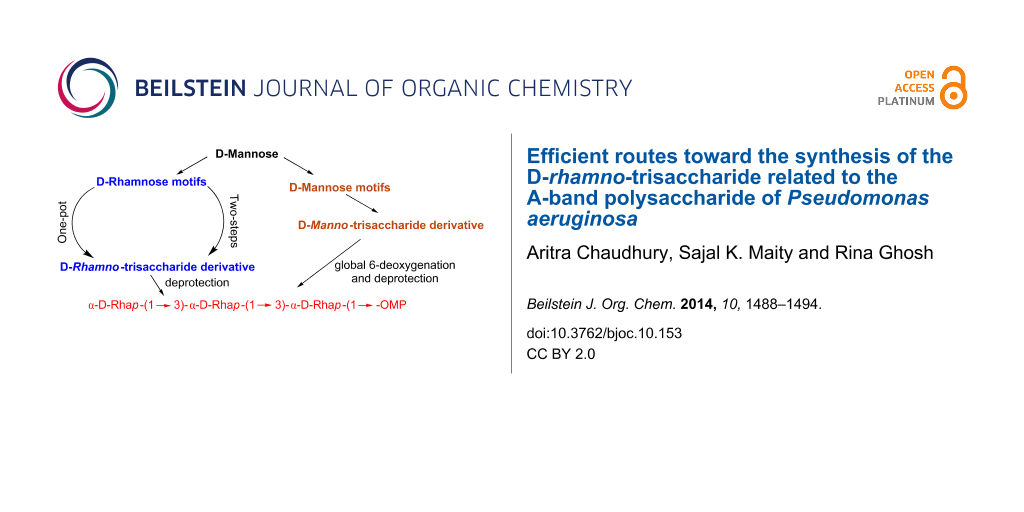
Abstract
The present work describes efficient avenues for the synthesis of the trisaccharide repeating unit [α-D-Rhap-(1→3)-α-D-Rhap-(1→3)-α-D-Rhap] associated with the A-band polysaccharide of Pseudomonas aeruginosa. One of the key steps involved 6-O-deoxygenation of either partially or fully acylated 4,6-O-benzylidene-1-thiomannopyranoside by radical-mediated redox rearrangement in high yields and regioselectivity. The D-rhamno-thioglycosides so obtained allowed efficient access to the trisaccharide target via stepwise glycosylation as well as a one-pot glycosylation protocol. In a different approach, a 4,6-O-benzylidene D-manno-trisaccharide derivative was synthesized, which upon global 6-O-deoxygenation followed by deprotection generated the target D-rhamno-trisaccharide. The application of the reported regioselective radical-mediated deoxygenation on 4,6-O-benzylidene D-manno thioglycoside (hitherto unexplored) has potential for ramification in the field of synthesis of oligosaccharides based on 6-deoxy hexoses.

Graphical Abstract
Introduction
With the firm establishment of the critical roles played by oligosaccharides in diverse biological processes [1-4], the field of oligosaccharide synthesis has seen a rapid development over the last two decades [5-7]. The D-rhamnoside motif is of particular interest with its presence established in the LPS/EPS systems of various bacterial strains which are pathogenic towards both plants and animals. These species include the Burkholderia cepacia complex [8], P. aeruginosa [9], Helicobacter pylori [10], Citrobacter freundii [11], Campylobacter fetus [12], Stenotrophonas maltophilia [13], Xanthomonas campestri [14] and Brucella sp. [15].
P. aeruginosa has long been established as an opportunistic pathogen which infects humans having compromised immunity with fatal consequences in a majority of cases. Its high persistence against a wide variety of antibiotics is also well documented. It has also been observed how the colonized form of this species in cystic fibrosis lungs, through non-expression of O-antigens, offer high persistence often rendering O-antigen-based vaccines ineffective. But the conservation of the A-band polysaccharide even in the colonized form makes this repeating unit a viable candidate for A-band polysaccharide-based vaccines which can avoid the vulnerability applicable to their O-antigen-based counterparts [16]. Hence, the A-band polysaccharide [16] of P. aeruginosa which has been characterized previously as a repeating combination of [→2)-α-D-Rhap-(1→3)-α-D-Rhap-(1→3)-α-D-Rhap] (Figure 1) provides for a synthetic target whose efficient synthesis is worth pursuing. It is to be noted that synthesis of a tetrasaccharide, α-D-Rhap-(1→2)-α-D-Rhap-(1→3)-α-D-Rhap-(1→3)-α-D-Rhap [17] and a trisaccharide, α-D-Rhap-(1→3)-α-D-Rhap-(1→2)-α-D-Rhap [18] related to the A-band polysaccharide of P. aeruginosa were made with a view to develop glycoconjugate vaccines, but none have ultimately materialized into valid vaccine candidates.
![[1860-5397-10-153-1]](/bjoc/content/figures/1860-5397-10-153-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Repeating unit of the A-band polysaccharide of P. aeruginosa.
Figure 1: Repeating unit of the A-band polysaccharide of P. aeruginosa.
