Abstract
New compounds with carbohydrate-similar structure (carbohydrate mimetics) are presented in this article. Starting from enantiopure nitrones and lithiated TMSE-allene we prepared three 1,2-oxazine derivatives which underwent a highly stereoselective Lewis acid-induced rearrangement to give bicyclic products in good yield. Subsequent reductive transformations delivered a library of new poly(hydroxy)aminooxepane derivatives. The crucial final palladium-catalyzed hydrogenolysis of the 1,2-oxazine moiety was optimized resulting in a reasonably efficient approach to a series of new seven-membered carbohydrate mimetics.

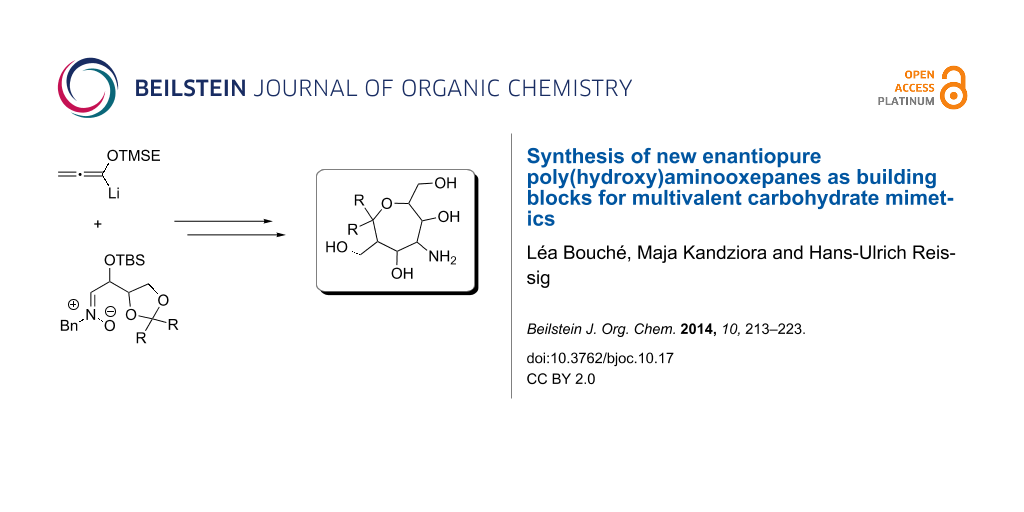
Graphical Abstract
Introduction
Since carbohydrates play a crucial role in biochemistry, compounds mimicking their structure and/or function (carbohydrate mimetics) have attracted great attention in academic research and in drug development [1-3]. These mimetics should not have the drawbacks of carbohydrates such as low binding affinity or instability [4,5]. Many carbohydrates and their mimetics contain pyran rings, however, the corresponding ring-expanded compounds, oxepanes, have been investigated only in a limited number of studies. Several oxepane units can be found in natural products, in particular as toxins in marine organisms or plants [6,7]. A few reports in the literature deal with seven-membered polyhydroxylated ethers like septanosides (containing an anomeric center) [8] as well as compounds without an acetal moiety. Known methods for the construction of the seven-membered polyhydroxylated oxacycle, are for instance cycloaddition [9-11] or cyclodehydration of commercially available alcohols [12-14]. The disadvantages of these methods are the lack of selectivity as well as of stereocontrol and hence other routes are needed. One interesting option is the pathway via oxepines [15-18] and the subsequent dihydroxylation or direct reduction of their C=C double bond to give the corresponding oxepane derivatives [19,20]. Alternatively, oxepanes were also synthesized by the ring enlargement of their six-membered homologues [21,22]. Disappointingly, these methods often involve long reaction sequences and display sometimes restricted flexibility. A search for new stereoselective approaches is therefore highly desirable.
During the last years our group systematically studied a new approach to carbohydrate mimetics D [23-28]. The general approach is shown in Scheme 1: the chiral pool-derived nitrones A [29,30] undergo a [3 + 3]-cyclization with lithiated [2-(trimethylsilyl)ethoxy]allene (TMSE-allene) [31] as C-3 building block [32] to form the 3,6-dihydro-2H-1,2-oxazines B; subsequent Lewis acid-promoted reactions [23] lead to the highly functionalized bicyclic 1,2-oxazinones C which can be regarded as protected aminopolyol precursors offering the option for plenty of selective transformations [27,28]; after reductive steps the polyhydroxylated cyclic ethers D are obtained. The advantages of this route are the stereocontrol and the flexibility concerning the chain length. In 2005, Al-Harrasi synthesized the first tert-butyldimethylsilyl (TBS)-protected aminopyrans (n = 0) and aminooxepanes (n = 1) via this reaction route [23]. Several of the poly(hydroxy)aminopyrans [24] were connected to gold nanoparticles and the resulting multivalent conjugates showed extremely high binding to P- and L-selectine [33,34]. For the planned biological testing of the corresponding aminooxepanes as components of multivalent conjugates, we required the fully deprotected compounds. Moreover, it was desirable to have additional derivatives with different configurations or functional groups. In this article, we therefore describe the full details of our route to a series of new enantiopure poly(hydroxy)aminooxepanes with variations at the 2-, 5- and 7-position of the seven-membered cyclic ether D.
![[1860-5397-10-17-i1]](/bjoc/content/inline/1860-5397-10-17-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General approach to enantiopure the poly(hydroxy)aminopyrans D (n = 0) and the aminooxepanes D (n = 1) by [3 + 3]-cyclization of the (Z)-nitrones A with lithiated TMSE-allene followed by Lewis acid-induced rearrangement of 3,6-dihydro-2H-1,2-oxazines B. [TBS = tert-butyldimethylsilyl, TMSE = 2-(trimethylsilyl)ethyl]
Scheme 1: General approach to enantiopure the poly(hydroxy)aminopyrans D (n = 0) and the aminooxepanes D (n =...
Results and Discussion
The enantiopure (Z)-nitrones 3, 6 and 8 were synthesized essentially following known pathways [35] (Scheme 2 and Scheme 3). The new D-erythrose-derived nitrone 3 containing a p-bromophenyl moiety in the dioxolane ring was obtained in a straightforward manner (Scheme 2). Commercially available D-isoascorbic acid (1) was smoothly transformed in four steps into the ethyl ester 2. After protection of the free 1,2-diol unit of 1 employing p-bromobenzaldehyde dimethylacetal, the ring double bond was oxidatively cleaved to give the sodium carboxylate which was directly transformed into the corresponding ethyl ester [36]. Protection with a TBS-group proceeded quantitatively to gain 2 in very good overall efficacy. Final transformation into the desired nitrone 3 was achieved in 83% yield (over two steps) by reduction of the ester using DIBAL-H at low temperature, followed by condensation with N-benzylhydroxylamine according to a Dondoni protocol [29]. This route allows the synthesis of enantiopure nitrone 3 in multi-gram scale.
