Chem. Ber. 1979, 112, 799–806. doi:10.1002/cber.19791120304
Return to citation in text: [1] -
Jain, P. C.; Anand, N. Indian J. Chem. 1968, 6, 616–618.
Return to citation in text: [1] -
Goodman, I.; Salce, L.; Hitchings, G. H. J. Med. Chem. 1968, 11, 516–521. doi:10.1021/jm00309a024
Return to citation in text: [1] -
Llona-Minguez, S.; Baiget, J.; Mackay, S. P. Pharm. Pat. Anal. 2013, 2, 481–498. doi:10.4155/ppa.13.31
Return to citation in text: [1] -
Ito, M.; Hamano, T.; Komatsu, T.; Asamitsu, K.; Yamakawa, T.; Okamoto, T. Mod. Rheumatol. 2014. doi:10.3109/14397595.2013.879416
Return to citation in text: [1] -
Gibson, C. L.; La Rosa, S.; Ohta, K.; Boyle, P. H.; Leurquin, F.; Lemaçon, A.; Suckling, C. J. Tetrahedron 2004, 60, 943–959. doi:10.1016/j.tet.2003.11.030
Return to citation in text: [1] -
Unpublished results.
Return to citation in text: [1] [2] [3] -
Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783
Return to citation in text: [1] [2] [3] -
Li, J.; Lowary, T. L. Org. Lett. 2008, 10, 881–884. doi:10.1021/ol703041y
Return to citation in text: [1] [2] -
Springthorpe, B.; Bailey, A.; Barton, P.; Birkinshaw, T. N.; Bonnert, R. V.; Brown, R. C.; Chapman, D.; Dixon, J.; Guile, S. D.; Humphries, R. G.; Hunt, S. F.; Ince, F.; Ingall, A. H.; Kirk, I. P.; Leeson, P. D.; Leff, P.; Lewis, R. J.; Martin, B. P.; McGinnity, D. F.; Mortimore, M. P.; Paine, S. W.; Pairaudeau, G.; Patel, A.; Rigby, A. J.; Riley, R. J.; Teobald, B. J.; Tomlinson, W.; Webborn, P. J. H.; Willis, P. A. Bioorg. Med. Chem. Lett. 2007, 17, 6013–6018. doi:10.1016/j.bmcl.2007.07.057
Return to citation in text: [1] -
Deardorff, D. R.; Linde, R. G., II; Martin, A. M.; Shulman, M. J. J. Org. Chem. 1989, 54, 2759–2762. doi:10.1021/jo00272a059
Return to citation in text: [1] -
Connell, R. D.; Rein, T.; Aakermark, B.; Helquist, P. J. Org. Chem. 1988, 53, 3845–3849. doi:10.1021/jo00251a035
Return to citation in text: [1] -
Dauvergne, J.; Happe, A. M.; Jadhav, V.; Justice, D.; Matos, M.-C.; McCormack, P. J.; Pitts, M. R.; Roberts, S. M.; Singh, S. K.; Snape, T. J.; Whittall, J. Tetrahedron 2004, 60, 2559–2567. doi:10.1016/j.tet.2004.01.046
Return to citation in text: [1] -
Zhou, J.; List, B. J. Am. Chem. Soc. 2007, 129, 7498–7499. doi:10.1021/ja072134j
Return to citation in text: [1] -
Kajita, T.; Matsumoto, T. Process for the preparation of optically active trans-cyclohexylamine compounds. WO Patent WO2000000459A1, Jan 6, 2000.
Return to citation in text: [1] -
Gomtsyan, A.; Daanen, J. F.; Gfesser, G. A.; Kort, M. E.; Lee, C.-H.; McDonald, H. A.; Puttfarcken, P. S.; Voight, E. A.; Kym, P. R. Preparation of urea compounds TRPV1 antagonists for treating pain. U.S. Patent US20120245163A1, Sept 27, 2012.