Thus, we targeted the trisaccharide [α-D-Rhap-(1→3)-α-D-Rhap-(1→3)-α-D-Rhap] as our synthetic goal toward construction of a probable vaccine candidate against P. aeruginosa.
The synthesis of the D-rhamnose-based oligosaccharide from the D-mannose motif has received substantial attention over the last decade. The problem of β-D-rhamnoside synthesis has been greatly addressed by Crich et al. [19-23]. But, Crich’s global deoxygenating strategy, despite its ultimate efficiency, still requires synthetic modification on the conventional 4,6-O-benzylidene framework involving reagents which are rather expensive. Moreover, additional steps are required for the preparation of these adequately derivatized mannose-based systems from which the D-rhamnose motif may be accessed. Other reports on the D-rhamnose-based synthesis have also surfaced in recent times [24,25]. Apart from this, the synthesis of monomeric D-rhamnose has been highly streamlined by Roy et al. in 2007 [26], and further improvement on this method was reported by Kiefel et al. in 2011 [27]. However, the deoxygenation protocol involving halogenation under Mitsunobu conditions was found to be inefficient when applied to thioglycosides directly. Moreover, the use of stoichiometric amounts of the toxic tin hydride for radical-based reductive dehalogenation as required by the above method appeared undesirable especially in the preparative stages where reactions have to be set up on a large scale. On the other hand, Kiefel’s method requires four steps for the conversion of D-mannose to D-rhamnose on which further manipulations are required to reach the adequately designed derivatives. This makes the starting material preparations rather long drawn.
Keeping these limitations in mind we selected the protocol devised by Pedersen et al. [28] and subsequently studied by Dang et al. [29-32] which seemed to address all the inadequacies described above. However, the compatibility of the method with thioglycosides had to be verified. Upon application, and much to our delight, it was found that thioglycosides responded equally well compared to their O-glycosidic counterparts. It may be mentioned here that the response of thioglycosides towards this method was not reported in the original observation [28]. To the best of our knowledge, this method has not yet been applied directly on thioglycosides. So, having established an easy means to access D-rhamno-thioglycosides from their D-manno-counterparts, we set about devising efficient routes to reach the target trisaccharide. The following section bears elaboration of our efforts.
Results and Discussion
Deoxygenation at the C-6 position of D-mannose derivatives bearing conventional or Crich’s modified 4,6-O-benzylidene protection [19-23] has been the principal philosophy behind the synthesis of the D-rhamnose [19-23,30,33-39] motif. This is a natural choice not only because it allows simultaneous selective blockage of the O-4 and O-6 positions but also sets up the model on which various deoxygenation protocols may be tried. The linkage pattern and stereochemistry at the glycosidic positions on the target dictate the presence of acyl protection on the O-2 position as shown in Scheme 1.
![[1860-5397-10-153-i1]](/bjoc/content/inline/1860-5397-10-153-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the monomeric building blocks; reagents and conditions: i) Pyr., BzCl, 0 °C–rt; ii) PhC(OMe)3, CSA, MeCN, 0 °C–rt, 80% AcOH (aq), 25 °C; iii) DTBP, TIPST, octane, reflux; iv) TCCA, acetone–H2O, rt; v) CCl3CN, DBU, DCM; vi) MeC(OEt)3, CSA, MeCN, 0 °C–rt, 80% AcOH (aq), 25 °C.
Scheme 1: Preparation of the monomeric building blocks; reagents and conditions: i) Pyr., BzCl, 0 °C–rt; ii) ...
We began our synthesis targeting two pivotal intermediates 1a and 1b which were obtained using previously reported methods [40,41] and then proceeding forward to the monomeric building blocks required according to the retrosynthetic analysis (Figure 2).
![[1860-5397-10-153-2]](/bjoc/content/figures/1860-5397-10-153-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The procedures used and the results obtained to reach the intermediate targets have been summarized in Scheme 1. Accordingly, compounds 2 and 3b [42] were obtained uneventfully from 1a in 92% and 75%, respectively (Scheme 1). All attempts to place an acetyl protection at the O-2 position of 1b with high yield failed. The poor yield was presumed to be due to the migration of the 2-O-acetyl group to the O-3 position leading to a mixture which was hard to separate. Hence we switched to O-benzoyl protection which was found to be less susceptible to migration [25]. The 2-O-benzoylated compound 4 was prepared by conversion of 1b into its corresponding methyl 2,3-orthobenzoate derivative followed by the selective cleavage of the same with 80% aq AcOH. Compound 3a which is the phenyl thioglycoside analogue of 4 was also accessed similarly. Both the compounds were next converted to their rhamnoside counterparts 6 and 7 [44], respectively by treatment with di-tert-butyl peroxide (DTBP) and triisopropylsilanethiol (TIPST) under reflux in octane over 2–3 h [30] in overall 74% and 64% yields, respectively, over two steps. It is worth noting here that during column chromatographic purification after AcOH-mediated cleavage of the orthobenzoate derivative some losses were incurred due to the migration of the 2-O-benzoyl group to the O-3 position. However the susceptibility of the benzoyl group to migration was much lower than that of its acetate analogue.