![[1860-5397-10-17-i2]](/bjoc/content/inline/1860-5397-10-17-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of (Z)-nitrone 3. Conditions: a) 1. p-Bromobenzaldehyde dimethylacetal, TFA, DMF, rt, 5 d; 2. 30% H2O2, K2CO3, H2O, 0 → 20 °C, 16 h; 3. EtI, MeCN, reflux, 20 h; b) TBSCl, imidazole, DMAP, CH2Cl2, 3 d, rt; c) 1. DIBAL-H, CH2Cl2, −78 °C, 3.5 h; 2. N-benzylhydroxylamine, MgSO4, CH2Cl2, rt, 16 h. [TFA = trifluoroacetic acid, DMAP = 4-(N,N-dimethylamino)pyridine, DIBAL-H = diisobutylaluminium hydride]
Scheme 2: Synthesis of (Z)-nitrone 3. Conditions: a) 1. p-Bromobenzaldehyde dimethylacetal, TFA, DMF, rt, 5 d...
![[1860-5397-10-17-i3]](/bjoc/content/inline/1860-5397-10-17-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1,2-oxazines syn-7, syn-9 and syn-10. Conditions: a) n-BuLi, THF, −40 °C, 15 min; b) 1. THF, −78 °C, 1.5 to 3 h; 2. H2O, 1 h, −78 °C → rt. The syn:anti ratios were determined by 1H NMR spectroscopic analysis of the crude products and are all >95:5 (see Supporting Information File 2).
Scheme 3: Synthesis of 1,2-oxazines syn-7, syn-9 and syn-10. Conditions: a) n-BuLi, THF, −40 °C, 15 min; b) 1...
The enantiopure (Z)-configured nitrones 6, 8 and 3 were treated with in situ lithiated TMSE-allene at −78 °C furnishing the expected 1,2-oxazines syn-7 [37], syn-9 and syn-10 in high yields and with excellent diastereoselectivities (Scheme 3). The nitrone 3 was used as a 96:4 mixture (with respect to the stereogenic center of the 1,3-dioxolane ring) and it could therefore provide four diasteromers of 1,2-oxazine syn-10 (two major syn- and two minor anti-diasteromeres), but only a single syn-stereoisomer (with respect to C-3 of the 1,2-oxazine ring) was isolated after column chromatography. A product derived from the minor isomer of nitrone 3 was not detected and was apparently lost during the reaction or the purification. Since the stereogenic center of the 1,3-dioxolane ring is converted into an sp2-hybridized carbon during the Lewis acid-induced ring opening, the configuration of syn-10 was not assigned at this center.
We discussed the mechanism of this [3 + 3]-cyclization in detail including possible side-reactions in an earlier report [31]. The observed high syn-diastereoselectivities are in accordance with our previously published results and were supported by an X-ray analysis of syn-7 [37]. We conclude from our observations that the formation of 1,2-oxazines from nitrones 3, 6 and 8 is mainly steered by the stereogenicity at the α-carbon (1,2-induction) [30,31], whereas the stereogenic center at the β-carbon (1,3-induction) has no or negligible influence. A few syn-selective additions of other nucleophiles to nitrones with two stereogenic centers similar to 6 or 8 were reported in the literature [38,39]. A preferred transition structure as depicted in Figure 1 plausibly explains our results. The silyloxy group at the α-carbon occupies an orthogonal position, the substituent R is close to the nitrone hydrogen substituent and the lithiated allene attacks from the Re-side of the nitrone (Felkin–Anh model for nitrones as proposed by Dondoni et al. [30]). An additional coordination of the lithium cation to the nitrone oxygen is possible.
![[1860-5397-10-17-1]](/bjoc/content/figures/1860-5397-10-17-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Proposed transition structure for the addition of lithiated TMSE-allene 5 to chiral nitrones 3, 6 and 8.
Figure 1: Proposed transition structure for the addition of lithiated TMSE-allene 5 to chiral nitrones 3, 6 a...
The Lewis acid-induced rearrangements of 1,2-oxazines syn-7, syn-9 and syn-10 were achieved in moderate to good yields using TMSOTf as promoter (Scheme 4). A higher yield of 73% was achieved for the p-bromophenyl derivative 13 (isolated as a single diastereomer) which may be due to the stabilized carbenium ion formed at the benzylic position.
![[1860-5397-10-17-i4]](/bjoc/content/inline/1860-5397-10-17-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of ketones 11, 12 and 13 with a bicyclic 1,2-oxazine skeleton by Lewis acid-induced rearrangement. Conditions: a) 1. TMSOTf, CH2Cl2, −5 °C, 5 to 6 h; 2. aq NH3, rt, 10 min. [TMSOTf = trimethylsilyl trifluoromethanesulfonate]
Scheme 4: Synthesis of ketones 11, 12 and 13 with a bicyclic 1,2-oxazine skeleton by Lewis acid-induced rearr...
This rearrangement presents a relatively rare example of an intramolecular aldol-like reaction of an enol ether with an activated acetal (which may also be regarded as a special case of a Prins reaction) forming a seven-membered ring. We propose a Zimmerman–Traxler-type transition state (Scheme 5), placing the aryl group R2 (R1 = H) in the sterically more favorable pseudo-equatorial position in the reaction leading to compound 13 (for other Lewis acid-mediated formations of seven membered oxacycles, see references [40,41]).
![[1860-5397-10-17-i5]](/bjoc/content/inline/1860-5397-10-17-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed extended chair-like conformation with Zimmerman–Traxler-type transition state.
Scheme 5: Proposed extended chair-like conformation with Zimmerman–Traxler-type transition state.
The relative configuration of the newly formed stereogenic center at the C-2 position of 13 was successfully determined by NOE experiments. By irradiation of the 2-H proton (Figure 2) several dipolar couplings with 1-H, 4-H and with one proton of the p-bromophenyl group were observed. For this reason, it is assumed that the 2-H proton of 13 is cis-orientated to the 1-H and 4-H protons and consequently shows an (R)-configuration.
![[1860-5397-10-17-2]](/bjoc/content/figures/1860-5397-10-17-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: GOESY–NMR spectrum (CDCl3, 500 MHz) of bicyclic 1,2-oxazine 13: irradiation of the 2-H proton. [GOESY = gradient enhanced nuclear Overhauser effect spectroscopy]
Figure 2: GOESY–NMR spectrum (CDCl3, 500 MHz) of bicyclic 1,2-oxazine 13: irradiation of the 2-H proton. [GOE...