Return to citation in text: [1] -
Lu, X.; Lin, S. J. Org. Chem. 2005, 70, 9651–9653. doi:10.1021/jo051561h
Return to citation in text: [1] -
Cieplak, A. S. J. Am. Chem. Soc. 1981, 103, 4540–4552. doi:10.1021/ja00405a041
Return to citation in text: [1] [2] -
Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1972, 94, 7159–7161. doi:10.1021/ja00775a053
Return to citation in text: [1] -
Scott, J. P.; Alam, M.; Bremeyer, N.; Goodyear, A.; Lam, T.; Wilson, R. D.; Zhou, G. Org. Process Res. Dev. 2011, 15, 1116–1123. doi:10.1021/op200002u
Return to citation in text: [1] -
Aicher, T. D.; Chicarelli, M. J.; Hinklin, R. J.; Tian, H.; Wallace, O. B.; Chen, Z.; Mabry, T. E.; McCowan, J. R.; Snyder, N. J.; Winneroski, L. L., Jr.; Allen, J. G. Preparation of cycloalkyl lactam derivatives, particularly N-substituted pyrrolidin-2-ones, as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1. WO Patent WO2006049952A1, May 11, 2006.
Return to citation in text: [1] -
Sagar Reddy, G. V.; Rao, G. V.; Subramanyam, R. V. K.; Iyengar, D. S. Synth. Commun. 2000, 30, 2233–2237. doi:10.1080/00397910008087402
Return to citation in text: [1] -
Zhang, Q.; Ma, X.; Ward, A.; Hong, W.-X.; Jaakola, V.-P.; Stevens, R. C.; Finn, M. G.; Chang, G. Angew. Chem., Int. Ed. 2007, 46, 7023–7025. doi:10.1002/anie.200701556
Return to citation in text: [1] -
Lednicer, D.; Emmert, D. E.; Lahti, R.; Rudzik, A. D. J. Med. Chem. 1972, 15, 1239–1243. doi:10.1021/jm00282a009
Return to citation in text: [1] -
Paryzek, Z.; Koenig, H.; Tabaczka, B. Synthesis 2003, 2023–2026. doi:10.1055/s-2003-41024
Return to citation in text: [1]
| 40. | Cieplak, A. S. J. Am. Chem. Soc. 1981, 103, 4540–4552. doi:10.1021/ja00405a041 |
| 41. | Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1972, 94, 7159–7161. doi:10.1021/ja00775a053 |
| 40. | Cieplak, A. S. J. Am. Chem. Soc. 1981, 103, 4540–4552. doi:10.1021/ja00405a041 |
| 1. | Seela, F.; Peng, X. Curr. Top. Med. Chem. 2006, 6, 867–892. doi:10.2174/156802606777303649 |
| 8. | Aronov, A. M.; McClain, B.; Moody, C. S.; Murcko, M. A. J. Med. Chem. 2008, 51, 1214–1222. doi:10.1021/jm701021b |
| 7. | McCarty, R. M.; Somogyi, A.; Lin, G.; Jacobsen, N. E.; Bandarian, V. Biochemistry 2009, 48, 3847–3852. doi:10.1021/bi900400e |
| 30. | Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783 |
| 5. | Morris, R. C.; Elliott, M. S. Mol. Genet. Metab. 2001, 74, 147–159. doi:10.1006/mgme.2001.3216 |
| 6. | Phillips, G.; Swairjo, M. A.; Gaston, K. W.; Bailly, M.; Limbach, P. A.; Iwata-Reuyl, D.; de Crécy-Lagard, V. ACS Chem. Biol. 2011, 7, 300–305. doi:10.1021/cb200361w |
| 14. | Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j |
| 47. | Paryzek, Z.; Koenig, H.; Tabaczka, B. Synthesis 2003, 2023–2026. doi:10.1055/s-2003-41024 |