The same problem persisted even in the rhamnosides 6 and 7. Hence, to minimize the loss during purification, the chromatographic purification was reserved until the end of the deoxygenation step. Thus, the intermediate compounds 3a and 4 were used without purification by column chromatography. Compound 2 was deoxygenated to its rhamnosyl counterpart 2a in a high yield (88%). Next, the thioglycoside was hydrolyzed to the hemiacetal 2b with trichloroisocyanuric acid (TCCA) in acetone–H2O [43] in 85% yield and was further converted almost quantitatively to the corresponding trichloroacetimidate 5 [44]. Having arrived at the monomeric building blocks 5, 6, and 7 we carried forward to the rhamnose-based disaccharide 8 which being a thioglycoside would subsequently serve as the glycosyl donor in the next step. Accordingly, 5 and 6 were coupled almost quantitatively to give 8. The disaccharide so obtained was then coupled with 7 to give the protected trisaccharide 9 in 90% yield using a N-iodosuccinimide-trimethylsilyl trifluoromethanesulfonate (NIS-TMSOTf) combination as the activating reagent. The target trisaccharide was then obtained via deprotection under the Zémplen conditions to give 10 quantitatively, as is summarized in Scheme 2.
![[1860-5397-10-153-i2]](/bjoc/content/inline/1860-5397-10-153-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Sequential stepwise synthesis of the trisaccharide; reagents and conditions: i) TMSOTf, DCM, molecular sieves 4 Å, N2, −50 °C; ii) NIS, TMSOTf, DCM, molecular sieves 4 Å, N2, −10 °C; iii) NaOMe, MeOH, rt, 12 h.
Scheme 2: Sequential stepwise synthesis of the trisaccharide; reagents and conditions: i) TMSOTf, DCM, molecu...
However, in the interest of time and economy of resources consumed, a one-pot synthesis is always desirable. Accordingly, further optimization of the glycosylation protocol was achieved by carrying out the whole glycosylation process in one-pot leading to the target trisaccharide 9 in 79% yield (Scheme 3). In this case the disaccharide synthesis was set up as described previously, and then the second acceptor 7 was introduced into the reaction mixture at −50 °C after which NIS and TMSOTf were added successively at the same temperature. The temperature was then raised upto −10 °C, and the reaction mixture was stirred for another 1 h at that temperature to give the target trisaccharide as summarized in Scheme 3.
![[1860-5397-10-153-i3]](/bjoc/content/inline/1860-5397-10-153-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the trisaccharide by sequential one-pot glycosylation reactions; reagents and conditions: i) (a) TMSOTf, DCM, molecular sieves 4 Å, N2, −50 °C; (b) NIS, TMSOTf, −50 °C, DCM, molecular sieves 4 Å, N2, −10 °C.
Scheme 3: Synthesis of the trisaccharide by sequential one-pot glycosylation reactions; reagents and conditio...
At this point, our search for overall synthetic efficiency was greatly augmented by a change of approach. Since, multiple deoxygenations had been observed by Dang et al. [30] on trehalose derivatives previously, we figured that we could access the rhamnose-based trisaccharide directly from a suitably derivatized manno-trisaccharide bearing the three 4,6-O-benzylidene protecting groups by a single global deoxygenation step. Accordingly, we coupled the monomeric units 2, 3b [45] and 4 to reach the mannose-based trisaccharide 12 (Scheme 4). The deoxygenation protocol, being incompatible with the O-benzyl protecting group, required an acyl protection profile on O-2 and O-3 positions. Such an arrangement was also in agreement with the stereochemical requirements at the anomeric positions of our target trisaccharide.
Since compounds 2 and 3b bear structural similarity with each other it was expected that they would have similar reactivity towards glycosylative activation. Hence, a preactivation-based glycosylation protocol was devised for the first step leading to the disaccharide 11 using 1-benzenesulfinyl piperidine–triflic anhydride (BSP–Tf2O) [46]. The subsequent step was carried out using NIS–TMSOTf to give the mannose-based trisaccharide 12. This trisaccharide was next deoxygenated globally to give the rhamnose-based trisaccharide 13 with high yield (80%). This was then deprotected under the Zémplen conditions to yield 10 quantitatively as is summarized in Scheme 4.
![[1860-5397-10-153-i4]](/bjoc/content/inline/1860-5397-10-153-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the target trisaccharide via global deoxygenation strategy; reagents and conditions: i) BSP, Tf2O, –60 °C, DCM, molecular sieves 4 Å, N2, –78 °C; ii) NIS, TMSOTf, DCM, molecular sieves 4 Å, N2, −10 °C; iii) DTBP, TIPST, octane, reflux, 3 h; iv) NaOMe, MeOH, rt, 12 h.
Scheme 4: Synthesis of the target trisaccharide via global deoxygenation strategy; reagents and conditions: i...