Ketones 11, 12 and 13 were subsequently smoothly reduced with sodium borohydride in ethanol. In all cases, only one diastereomer (hydroxy group at C-10 with an S-configuration) was isolated (Scheme 6). These results are in accordance with previous observations obtained for the corresponding TBS-protected derivatives [42] suggesting that a preferred conformation of the seven-membered ring favors the hydride attack only from the Re-side. The Si-side (back-side) attack is probably hindered by the bulky OTBS group (also see Scheme 5). The configurational assignments are in accordance with NOE experiments performed with the alcohol derived from ketone 13 (precursor of 16), where dipolar couplings have been observed between the 10-H and 9-H protons (CH2 next to the N–O bond). Finally, the TBS-groups were removed under standard conditions furnishing triols 14, 15 and 16 in high yields.
![[1860-5397-10-17-i6]](/bjoc/content/inline/1860-5397-10-17-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of triols 14, 15 and 16 by reduction of the carbonyl group and deprotection. Conditions: a) NaBH4, EtOH, 0 °C, 40 min to 16 h; b) TBAF (1 M THF), THF, 0 °C, 10 min to 3.5 h. [TBAF = tetra-n-butylammonium fluoride]
Scheme 6: Synthesis of triols 14, 15 and 16 by reduction of the carbonyl group and deprotection. Conditions: ...
The free primary hydroxy group of bicyclic ketone 11 was propargylated in 71% yield employing propargylic bromide under standard conditions (Scheme 7). After reduction of the carbonyl group [43] and TBS cleavage, propargylic ether 18 was obtained (88% yield over two steps) [44]. The alkyne moiety provides options for further transformations, e.g. Sonogashira reactions, Glaser couplings or 1,3-dipolar cycloadditions (click reactions) [45].
![[1860-5397-10-17-i7]](/bjoc/content/inline/1860-5397-10-17-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of propargylic ether 18. Conditions: a) propargyl bromide, NaOH, TBAI, H2O/CH2Cl2, −20 °C → rt, 7 d; b) NaBH4, EtOH, 0 °C, 4 h; c) TBAF (1 M THF), THF, 0 °C → rt, 3 d. [TBAI = tetra-n-butylammonium iodide]
Scheme 7: Synthesis of propargylic ether 18. Conditions: a) propargyl bromide, NaOH, TBAI, H2O/CH2Cl2, −20 °C...
We were also interested in the preparation of the bicyclic azide 24 and hence the primary hydroxy group of 11 was converted into mesylate 19 followed by a reduction of the carbonyl group (Scheme 8). The attempted nucleophilic substitution by sodium azide gave a mixture of two products: the desired TBS-protected azide 21 and an unexpected side product 22. To overcome the formation of this side product, the introduction of the azido group was directly performed with mesylate 19. Since the nucleophilic substitution of 19 into 23 gave only moderate yields with low reproducibility (from 39–67%), this step was optimized using Mitsunobu conditions with diphenylphosphoryl azide according to a protocol by Bose [46]. The desired bicyclic azide 23 was now isolated in 79% yield and its subsequent reduction and deprotection with TBAF gave compound 24 in essentially quantitative yield (over two steps). After protection of the two hydroxy substituents of 24 with TBS-groups, a Staudinger reaction was performed, which furnished bicyclic 1,2-oxazine derivative 25 with a primary amino group in 80% yield (over two steps).
![[1860-5397-10-17-i8]](/bjoc/content/inline/1860-5397-10-17-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of tricyclic compound 20, bicyclic azide 24 and bicyclic amine 25. Conditions: a) MsCl, Et3N, CH2Cl2, 0 °C → rt, 5 h; b) NaBH4, EtOH, 0 °C, 6 to 16 h; c) NaN3, DMF, reflux, 16 to 46 h; d) Et3N, DMF, reflux, 16 h; e) TBAF (1 M THF), THF, 0 °C → rt, 1 h; f) TPP, DIAD, DPPA, THF, −20 °C → rt, 2 d; g) TBSOTf, Et3N, CH2Cl2, 0 °C → rt, 3.5 h; h) TPP, THF/H2O, rt, 16 h. [Ms = methanesulfonate, TPP = triphenylphosphane, DIAD = diisopropyl azodicarboxylate, DPPA = diphenylphosphoryl azide]
Scheme 8: Synthesis of tricyclic compound 20, bicyclic azide 24 and bicyclic amine 25. Conditions: a) MsCl, Et...
Next we wanted to synthesize the unexpectedly formed unique tricyclic 1,2-oxazine 20 with higher efficacy. After reduction of 19 with sodium borohydride the intermediate alcohol was heated with triethylamine (to avoid the deprotection of the TBS-group) and compound 20 was obtained in 75% yield (over three steps).
The previously isolated unprotected triols 14, 15 and 20 were subjected to standard hydrogenolysis conditions using palladium on charcoal in order to remove the N-benzyl group and to cleave the N–O bond [47,48] in one step. This process is often challenging due to the difficult control of the various reaction parameters and also because of the high polarity of the newly formed compounds. According to the experience of our group this step often leads to irreproducible results in particular in the case of the oxepane derivatives. This was confirmed by reductions performed with 14 and 15. In Scheme 9, we present the best results obtained under these “standard conditions”. The aminooxepanes 26 and 27 [49] were isolated in 63% and 88% yield, respectively. Gratifyingly, the unique tricyclic 1,2-oxazine 20 was very efficiently converted into bicyclic aminooxepane derivative 28 (96% yield) without formation of side products.
![[1860-5397-10-17-i9]](/bjoc/content/inline/1860-5397-10-17-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Hydrogenolyses of bicyclic and tricyclic 1,2-oxazines 14, 15 and 20 to aminooxepanes 26, 27 and 28. Conditions: a) H2, Pd/C, MeOH, rt, 18 h.
Scheme 9: Hydrogenolyses of bicyclic and tricyclic 1,2-oxazines 14, 15 and 20 to aminooxepanes 26, 27 and 28....
The irreproducible results with 14 and 15 are probably due to the formation of side products such as 29 and 30 isolated in low quantities (proposed structures are presented in Figure 3). The 1H NMR spectra of all observed side products show signals at ca. 4.5 ppm, typically appearing as two symmetric doublets (AX system) with a coupling constant of 5.7 Hz. These observations suggest the presence of a methylene bridge at the aminooxepane skeleton. The position of the methylene bridge was only determined for side product 27 using HMBC NMR spectroscopy which shows a small coupling between one proton of the methylene bridge and C-3 (position of the alcohol). We assume that they are formed by in situ generated formaldehyde (dehydrogenation of methanol) [50-53] and subsequent aminal formation with aminooxepanes 26 and 27. The aminoalcohol 28 seems not to form the corresponding compounds. We suppose that bicyclic compound 28 is more strained and hence the formation of a third ring may be unfavorable.