| 2. | Suhadolnik, R. J. Pyrrolopyrimidine Nucleosides. "Nucleoside Antibiotics"; Wiley-Interscience: New York, 1970. |
| 3. | Ritch, P. S.; Glazer, R. I. Pyrrolo[2,3-d]pyrimidine nucleosides. Developments in Cancer Chemotherapy; CRC Press: Florida, 1984. |
| 4. | Martin, J. C. Nucleotide Analogues as Antiviral Agents; American Chemical Society: Washington, 1989. doi:10.1021/bk-1989-0401 |
| 18. | Arndt, D.; Graffi, A. Z. Chem. 1977, 17, 224–225. |
| 19. | Anderson, B. G.; Bauta, W. E.; Cantrell, W. R., Jr.; Engles, T.; Lovett, D. P. Org. Process Res. Dev. 2008, 12, 1229–1237. doi:10.1021/op800182x |
| 20. | Arndt, D.; Graffi, A. S-Glycosides of arabinofuranose. German Democratic Republic Patent DD107278A1, 1974. |
| 21. | Claiborne, C. F.; Critchley, S.; Langston, S. P.; Olhava, E. J.; Peluso, S.; Weatherhead, G. S.; Vyskocil, S.; Visiers, I.; Mizutani, H.; Cullis, C. Preparation of carbocyclic purine nucleoside analogs as antitumor agents and inhibitors of E1 activating enzymes. WO Patent WO2008019124A1, Feb 14, 2008. |
| 22. | Fujishima, T.; Uchida, K.; Yoshino, H. N6-(β-D-Ribofuranosyl)adenine. Japanese Patent JP50034040B, 1975. |
| 23. | Lüipke, U.; Seela, F. Chem. Ber. 1979, 112, 799–806. doi:10.1002/cber.19791120304 |
| 24. | Jain, P. C.; Anand, N. Indian J. Chem. 1968, 6, 616–618. |
| 25. | Goodman, I.; Salce, L.; Hitchings, G. H. J. Med. Chem. 1968, 11, 516–521. doi:10.1021/jm00309a024 |
| 13. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 45. | Zhang, Q.; Ma, X.; Ward, A.; Hong, W.-X.; Jaakola, V.-P.; Stevens, R. C.; Finn, M. G.; Chang, G. Angew. Chem., Int. Ed. 2007, 46, 7023–7025. doi:10.1002/anie.200701556 |
| 17. | Klepper, F.; Jahn, E.-M.; Hickmann, V.; Carell, T. Angew. Chem., Int. Ed. 2007, 46, 2325–2327. doi:10.1002/anie.200604579 |
| 28. | Gibson, C. L.; La Rosa, S.; Ohta, K.; Boyle, P. H.; Leurquin, F.; Lemaçon, A.; Suckling, C. J. Tetrahedron 2004, 60, 943–959. doi:10.1016/j.tet.2003.11.030 |
| 46. | Lednicer, D.; Emmert, D. E.; Lahti, R.; Rudzik, A. D. J. Med. Chem. 1972, 15, 1239–1243. doi:10.1021/jm00282a009 |
| 9. | Cheng, C. S.; Hinshaw, B. C.; Panzica, R. P.; Townsend, L. B. J. Am. Chem. Soc. 1976, 98, 7870–7872. doi:10.1021/ja00440a094 |
| 10. | Kondo, T.; Okamoto, K.; Ohgi, T.; Goto, T. Tetrahedron 1986, 42, 207–213. doi:10.1016/S0040-4020(01)87419-6 |
| 11. | Migawa, M. T.; Hinkley, J. M.; Hoops, G. C.; Townsend, L. B. Synth. Commun. 1996, 26, 3317–3322. doi:10.1080/00397919608004641 |
| 12. | Ramzaeva, N.; Becher, G.; Seela, F. Synthesis 1998, 1327–1330. doi:10.1055/s-1998-6088 |
| 13. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 14. | Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j |
| 15. | Brückl, T.; Thoma, I.; Wagner, A. J.; Knochel, P.; Carell, T. Eur. J. Org. Chem. 2010, 6517–6519. doi:10.1002/ejoc.201000987 |