1H NMR in MeOH-d4 of the target trisaccharide 10 showed the anomeric protons of the consecutive rhamnose residues from the non-reducing end appearing at δ 5.09, 5.06 and 5.30 ppm, respectively. 13C NMR in the same solvent revealed that the chemical shifts of the anomeric carbons of the two consecutive rhamnose units from the non-reducing end coincided at δ 102.3 ppm (as also evidenced from HSQC data) with the corresponding 1JC1-H1 of 170.4 Hz and that of the anomeric carbon of the reducing end monomer unit appeared at 98.9 ppm with the corresponding 1JC1-H1 of 170.2 Hz. The value at δ 102.3 ppm can be corroborated with the reported [16] C1-chemical shifts at δ 103.19 and 103.43 with 1JC1-H1 of 173 Hz, exhibited in D2O corresponding to the two [→3)-α-D-Rhap-(1→] units in the A-band polysaccharide of P. aeruginosa. The small difference in the chemical shifts and the corresponding 1JC1-H1 values may be attributed to the difference in the solvents (MeOH-d4 and D2O), chosen in these two cases for recording NMR. Comparing and considering the NMR data in these two cases we surmised that all the three anomeric carbons are α-configured [47].
Conclusion
In short, we have described three different efficient routes to access the A-band trisaccharide associated with P. aeruginosa. The radical-based 6-deoxygenation protocol on 4,6-O-benzylidene (hitherto unexplored on thioglycosides) was utilized to arrive at the D-rhamno-thioglycoside derivatives from their D-manno-counterparts. This, along with a similar global 6-deoxygenation strategy on D-manno-trisaccharide derivative (bearing 4,6-O-benzylidene protection on each mannose unit) offer great potential in future oligosaccharide syntheses based on 6-deoxy hexoses.
Supporting Information
| Supporting Information File 1: Experimental details for the preparation of compounds 2a, 2b, 3a, 4, 6–13 and the corresponding characterization data. | ||
| Format: PDF | Size: 319.7 KB | Download |
| Supporting Information File 2: 1H and 13C NMR of compounds 2a, 4, 6, 8–13 and 2D NMR (COSY, HSQC and HMBC) of compound 10. | ||
| Format: PDF | Size: 4.8 MB | Download |
Acknowledgements
The authors are grateful to CSIR, New Delhi, India for providing a fellowship (SRF) to AC and funding of project (No. 2382/01/10/2010 EMR II) to RG. Financial assistance from DST-SERB, India to RG and supports from CAS-UGC, India and FIST-DST, India to the Department of Chemistry, Jadavpur University, Kolkata are also acknowledged.
References
-
Varki, A. Glycobiology 1993, 3, 97–130. doi:10.1093/glycob/3.2.97
Return to citation in text: [1] -
Varki, A.; Cummings, D. C.; Esko, J. D.; Freeze, H. H.; Stanley, P.; Bertozzi, C. R.; Hart, G. W.; Etzler, M. E. Essentials of Glycobiology; Cold Spring Harbor Press: New York, 2009.
Return to citation in text: [1] -
Seeberger, P. H.; Werz, D. B. Nature 2007, 446, 1046–1051. doi:10.1038/nature05819
Return to citation in text: [1] -
Fuster, M. M.; Esko, J. D. Nat. Rev. Cancer 2005, 5, 526–542. doi:10.1038/nrc1649
Return to citation in text: [1] -
Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036
Return to citation in text: [1] -
Nicolaou, K. C.; Mitchell, H. J. Angew. Chem., Int. Ed. 2001, 40, 1576–1624. doi:10.1002/1521-3773(20010504)40:9<1576::AID-ANIE15760>3.0.CO;2-G
Return to citation in text: [1] -
Hsu, C.-H.; Hung, S.-C.; Wu, C.-Y.; Wong, C.-H. Angew. Chem., Int. Ed. 2011, 50, 11872–11923. doi:10.1002/anie.201100125
Return to citation in text: [1] -
Herasimenka, Y.; Cescutti, P.; Impallomeni, G.; Campana, S.; Taccetti, G.; Ravenni, N.; Zanetti, F.; Rizzo, R. J. Cystic Fibrosis 2007, 6, 145–152. doi:10.1016/j.jcf.2006.06.004
Return to citation in text: [1] -
Yokota, S.-i.; Kaya, S.; Sawada, S.; Kawamura, T.; Araki, Y.; Ito, E. Eur. J. Biochem. 1987, 167, 203–209. doi:10.1111/j.1432-1033.1987.tb13324.x
Return to citation in text: [1] -
Kocharova, N. A.; Knirel, Y. A.; Widmalm, G.; Jansson, P. E.; Moran, A. P. Biochemistry 2000, 39, 4755–4760. doi:10.1021/bi992635k
Return to citation in text: [1] -