![[1860-5397-10-17-3]](/bjoc/content/figures/1860-5397-10-17-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Proposed structures of the observed side products 29 and 30 during the hydrogenolyses of 14 and 15.
Figure 3: Proposed structures of the observed side products 29 and 30 during the hydrogenolyses of 14 and 15.
In order to avoid this side-reaction we performed hydrogenolysis experiments in alternative solvents, e.g. with ethanol or isopropanol, but small amounts of the corresponding side products were also observed in these experiments. Unpolar solvents were not suitable for the hydrogenolysis, probably due to the low solubility and conversion of the fairly polar starting materials or intermediates. After these problems we tried to diminish the nucleophilicity of the amino group by addition of acid since this protocol has proved to be advantageous in other reduction processes of amines [54]. Acetic acid in methanol (1:5) was a good medium for our reductions and substrate 14 was now smoothly reduced under hydrogen atmosphere with palladium on charcoal. After filtration through a pad of acidic DOWEX® resin followed by elution with aqueous ammonia the clean poly(hydroxy)aminooxepane 26 was obtained in 90% yield (Scheme 10). Under these optimized reaction conditions other hydrogenolyses were studied. p-Bromophenyl-substituted triol 16 was transformed into the corresponding poly(hydroxy)aminooxepane derivative 31 in 73% yield. As expected, the bromo substituent of the aryl group was also reductively removed delivering a phenyl group in product 31. Propargylic ether 18 was converted in quantitative yield into n-propyl ether 32. Remarkably, the hydrogenolysis of bicyclic azide 24 smoothly furnished the diaminooxepane 33 in 72% yield. The results obtained using these new conditions are promising for other N–O cleavages of polar 1,2-oxazine derivatives. Furthermore, due to the newly formed amino group the prepared poly(hydroxy)aminooxepanes offer the option to selectively perform reductive aminations or Schotten–Baumann reactions. All these processes can lead to new carbohydrate mimetics, also in a multivalent fashion.
![[1860-5397-10-17-i10]](/bjoc/content/inline/1860-5397-10-17-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Hydrogenolyses of bicyclic 1,2-oxazines to aminooxepanes 26, 31 and 32 and to diaminooxepane 33 under optimized conditions. Conditions: a) 1. H2, Pd/C, MeOH/AcOH = 5:1, rt, 17 h; 2. DOWEX® H+; 3. aq NH3.
Scheme 10: Hydrogenolyses of bicyclic 1,2-oxazines to aminooxepanes 26, 31 and 32 and to diaminooxepane 33 und...
Conclusion
In summary, the presented route employs the following key steps: Dondoni protocol for nitrone synthesis, [3 + 3]-cyclization with lithiated TMSE-protected allene, Lewis acid-induced rearrangement and reductive processes including a final hydrogenolysis. The new highly functionalized poly(hydroxy)aminooxepanes were thereby successfully synthesized in a highly stereocontrolled manner. The obtained compounds are potential carbohydrate mimetics and supplement the already studied six-membered homologues [33,34]. The introduction of new functional groups such as alkynyl or azido substituents at the bicyclic skeleton will also allow further transformations such as click reactions or palladium-catalyzed couplings. We expect that the newly prepared aminooxepanes or their multivalent conjugates will have interesting properties and possibly biological activities, e.g. as selectine inhibitors, which is currently investigated.
Experimental
General methods: See Supporting Information File 1
Typical procedure for the preparation of syn-1,2-oxazines by addition of lithiated TMSE-allene to chiral nitrones (procedure 1)
(3S,1’S,4’’S)-2-Benzyl-1’-[(tert-butyldimethylsiloxy)-(2’’,2’’-dimethyl-1’’,3’’-dioxolan-4’’-yl)methyl]-4-[2’’’-(trimethylsilyl)ethoxy]-3,6-dihydro-2H-1,2-oxazine (syn-7): Analogous to literature [31], under argon atmosphere TMSE-allene 4 (5.00 g, 32.0 mmol) was dissolved in dry THF (60 mL). The slightly yellow solution was cooled to −40 °C and n-BuLi (2.5 M in hexanes; 10.2 mL, 25.6 mmol) was slowly added. After 15 min, the resulting strong-yellow mixture was cooled to −78 °C, and a solution of nitrone 6 (4.00 g, 10.5 mmol) in dry THF (60 mL) was added dropwise. The reaction mixture turned pink, then orange and later dark red. After stirring at this temperature for 2.5 h, the reaction mixture was quenched with water (40 mL) and left to warm to rt and then extracted with Et2O (3 × 65 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered through cotton and the solvent was removed in vacuo. Crude material [orange solid, 4.00 g; syn:anti-7 >95:5 and corresponding diene (see Supporting Information File 1) <2%] was purified by column chromatography (silica gel, hexanes/EtOAc 17:0.5) to yield syn-7 (3.69 g, 65%, lit. [23,37] 71%, syn:anti-7 94:6) as a pale-yellow solid; mp 102–105 °C; TLC (silica gel, hexanes/EtOAc 8:1) Rf 0.34; 1H NMR (700 MHz, CDCl3) δ 0.02 (s, 3H, SiMe), 0.06 (s, 9H, SiMe3), 0.08 (s, 3H, SiMe), 0.87 (s, 9H, Sit-Bu), 1.00, 1.09 (AB part of ABXY system, JAX = 5.2 Hz, JBY = 6.7 Hz, JAY = 10.2 Hz, JBX = 10.8 Hz, JAB = 14.0 Hz, 1H each, 2’’’-H), 1.28, 1.34 (2 s, 3H each, Me), 2.78 (mc, 1H, 3-H), 3.12, 3.19 (AB part of ABX-system, JAX = 6.6 Hz, JBX ≈ JAB = 7.8 Hz, 1H each, 5’’-H), 3.78, 3.83 (XY part of ABXY system, JAX = 5.2 Hz, JBY = 6.7 Hz, JXY = 8.5 Hz, JAY = 10.2 Hz, JBX = 10.8 Hz, 1H each, 1’’’-H), 3.86 (d, J = 12.5 Hz, 1H, NCH2), 3.87 (dd, J = 3.5, 8.0 Hz, 1H, 1’-H), 4.14* (d, J = 12.5 Hz, 1H, NCH2), 4.14* (dd, J = 3.5, 15.1 Hz, 1H, 6-H), 4.25 (dt, J ≈ 6.6, 8.0 Hz, 1H, 4’’-H), 4.46 (td, J ≈ 2.0, 15.1 Hz, 1H, 6-H), 4.81 (dd, J = 2.0, 3.5 Hz, 1H, 5-H), 7.28, 7.33, 7.38 (3 mc, 1H, 2H, 2H, Ph) ppm, * overlapping signals; ESI–TOF (m/z): [M + H]+ calcd for C28H50NO5Si2, 536.3222; found, 536.3289. The analytical data are in accordance with literature [23,37]. Characteristic signals of anti-7: 1H NMR (700 MHz, CDCl3): δ 1.33, 1.36 (2 s, 3H each), 4.08 (t, J ≈ 6.3 Hz, 1H, 6-H), 4.29 (mc, 1H, 4’’-H), 4.76 (mc, 1H, 5-H) ppm.