| 16. | Ming, X.; Seela, F. Chem. Biodiversity 2010, 7, 2616–2621. doi:10.1002/cbdv.201000239 |
| 42. | Scott, J. P.; Alam, M.; Bremeyer, N.; Goodyear, A.; Lam, T.; Wilson, R. D.; Zhou, G. Org. Process Res. Dev. 2011, 15, 1116–1123. doi:10.1021/op200002u |
| 43. | Aicher, T. D.; Chicarelli, M. J.; Hinklin, R. J.; Tian, H.; Wallace, O. B.; Chen, Z.; Mabry, T. E.; McCowan, J. R.; Snyder, N. J.; Winneroski, L. L., Jr.; Allen, J. G. Preparation of cycloalkyl lactam derivatives, particularly N-substituted pyrrolidin-2-ones, as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1. WO Patent WO2006049952A1, May 11, 2006. |
| 1. | Seela, F.; Peng, X. Curr. Top. Med. Chem. 2006, 6, 867–892. doi:10.2174/156802606777303649 |
| 26. | Llona-Minguez, S.; Baiget, J.; Mackay, S. P. Pharm. Pat. Anal. 2013, 2, 481–498. doi:10.4155/ppa.13.31 |
| 27. | Ito, M.; Hamano, T.; Komatsu, T.; Asamitsu, K.; Yamakawa, T.; Okamoto, T. Mod. Rheumatol. 2014. doi:10.3109/14397595.2013.879416 |
| 44. | Sagar Reddy, G. V.; Rao, G. V.; Subramanyam, R. V. K.; Iyengar, D. S. Synth. Commun. 2000, 30, 2233–2237. doi:10.1080/00397910008087402 |
| 32. | Springthorpe, B.; Bailey, A.; Barton, P.; Birkinshaw, T. N.; Bonnert, R. V.; Brown, R. C.; Chapman, D.; Dixon, J.; Guile, S. D.; Humphries, R. G.; Hunt, S. F.; Ince, F.; Ingall, A. H.; Kirk, I. P.; Leeson, P. D.; Leff, P.; Lewis, R. J.; Martin, B. P.; McGinnity, D. F.; Mortimore, M. P.; Paine, S. W.; Pairaudeau, G.; Patel, A.; Rigby, A. J.; Riley, R. J.; Teobald, B. J.; Tomlinson, W.; Webborn, P. J. H.; Willis, P. A. Bioorg. Med. Chem. Lett. 2007, 17, 6013–6018. doi:10.1016/j.bmcl.2007.07.057 |
| 30. | Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783 |
| 36. | Zhou, J.; List, B. J. Am. Chem. Soc. 2007, 129, 7498–7499. doi:10.1021/ja072134j |
| 37. | Kajita, T.; Matsumoto, T. Process for the preparation of optically active trans-cyclohexylamine compounds. WO Patent WO2000000459A1, Jan 6, 2000. |
| 38. | Gomtsyan, A.; Daanen, J. F.; Gfesser, G. A.; Kort, M. E.; Lee, C.-H.; McDonald, H. A.; Puttfarcken, P. S.; Voight, E. A.; Kym, P. R. Preparation of urea compounds TRPV1 antagonists for treating pain. U.S. Patent US20120245163A1, Sept 27, 2012. |
| 30. | Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783 |
| 34. | Connell, R. D.; Rein, T.; Aakermark, B.; Helquist, P. J. Org. Chem. 1988, 53, 3845–3849. doi:10.1021/jo00251a035 |
| 35. | Dauvergne, J.; Happe, A. M.; Jadhav, V.; Justice, D.; Matos, M.-C.; McCormack, P. J.; Pitts, M. R.; Roberts, S. M.; Singh, S. K.; Snape, T. J.; Whittall, J. Tetrahedron 2004, 60, 2559–2567. doi:10.1016/j.tet.2004.01.046 |
| 33. | Deardorff, D. R.; Linde, R. G., II; Martin, A. M.; Shulman, M. J. J. Org. Chem. 1989, 54, 2759–2762. doi:10.1021/jo00272a059 |
© 2014 Llona-Minguez and Mackay; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)