Kocharova, N. A.; Borisova, S. A.; Zatonsky, G. V.; Shashkov, A. S.; Knirel, Y. A.; Kholodkova, E. V.; Staniskavsky, E. S. Carbohydr. Res. 1998, 306, 331–333. doi:10.1016/S0008-6215(97)10049-0
Return to citation in text: [1] -
Senchenkova, S. N.; Shashkov, A. S.; Knirel, Y. A.; Mcgovern, J. J.; Moran, A. P. Eur. J. Biochem. 1996, 239, 434–438. doi:10.1111/j.1432-1033.1996.0434u.x
Return to citation in text: [1] -
Winn, A. M.; Wilkinson, S. G. Carbohydr. Res. 1996, 294, 109–115. doi:10.1016/S0008-6215(96)90623-0
Return to citation in text: [1] -
Molinaro, A.; Silipo, A.; Lanzetta, R.; Newman, M.-A.; Dow, M. J.; Parrilli, M. Carbohydr. Res. 2003, 338, 277–281. doi:10.1016/S0008-6215(02)00433-0
Return to citation in text: [1] -
Kihlberg, J.; Bundle, D. R. Carbohydr. Res. 1992, 216, 67–78. doi:10.1016/0008-6215(92)84151-H
Return to citation in text: [1] -
Arsenault, T. L.; Hughes, D. W.; MacLean, D. B.; Szarek, W. A.; Kropinski, A. M. B.; Lam, J. S. Can. J. Chem. 1991, 69, 1273–1280. doi:10.1139/v91-190
Return to citation in text: [1] [2] [3] -
Tsvetkov, Y. E.; Backinowsky, L. V.; Kochetkov, N. K. Carbohydr. Res. 1989, 193, 75–90. doi:10.1016/0008-6215(89)85108-0
Return to citation in text: [1] -
Zou, W.; Sen, A. K.; Szarek, W. A.; MacLean, D. B. Can. J. Chem. 1993, 71, 2194–2200. doi:10.1139/v93-275
Return to citation in text: [1] -
Picard, S.; Crich, D. Chimia 2011, 65, 59–64. doi:10.2533/chimia.2011.59
Return to citation in text: [1] [2] [3] -
Crich, D.; Bowers, A. A. Org. Lett. 2006, 8, 4327–4330. doi:10.1021/ol061706m
Return to citation in text: [1] [2] [3] -
Crich, D.; Bowers, A. A. J. Org. Chem. 2006, 71, 3452–3463. doi:10.1021/jo0526688
Return to citation in text: [1] [2] [3] -
Crich, D.; Yao, Q. J. Am. Chem. Soc. 2004, 126, 8232–8236. doi:10.1021/ja048070j
Return to citation in text: [1] [2] [3] -
Crich, D.; Li, L. J. Org. Chem. 2009, 74, 773–781. doi:10.1021/jo8022439
Return to citation in text: [1] [2] [3] -
Bedini, E.; Carabellese, A.; Barone, G.; Parrilli, M. J. Org. Chem. 2005, 70, 8064–8070. doi:10.1021/jo051153d
Return to citation in text: [1] -
Bedini, E.; Carabellese, A.; Corsaro, M. M.; De Castro, C.; Parrilli, M. Carbohydr. Res. 2004, 339, 1907–1915. doi:10.1016/j.carres.2004.06.010
Return to citation in text: [1] [2] -
Fauré, R.; Shiao, T. C.; Damerval, S.; Roy, R. Tetrahedron Lett. 2007, 48, 2385–2388. doi:10.1016/j.tetlet.2007.01.122
Return to citation in text: [1] -
Zunk, M.; Kiefel, M. J. Tetrahedron Lett. 2011, 52, 1296–1299. doi:10.1016/j.tetlet.2011.01.064
Return to citation in text: [1] -
Jeppesen, L. M.; Lundt, I.; Pedersen, C. Acta Chem. Scand. 1973, 27, 3579–3585. doi:10.3891/acta.chem.scand.27-3579
Return to citation in text: [1] [2] -
Roberts, B. P.; Smits, T. M. Tetrahedron Lett. 2001, 42, 3663–3666. doi:10.1016/S0040-4039(01)00540-8
Return to citation in text: [1] -
Dang, H.-S.; Roberts, B. P.; Sekhon, J.; Smits, T. M. Org. Biomol. Chem. 2003, 1, 1330–1341. doi:10.1039/b212303g
Return to citation in text: [1] [2] [3] [4] -
Fielding, A. J.; Franchi, P.; Roberts, B. P.; Smits, T. M. J. Chem. Soc., Perkin Trans. 2 2002, 155–163. doi:10.1039/B106140M
Return to citation in text: [1] -
Cai, Y.; Dang, H.-S.; Roberts, B. P. J. Chem. Soc., Perkin Trans. 1 2002, 2449–2458. doi:10.1039/b208200b
Return to citation in text: [1] -
Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1035–1044. doi:10.1021/jo01256a059
Return to citation in text: [1] -
Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1045–1053. doi:10.1021/jo01256a060
Return to citation in text: [1] -
Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1053–1058. doi:10.1021/jo01256a061
Return to citation in text: [1] -
Chana, J. S.; Collins, P. M.; Farnia, F.; Peacock, D. J. J. Chem. Soc., Chem. Commun. 1988, 94–96. doi:10.1039/c39880000094
Return to citation in text: [1] -
Binkley, R. W.; Goewey, G. S.; Johnston, J. C. J. Org. Chem. 1984, 49, 992–996. doi:10.1021/jo00180a008
Return to citation in text: [1] -
Hanessian, S. Org. Synth. 1987, 65, 243–250.