Typical procedure for the Lewis acid-induced rearrangement (procedure 2)
(1R,4S,5S,6S)-7-Benzyl-5-(tert-butyldimethylsiloxy)-4-(hydroxymethyl)-2,2-dimethyl-3,8-dioxa-7-azabicyclo[4.3.1]decan-10-one (11): Analogous to literature [23,37], under argon atmosphere 1,2-oxazine syn-7 (2.00 g, 3.73 mmol, syn:anti 94:6) was dissolved in dry CH2Cl2 (32 mL). The solution was cooled to −5 °C and TMSOTf (1.35 mL, 7.46 mmol) was slowly added. After 5.5 h stirring at 0 °C, the dark red mixture was quenched with a solution of aq ammonia (5%) turning yellow. Work-up was performed with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered through cotton and the solvent was removed in vacuo. The crude material (orange oil, 2.05 g) was purified by column chromatography (silica gel, hexanes/EtOAc 5:1 to 4:1) to yield 11 (851 mg, 53%, lit. [23] 55%) as a pale-yellow oil; TLC (silica gel, hexanes/EtOAc 2:1) Rf 0.48; 1H NMR (500 MHz, CDCl3) δ −0.09, −0.02 (2 s, 3H each, SiMe), 0.86 (s, 9H, Sit-Bu), 1.37, 1.38 (2 s, 3H each, Me), 1.72 (bs, 1H, OH), 2.57 (mc, 1H, 1-H), 3.37 (mc, 1H, 6-H), 3.56, 3.67 (AB part of ABX system, JAX = 4.5 Hz, JBX = 8.1 Hz, JAB = 10.9 Hz, 1H each, 4-CH2), 3.94 (A part of AB system, JAB = 13.4 Hz, 1H, NCH2), 4.11–4.13 (m, 4H, 5-H, 9-H, NCH2), 4.58 (dd, J = 4.5, 8.1 Hz, 1H, 4-H), 7.26–7.29 (m, 5H, Ph) ppm; ESI-TOF (m/z): [M + H]+ calcd for C23H38NO5Si, 436.2514; found, 436.2553. The analytical data are in accordance with literature [23,37].
Typical procedure for ketone reduction with NaBH4 (procedure 3)
(1S,4S,5S,6R,10S)-7-Benzyl-5-(tert-butyldimethylsiloxy)-4-(hydroxymethyl)-2,2-dimethyl-3,8-dioxa-7-azabicyclo[4.3.1]decan-10-ol: Under argon atmosphere ketone 11 (703 mg, 1.61 mmol) was dissolved in dry EtOH (26 mL). The solution was cooled to 0 °C and NaBH4 (119 mg, 3.15 mmol) was added in portions. After stirring at 0 °C for 4 h, the solvent was removed in vacuo. The crude material was dissolved in CH2Cl2 (20 mL) and then extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered through cotton and the solvent was removed in vacuo to yield the corresponding alcohol (704 mg, quant.) as a colorless solid; melting range 132–137 °C; [α]D22 −61.1 (c 0.63, CHCl3); TLC (silica gel, hexanes/EtOAc 2:1) Rf 0.58; 1H NMR (700 MHz, CDCl3) δ −0.09, −0.04 (2 s, 3H each, SiMe), 0.88 (s, 9H, Sit-Bu), 1.32, 1.53 (2 s, 3H each, Me), 1.75 (bs, 1H, 4-OH), 2.17 (mc, 1H, 1-H), 3.29 (dd, J = 2.5, 4.8 Hz, 1H, 6-H), 3.56 (mc, 1H, 4-CH2), 3.70 (B part of ABX system, JAX = 8.5 Hz, JAB = 10.7 Hz, 1H, 4-CH2), 3.81 (A part of ABX-system, JAX = 3.0 Hz, JAB = 12.0 Hz, 1H, 9-H), 3.87 (d, J = 13.9 Hz, 1H, NCH2), 3.93 (B part of ABX system, JAB = 12.0 Hz, 1H, 9-H)*, 3.94 (d, J = 11.4 Hz, 1H, 10-OH), 4.17 (d, J = 13.9 Hz, 1H, NCH2), 4.33 (dt, J ≈ 1.0, 2.5 Hz, 1H, 5-H), 4.40-4.44 (m, 2H, 4-H, 10-H), 7.27–7.28, 7.32–7.36 (2 m, 5H, Ph) ppm, *no BX coupling present; 13C NMR (175 MHz, CDCl3) δ −4.9, −4.4 (2 q, SiMe), 18.1 (s, SiCMe3), 24.9 (q, Me), 25.9 (q, SiCMe3), 34.6 (q, Me), 47.4 (d, C-1), 58.4 (t, NCH2), 63.6 (t, 4-CH2), 64.5 (d, C-5), 68.6 (t, C-9), 71.1 (d, C-6), 72.8 (d, C-10), 75.7 (d, C-4), 78.3 (s, C-2), 127.6, 128.3, 128.6, 137.3 (3 d, s, Ph) ppm; IR (ATR) ![[Graphic 1]](/bjoc/content/inline/1860-5397-10-17-i11.svg?max-width=637&scale=1.18182) : 3570, 3435 (OH), 3085–3030 (=C-H), 2990–2855 (C-H), 1250, 1215 (C-O), 1060, 1040 (C-O-C) cm−1; ESI–TOF (m/z): [M + H]+ calcd for C23H40NO5Si, 438.2670; found, 438.2702; anal. calcd for C23H39NO5Si (437.6): C, 63.12; H, 8.98; N, 3.20; found: C, 62.68; H, 8.96; N, 3.09.
: 3570, 3435 (OH), 3085–3030 (=C-H), 2990–2855 (C-H), 1250, 1215 (C-O), 1060, 1040 (C-O-C) cm−1; ESI–TOF (m/z): [M + H]+ calcd for C23H40NO5Si, 438.2670; found, 438.2702; anal. calcd for C23H39NO5Si (437.6): C, 63.12; H, 8.98; N, 3.20; found: C, 62.68; H, 8.96; N, 3.09.
Typical procedure for deprotection using TBAF (procedure 4)
(1S,4S,5S,6R,10S)-7-Benzyl-4-(hydroxymethyl)-2,2-dimethyl-3,8-dioxa-7-azabicyclo[4.3.1]decan-5,10-diol (14): The TBS-protected alcohol (200 mg, 0.457 mmol) was dissolved in THF (11 mL) and the solution was cooled to 0 °C. After addition of TBAF (1 M in THF; 0.9 mL, 0.90 mmol), the reaction mixture was stirred at this temperature for 10 min. Then the mixture was quenched with water (5 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered through cotton and the solvent was removed in vacuo. The crude material (yellow oil, 224 mg) was purified by column chromatography (silica gel, hexanes/EtOAc 1:2 to pure EtOAc) to yield 14 (135 mg, 91%) as a colorless solid; mp 50–53 °C; [α]D22 −52.5 (c 0.6, MeOH); TLC (silica gel, hexanes/EtOAc 1:3): Rf 0.27; 1H NMR (700 MHz, CD3OD) δ 1.31, 1.49 (2 s, 3H each, Me), 2.13 (mc, 1H, 1-H), 3.34 (dd, J = 3.0, 5.0 Hz, 1H, 6-H), 3.63, 3.71 (AB part of ABX system, JAX = 3.5 Hz, JBX = 6.5 Hz, JAB = 11.0 Hz, 1H each, 4-CH2), 3.74 (dd, J = 3.0, 12.8 Hz, 1H, 9-H), 3.91 (d, J = 14.1 Hz, 1H, NCH2), 3.96 (d, J = 12.8 Hz, 1H, 9-H), 4.12 (d, J = 14.1 Hz, 1H, NCH2), 4.44–4.47 (m, 3H, 4-H, 5-H, 10-H), 7.22, 7.29, 7.35 (3 mc, 1H, 2H, 2H, Ph) ppm; 13C NMR (175 MHz, CD3OD) δ 24.6, 34.5 (2 q, Me), 48.7 (d, C-1), 49.9 (d, C-5), 59.4 (t, NCH2), 63.6 (t, 4-CH2), 65.9 (t, C-9), 70.0 (d, C-4), 74.7 (d, C-6), 76.3 (d, C-10), 79.1 (s, C-2), 128.1, 129.2, 129.4, 139.2 (3 d, s, Ph) ppm; IR (ATR) : 3360 (OH), 3085–3030 (=C-H), 2970–2860 (C-H), 1215 (C-O), 1060 (C-O-C) cm−1; ESI–TOF (m/z): [M + Na]+ calcd for C17H25NO5Na, 346.1630; found, 346.1622; anal. calcd for C17H25NO5 (323.4): C, 63.14; H, 7.79; N, 4.33; found: C, 63.20; H, 7.83; N, 4.31.
Synthesis of (1R,4S,5S,6S)-4-(azidomethyl)-7-benzyl-5-(tert-butyldimethylsiloxy)-2,2-dimethyl-3,8-dioxa-7-azabicyclo[4.3.1]decan-10-one (23): Under argon atmosphere TPP (99 mg, 0.38 mmol) was dissolved in dry THF (1.5 mL) and the solution was cooled to −20 °C. DIAD (76.5 mg, 0.378 mmol) was added and the reaction mixture became milky white. After 10 min stirring at that temperature, alcohol 11 (150 mg, 0.344 mmol in 0.6 mL of dry THF) was slowly added and the mixture was stirred for further 30 min at −20 °C. The now milky-yellow solution was progressively warmed to 0 °C and DPPA (89.0 μL, 0.413 mmol) was added. After stirring for 6 h at 0 °C and 1 d at rt, the reaction mixture was quenched with water (2 mL) and extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered through cotton and the solvent was removed in vacuo. Crude material (yellow oil, 479 mg) was purified by column chromatography (silica gel, hexanes to hexanes/EtOAc 8:1) to yield azide 23 (125 mg, 79%) as a colorless solid; mp 117–121 °C; [α]D22 +22.7 (c 1.01, CHCl3); TLC (silica gel, hexanes/EtOAc 3:1) Rf 0.73; 1H NMR (500 MHz, CDCl3) δ −0.11, −0.03 (2 s, 3H each, SiMe), 0.86 (s, 9H, Sit-Bu), 1.37, 1.42 (2 s, 3H each, Me), 2.55 (t, J ≈ 2.8 Hz, 1H, 1-H), 3.05 (dd, J = 4.3, 12.3 Hz, 1H, 4-CH2), 3.36 (dd, J = 1.1, 2.8 Hz, 1H, 6-H), 3.45 (dd, J = 8.7, 12.3 Hz, 1H, 4-CH2), 3.95 (A part of AB system, JAB = 13.5 Hz, 1H, NCH2), 4.05 (mc, 1H, 5-H), 4.14 (B part of AB system, JAB = 13.5 Hz, 1H, NCH2), 4.18 (mc, 2H, 9-H), 4.61 (ddd, J = 0.8, 4.3, 8.7 Hz, 1H, 4-H), 7.27-7.35 (m, 5H, Ph) ppm; 13C NMR (125 MHz, CDCl3) δ −4.8, −4.6 (2 q, SiMe), 18.2 (s, SiCMe3), 22.4 (q, Me), 25.8 (q, SiCMe3), 31.6 (q, Me), 53.2 (t, 4-CH2), 58.1 (d, C-1), 58.7 (t, NCH2), 67.9 (t, C-9), 69.4 (d, C-5), 74.2 (d, C-4), 75.0 (s, C-2), 75.1 (d, C-6), 127.9, 128.66, 128.71, 136.2 (3 d, s, Ph), 199.8 (s, C-10) ppm; IR (ATR) : 3090–3030 (=C-H), 2855–2995 (C-H), 2095 (N3), 1710 (C=O), 1495 (C=C), 1295, 1255 (C-O), 1190–1075 (C-O-C) cm−1; ESI–TOF (m/z): [M + H]+ calcd for C23H37N4O4Si, 461.2579; found, 461.2607.