Return to citation in text: [1] -
Hullar, T. L.; Siskin, S. B. J. Org. Chem. 1970, 35, 225–228. doi:10.1021/jo00826a046
Return to citation in text: [1] -
Niu, Y.; Wang, N.; Cao, X.; Ye, X.-S. Synlett 2007, 2116–2120. doi:10.1055/s-2007-984903
Return to citation in text: [1] -
Shivatare, S. S.; Chang, S.-H.; Tsai, T.-I.; Ren, C.-T.; Chuang, H.-Y.; Hsu, L.; Lin, C.-W.; Li, S.-T.; Wu, C.-Y.; Wong, C.-H. J. Am. Chem. Soc. 2013, 135, 15382–15391. doi:10.1021/ja409097c
Return to citation in text: [1] -
Franzyk, H.; Meldal, M.; Paulsen, H.; Bock, K. J. Chem. Soc., Perkin Trans. 1 1995, 2883–2898. doi:10.1039/p19950002883
Return to citation in text: [1] -
Basu, N.; Maity, S. K.; Chaudhury, A.; Ghosh, R. Carbohydr. Res. 2013, 369, 10–13. doi:10.1016/j.carres.2013.01.001
Return to citation in text: [1] -
Fauré, R.; Shiao, T. C.; Lagnoux, D.; Giguère, D.; Roy, R. Org. Biomol. Chem. 2007, 5, 2704–2708. doi:10.1039/b708365c
Return to citation in text: [1] [2] -
Chevalier, R.; Esnault, J.; Vandewalle, P.; Sendid, B.; Colombel, J.-F.; Poulain, D.; Mallet, J.-M. Tetrahedron 2005, 61, 7669–7677. doi:10.1016/j.tet.2005.05.098
Return to citation in text: [1] -
Crich, D.; Smith, M. J. Am. Chem. Soc. 2001, 123, 9015–9020. doi:10.1021/ja0111481
Return to citation in text: [1] -
Kasai, R.; Okihara, M.; Asakawa, J.; Mizutani, K.; Tanaka, O. Tetrahedron 1979, 35, 1427–1432. doi:10.1016/0040-4020(79)85038-3
Return to citation in text: [1]
| 30. | Dang, H.-S.; Roberts, B. P.; Sekhon, J.; Smits, T. M. Org. Biomol. Chem. 2003, 1, 1330–1341. doi:10.1039/b212303g |
| 43. | Basu, N.; Maity, S. K.; Chaudhury, A.; Ghosh, R. Carbohydr. Res. 2013, 369, 10–13. doi:10.1016/j.carres.2013.01.001 |
| 44. | Fauré, R.; Shiao, T. C.; Lagnoux, D.; Giguère, D.; Roy, R. Org. Biomol. Chem. 2007, 5, 2704–2708. doi:10.1039/b708365c |
| 1. | Varki, A. Glycobiology 1993, 3, 97–130. doi:10.1093/glycob/3.2.97 |
| 2. | Varki, A.; Cummings, D. C.; Esko, J. D.; Freeze, H. H.; Stanley, P.; Bertozzi, C. R.; Hart, G. W.; Etzler, M. E. Essentials of Glycobiology; Cold Spring Harbor Press: New York, 2009. |
| 3. | Seeberger, P. H.; Werz, D. B. Nature 2007, 446, 1046–1051. doi:10.1038/nature05819 |
| 4. | Fuster, M. M.; Esko, J. D. Nat. Rev. Cancer 2005, 5, 526–542. doi:10.1038/nrc1649 |
| 10. | Kocharova, N. A.; Knirel, Y. A.; Widmalm, G.; Jansson, P. E.; Moran, A. P. Biochemistry 2000, 39, 4755–4760. doi:10.1021/bi992635k |
| 19. | Picard, S.; Crich, D. Chimia 2011, 65, 59–64. doi:10.2533/chimia.2011.59 |
| 20. | Crich, D.; Bowers, A. A. Org. Lett. 2006, 8, 4327–4330. doi:10.1021/ol061706m |
| 21. | Crich, D.; Bowers, A. A. J. Org. Chem. 2006, 71, 3452–3463. doi:10.1021/jo0526688 |
| 22. | Crich, D.; Yao, Q. J. Am. Chem. Soc. 2004, 126, 8232–8236. doi:10.1021/ja048070j |
| 23. | Crich, D.; Li, L. J. Org. Chem. 2009, 74, 773–781. doi:10.1021/jo8022439 |
| 9. | Yokota, S.-i.; Kaya, S.; Sawada, S.; Kawamura, T.; Araki, Y.; Ito, E. Eur. J. Biochem. 1987, 167, 203–209. doi:10.1111/j.1432-1033.1987.tb13324.x |
| 24. | Bedini, E.; Carabellese, A.; Barone, G.; Parrilli, M. J. Org. Chem. 2005, 70, 8064–8070. doi:10.1021/jo051153d |