Typical procedure for hydrogenolysis with Pd/C in AcOH/MeOH (procedure 5)
(2S,3S,4R,5S,6S)-4-Amino-2,6-bis(hydroxymethyl)-7,7-dimethyloxepan-3,5-diol (26): Bicyclic compound 14 (100 mg, 0.309 mmol) was dissolved in dry MeOH (10 mL) and AcOH (2 mL). After addition of Pd/C (100 mg, 0.094 mmol Pd) the suspension was saturated with hydrogen at rt for 1 h followed by stirring under hydrogen pressure (balloon). After reaction completion 18 h, the mixture was filtered through Celite®, washed with EtOH and the solvents were removed in vacuo. The obtained salt was dissolved in water and filtered through a DOWEX® column (H2O) and washed with water until the complete acid was removed (control with pH paper). Then the column was washed with aq NH3 (2 to 5%) to yield the free amine 26 (65 mg, 90%) as a colorless solid; mp 121–125 °C; [α]D22 +10.2 (c 1.35, MeOH); TLC [silica gel, MeCN/aq NH3 (25%) 5:1] Rf 0.10; 1H NMR (700 MHz, CD3OD) δ 1.19, 1.34 (2 s, 3H each, Me), 1.77 (ddd, J = 3.0, 6.5, 9.5 Hz, 1H, 6-H), 2.89 (dd, J = 6.9, 8.8 Hz, 1H, 4-H), 3.52–3.55 (m, 2H, 2-CH2, 3-H), 3.59 (B part of ABX system, JBX = 5.0 Hz, JAB = 11.5 Hz, 1H, 2-CH2), 3.65, 3.68 (AB part of ABX system, JAX = 3.0 Hz, JBX = 6.5 Hz, JAB = 11.3 Hz, 1H each, 6-CH2), 3.71 (mc, 2H, 2-H, 5-H) ppm; 13C NMR (175 MHz, CD3OD) δ 21.1, 31.5 (2 q, Me), 60.0 (d, C-6), 63.6 (t, 2-CH2), 64.1 (d, C-4), 64.2 (t, 2-CH2), 71.3, 73.8 (2 d, C-2, C-5), 76.5 (d, C-3), 77.1 (s, C-7) ppm; IR (ATR) : 3530–3310 (OH, NH2), 2960–2860 (C-H), 1455, 1470, 1140 (C-O) cm−1; ESI–TOF (m/z): [M + Na]+ calcd for C10H21NO5Na, 258.1320; found, 258.1317; anal. calcd for C10H21NO5 (235.3): C, 51.05; H, 9.00; N, 5.95; found, C, 50.78; H, 8.97; N: 5.59.
Acknowledgements
This work was generously supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich SFB 765), the Center of International Cooperation of the Freie Universität Berlin and Bayer HealthCare. We thank Dr. A. Al-Harrasi for preliminary experiments and N. Danneberg, M. Menger and L. Selter for experimental assistance. We acknowledge valuable discussions and help during preparation of the manuscript by Dr. R. Zimmer and J. Salta.
References
-
Wong, C.-H. Carbohydrate-based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1.
Return to citation in text: [1] -
Sears, P.; Wong, C.-H. Angew. Chem. 1999, 111, 2446–2471. doi:10.1002/(SICI)1521-3757(19990816)111:16<2446::AID-ANGE2446>3.0.CO;2-4
Angew. Chem., Int. Ed. 1999, 38, 2300-2324. doi:10.1002/(SICI)1521-3773(19990816)38:16<2300::AID-ANIE2300>3.0.CO;2-6
Return to citation in text: [1] -
Koester, D. C.; Holkenbrink, A.; Werz, D. B. Synthesis 2010, 3217–3242. doi:10.1055/s-0030-1258228
Return to citation in text: [1] -
Lee, Y. C.; Lee, R. T. Acc. Chem. Res. 1995, 28, 321–327. doi:10.1021/ar00056a001
Return to citation in text: [1] -
Lundquist, J. J.; Toone, E. J. Chem. Rev. 2002, 102, 555–578. doi:10.1021/cr000418f
Return to citation in text: [1] -
Kakinuma, K.; Ikekawa, N.; Nakagawa, A.; Omura, S. J. Am. Chem. Soc. 1979, 101, 3402–3404. doi:10.1021/ja00506a056
Return to citation in text: [1] -
Funel-Le Bon, C.; Berrué, F.; Thomas, O. P.; Reyes, F.; Amade, P. J. Nat. Prod. 2005, 68, 1284–1287. doi:10.1021/np050100o
Return to citation in text: [1] -
Saha, J.; Peczuh, M. W. Synthesis and properties of septanose carbohydrates. In Advances in Carbohydrate Chemistry and Biochemistry; Derek, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2011; Vol. 66, pp 121–186. doi:10.1016/B978-0-12-385518-3.00003-1
Return to citation in text: [1] -
Sahabuddin, S.; Roy, A.; Drew, M. G. B.; Roy, B. G.; Achari, B.; Mandal, S. B. J. Org. Chem. 2006, 71, 5980–5992. doi:10.1021/jo0606554
Return to citation in text: [1] -
Tripathi, S.; Roy, B. G.; Drew, M. G. B.; Achari, B.; Mandal, S. B. J. Org. Chem. 2007, 72, 7427–7430. doi:10.1021/jo070846m
Return to citation in text: [1] -
Bhattacharjee, A.; Datta, S.; Chattopadhyay, P.; Ghoshal, N.; Kundu, A. P.; Pal, A.; Mukhopadhyay, R.; Chowdhury, S.; Bhattacharjya, A.; Patra, A. Tetrahedron 2003, 59, 4623–4639. doi:10.1016/S0040-4020(03)00634-3
Return to citation in text: [1] -
Hoberg, J. O. Tetrahedron 1998, 54, 12631–12670. doi:10.1016/S0040-4020(98)00596-1
Return to citation in text: [1] -
Kotkar, D.; Ghosh, P. K. J. Chem. Soc., Chem. Commun. 1986, 650–651. doi:10.1039/C39860000650
Return to citation in text: [1] -
Pavlik, C.; Onorato, A.; Castro, S.; Morton, M.; Peczuh, M. W.; Smith, M. B. Org. Lett. 2009, 11, 3722–3725. doi:10.1021/ol9013427
Return to citation in text: [1] -
Snyder, N. L.; Haines, H. M.; Peczuh, M. W. Tetrahedron 2006, 62, 9301–9320. doi:10.1016/j.tet.2006.07.021
Return to citation in text: [1] -
Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 44, 4057–4061. doi:10.1016/S0040-4039(03)00849-9
Return to citation in text: [1] -
Castro, S.; Johnson, C. S.; Surana, B.; Peczuh, M. W. Tetrahedron 2009, 65, 7921–7926. doi:10.1016/j.tet.2009.07.041
Return to citation in text: [1] -
Riley, D. L.; van Otterlo, W. A. L. Oxepines and Azepines. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 535–568. doi:10.1002/9783527634880
See for a recent review about oxepines in natural product synthesis.