| 25. | Bedini, E.; Carabellese, A.; Corsaro, M. M.; De Castro, C.; Parrilli, M. Carbohydr. Res. 2004, 339, 1907–1915. doi:10.1016/j.carres.2004.06.010 |
| 8. | Herasimenka, Y.; Cescutti, P.; Impallomeni, G.; Campana, S.; Taccetti, G.; Ravenni, N.; Zanetti, F.; Rizzo, R. J. Cystic Fibrosis 2007, 6, 145–152. doi:10.1016/j.jcf.2006.06.004 |
| 17. | Tsvetkov, Y. E.; Backinowsky, L. V.; Kochetkov, N. K. Carbohydr. Res. 1989, 193, 75–90. doi:10.1016/0008-6215(89)85108-0 |
| 47. | Kasai, R.; Okihara, M.; Asakawa, J.; Mizutani, K.; Tanaka, O. Tetrahedron 1979, 35, 1427–1432. doi:10.1016/0040-4020(79)85038-3 |
| 5. | Zhu, X.; Schmidt, R. R. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. doi:10.1002/anie.200802036 |
| 6. | Nicolaou, K. C.; Mitchell, H. J. Angew. Chem., Int. Ed. 2001, 40, 1576–1624. doi:10.1002/1521-3773(20010504)40:9<1576::AID-ANIE15760>3.0.CO;2-G |
| 7. | Hsu, C.-H.; Hung, S.-C.; Wu, C.-Y.; Wong, C.-H. Angew. Chem., Int. Ed. 2011, 50, 11872–11923. doi:10.1002/anie.201100125 |
| 18. | Zou, W.; Sen, A. K.; Szarek, W. A.; MacLean, D. B. Can. J. Chem. 1993, 71, 2194–2200. doi:10.1139/v93-275 |
| 14. | Molinaro, A.; Silipo, A.; Lanzetta, R.; Newman, M.-A.; Dow, M. J.; Parrilli, M. Carbohydr. Res. 2003, 338, 277–281. doi:10.1016/S0008-6215(02)00433-0 |
| 16. | Arsenault, T. L.; Hughes, D. W.; MacLean, D. B.; Szarek, W. A.; Kropinski, A. M. B.; Lam, J. S. Can. J. Chem. 1991, 69, 1273–1280. doi:10.1139/v91-190 |
| 46. | Crich, D.; Smith, M. J. Am. Chem. Soc. 2001, 123, 9015–9020. doi:10.1021/ja0111481 |
| 13. | Winn, A. M.; Wilkinson, S. G. Carbohydr. Res. 1996, 294, 109–115. doi:10.1016/S0008-6215(96)90623-0 |
| 16. | Arsenault, T. L.; Hughes, D. W.; MacLean, D. B.; Szarek, W. A.; Kropinski, A. M. B.; Lam, J. S. Can. J. Chem. 1991, 69, 1273–1280. doi:10.1139/v91-190 |
| 16. | Arsenault, T. L.; Hughes, D. W.; MacLean, D. B.; Szarek, W. A.; Kropinski, A. M. B.; Lam, J. S. Can. J. Chem. 1991, 69, 1273–1280. doi:10.1139/v91-190 |
| 12. | Senchenkova, S. N.; Shashkov, A. S.; Knirel, Y. A.; Mcgovern, J. J.; Moran, A. P. Eur. J. Biochem. 1996, 239, 434–438. doi:10.1111/j.1432-1033.1996.0434u.x |
| 30. | Dang, H.-S.; Roberts, B. P.; Sekhon, J.; Smits, T. M. Org. Biomol. Chem. 2003, 1, 1330–1341. doi:10.1039/b212303g |
| 11. | Kocharova, N. A.; Borisova, S. A.; Zatonsky, G. V.; Shashkov, A. S.; Knirel, Y. A.; Kholodkova, E. V.; Staniskavsky, E. S. Carbohydr. Res. 1998, 306, 331–333. doi:10.1016/S0008-6215(97)10049-0 |
| 15. | Kihlberg, J.; Bundle, D. R. Carbohydr. Res. 1992, 216, 67–78. doi:10.1016/0008-6215(92)84151-H |
| 45. | Chevalier, R.; Esnault, J.; Vandewalle, P.; Sendid, B.; Colombel, J.-F.; Poulain, D.; Mallet, J.-M. Tetrahedron 2005, 61, 7669–7677. doi:10.1016/j.tet.2005.05.098 |
| 28. | Jeppesen, L. M.; Lundt, I.; Pedersen, C. Acta Chem. Scand. 1973, 27, 3579–3585. doi:10.3891/acta.chem.scand.27-3579 |
| 26. | Fauré, R.; Shiao, T. C.; Damerval, S.; Roy, R. Tetrahedron Lett. 2007, 48, 2385–2388. doi:10.1016/j.tetlet.2007.01.122 |
| 27. | Zunk, M.; Kiefel, M. J. Tetrahedron Lett. 2011, 52, 1296–1299. doi:10.1016/j.tetlet.2011.01.064 |
| 25. | Bedini, E.; Carabellese, A.; Corsaro, M. M.; De Castro, C.; Parrilli, M. Carbohydr. Res. 2004, 339, 1907–1915. doi:10.1016/j.carres.2004.06.010 |
| 44. | Fauré, R.; Shiao, T. C.; Lagnoux, D.; Giguère, D.; Roy, R. Org. Biomol. Chem. 2007, 5, 2704–2708. doi:10.1039/b708365c |
| 40. | Niu, Y.; Wang, N.; Cao, X.; Ye, X.-S. Synlett 2007, 2116–2120. doi:10.1055/s-2007-984903 |
| 41. | Shivatare, S. S.; Chang, S.-H.; Tsai, T.-I.; Ren, C.-T.; Chuang, H.-Y.; Hsu, L.; Lin, C.-W.; Li, S.-T.; Wu, C.-Y.; Wong, C.-H. J. Am. Chem. Soc. 2013, 135, 15382–15391. doi:10.1021/ja409097c |
| 42. | Franzyk, H.; Meldal, M.; Paulsen, H.; Bock, K. J. Chem. Soc., Perkin Trans. 1 1995, 2883–2898. doi:10.1039/p19950002883 |
| 19. | Picard, S.; Crich, D. Chimia 2011, 65, 59–64. doi:10.2533/chimia.2011.59 |
| 20. | Crich, D.; Bowers, A. A. Org. Lett. 2006, 8, 4327–4330. doi:10.1021/ol061706m |
| 21. | Crich, D.; Bowers, A. A. J. Org. Chem. 2006, 71, 3452–3463. doi:10.1021/jo0526688 |
| 22. | Crich, D.; Yao, Q. J. Am. Chem. Soc. 2004, 126, 8232–8236. doi:10.1021/ja048070j |
| 23. | Crich, D.; Li, L. J. Org. Chem. 2009, 74, 773–781. doi:10.1021/jo8022439 |
| 19. | Picard, S.; Crich, D. Chimia 2011, 65, 59–64. doi:10.2533/chimia.2011.59 |
| 20. | Crich, D.; Bowers, A. A. Org. Lett. 2006, 8, 4327–4330. doi:10.1021/ol061706m |
| 21. | Crich, D.; Bowers, A. A. J. Org. Chem. 2006, 71, 3452–3463. doi:10.1021/jo0526688 |
| 22. | Crich, D.; Yao, Q. J. Am. Chem. Soc. 2004, 126, 8232–8236. doi:10.1021/ja048070j |
| 23. | Crich, D.; Li, L. J. Org. Chem. 2009, 74, 773–781. doi:10.1021/jo8022439 |
| 30. | Dang, H.-S.; Roberts, B. P.; Sekhon, J.; Smits, T. M. Org. Biomol. Chem. 2003, 1, 1330–1341. doi:10.1039/b212303g |
| 33. | Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1035–1044. doi:10.1021/jo01256a059 |
| 34. | Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1045–1053. doi:10.1021/jo01256a060 |
| 35. | Hanessian, S.; Plessas, N. R. J. Org. Chem. 1969, 34, 1053–1058. doi:10.1021/jo01256a061 |
| 36. | Chana, J. S.; Collins, P. M.; Farnia, F.; Peacock, D. J. J. Chem. Soc., Chem. Commun. 1988, 94–96. doi:10.1039/c39880000094 |
| 37. | Binkley, R. W.; Goewey, G. S.; Johnston, J. C. J. Org. Chem. 1984, 49, 992–996. doi:10.1021/jo00180a008 |
| 38. | Hanessian, S. Org. Synth. 1987, 65, 243–250. |
| 39. | Hullar, T. L.; Siskin, S. B. J. Org. Chem. 1970, 35, 225–228. doi:10.1021/jo00826a046 |
| 29. | Roberts, B. P.; Smits, T. M. Tetrahedron Lett. 2001, 42, 3663–3666. doi:10.1016/S0040-4039(01)00540-8 |
| 30. | Dang, H.-S.; Roberts, B. P.; Sekhon, J.; Smits, T. M. Org. Biomol. Chem. 2003, 1, 1330–1341. doi:10.1039/b212303g |
| 31. | Fielding, A. J.; Franchi, P.; Roberts, B. P.; Smits, T. M. J. Chem. Soc., Perkin Trans. 2 2002, 155–163. doi:10.1039/B106140M |
| 32. | Cai, Y.; Dang, H.-S.; Roberts, B. P. J. Chem. Soc., Perkin Trans. 1 2002, 2449–2458. doi:10.1039/b208200b |
| 28. | Jeppesen, L. M.; Lundt, I.; Pedersen, C. Acta Chem. Scand. 1973, 27, 3579–3585. doi:10.3891/acta.chem.scand.27-3579 |
© 2014 Chaudhury et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)