Return to citation in text: [1] -
Fyvie, W. S.; Morton, M.; Peczuh, M. W. Carbohydr. Res. 2004, 339, 2363–2370. doi:10.1016/j.carres.2004.07.009
Return to citation in text: [1] -
Wong, J. C. Y.; Lacombe, P.; Sturino, C. F. Tetrahedron Lett. 1999, 40, 8751–8754. doi:10.1016/S0040-4039(99)01881-X
Return to citation in text: [1] -
Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e
Return to citation in text: [1] -
Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2009, 74, 739–746. doi:10.1021/jo801967s
Return to citation in text: [1] -
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996
Return to citation in text: [1] [2] -
Pfrengle, F.; Reissig, H.-U. Chem.–Eur. J. 2010, 16, 11915–11925. doi:10.1002/chem.201001060
Return to citation in text: [1] -
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Angew. Chem. 2009, 121, 3211–3215. doi:10.1002/ange.200805724
Angew. Chem., Int. Ed. 2009, 48, 3165-3169 doi:10.1002/anie.200805724
Return to citation in text: [1] -
Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d
Return to citation in text: [1] [2] -
Bouché, L.; Reissig, H.-U. Pure Appl. Chem. 2012, 84, 23–36. doi:10.1351/PAC-CON-11-09-20
Return to citation in text: [1] [2] -
Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T. Synth. Commun. 1994, 24, 2537–2550. doi:10.1080/00397919408010565
Return to citation in text: [1] [2] -
Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804
Return to citation in text: [1] [2] [3] -
Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627
Return to citation in text: [1] [2] [3] [4] -
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h
Return to citation in text: [1] -
Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a
Return to citation in text: [1] [2] -
Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/C1OB05583F
Return to citation in text: [1] [2] -
Abushanab, E.; Vemishetti, P.; Leiby, R. W.; Singh, H. K.; Mikkilineni, A. B.; Wu, D. C.-J.; Saibaba, R.; Panzica, R. P. J. Org. Chem. 1988, 53, 2598–2602. doi:10.1021/jo00246a037
Return to citation in text: [1] -
The ethyl ester was obtained as mixture of diastereomers (52:48), see Supporting Information.
Return to citation in text: [1] -
Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Dondoni, A.; Junquera, F.; Merchan, F. L.; Merino, P.; Tejero, T. Synthesis 1994, 1450–1456. doi:10.1055/s-1994-25712
Return to citation in text: [1] -
Merino, P.; Lanaspa, A.; Merchan, F. L.; Tejero, T. J. Org. Chem. 1996, 61, 9028–9032. doi:10.1021/jo961293a
Return to citation in text: [1] -
Fujiwara, K.; Amano, A.; Tokiwano, T.; Murai, A. Tetrahedron 2000, 56, 1065–1080. doi:10.1016/S0040-4020(00)00009-0
Return to citation in text: [1] -
Phillips, A. J.; Morris, J. C.; Abell, A. D. Tetrahedron Lett. 2000, 41, 2723–2727. doi:10.1016/S0040-4039(00)00248-3
Return to citation in text: [1] -
These results were confirmed by X-ray analysis, see ref [37] and [49].
Return to citation in text: [1] -
We assume that the hydride reagent also attacks the Re-side, as in the previous reductions (see Scheme 4 and Scheme 5). NMR data of the corresponding alcohol are in accordance with this assignment.
Return to citation in text: [1] -
Conditions used were analogous to procedures in ref [24].
Return to citation in text: [1] -
Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Synthesis 2013, 45, 2752–2762. doi:10.1055/s-0033-1339509
See for Click reactions using 1,2-oxazines which were recently described by our group.
Return to citation in text: [1] -
Lal, B.; Pramanik, B. N.; Manhas, M. S.; Bose, A. K. Tetrahedron Lett. 1977, 18, 1977–1980. doi:10.1016/S0040-4039(01)83657-1
Return to citation in text: [1] -
Bressel, B.; Egart, B.; Al-Harrasi, A.; Pulz, R.; Reissig, H.-U.; Brüdgam, I. Eur. J. Org. Chem. 2008, 467–474. doi:10.1002/ejoc.200700792
Return to citation in text: [1] -
Jasiński, M.; Lentz, D.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2012, 3304–3316. doi:10.1002/ejoc.201200158
Return to citation in text: [1] -
Al-Harrasi, A.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Z. Kristallogr. NCS 2006, 221, 131–132.
See for an X-ray analysis of the corresponding TBS-protected oxepane of 27.
Return to citation in text: [1] [2] -
Smith, G. V.; Notheisz, F. Hydrogenolysis of Benzyl-Nitrogen Bonds. Heterogeneous Catalysis in Organic Chemistry; Academic Press, 1999; pp 171–172.
Return to citation in text: [1] -
Fu, X.; Cook, J. M. J. Am. Chem. Soc. 1992, 114, 6910–6912. doi:10.1021/ja00043a043
Return to citation in text: [1] -
Fu, X.; Cook, J. M. J. Org. Chem. 1993, 58, 661–672. doi:10.1021/jo00055a019
Return to citation in text: [1] -
The same type of side products was also obtained in our group: Zimmer, R.; Kandziora, M.; Salta, J. unpublished results.
Return to citation in text: [1] -
Su, J.-K.; Jia, Y.-M.; He, R.; Rui, P.-X.; Han, N.; He, X.; Xiang, J.; Chen, X.; Zhu, J.; Yu, C.-Y. Synlett 2010, 1609–1616. doi:10.1055/s-0029-1258085
Return to citation in text: [1]
| 40. | Fujiwara, K.; Amano, A.; Tokiwano, T.; Murai, A. Tetrahedron 2000, 56, 1065–1080. doi:10.1016/S0040-4020(00)00009-0 |
| 41. | Phillips, A. J.; Morris, J. C.; Abell, A. D. Tetrahedron Lett. 2000, 41, 2723–2727. doi:10.1016/S0040-4039(00)00248-3 |
| 43. | We assume that the hydride reagent also attacks the Re-side, as in the previous reductions (see Scheme 4 and Scheme 5). NMR data of the corresponding alcohol are in accordance with this assignment. |
| 1. | Wong, C.-H. Carbohydrate-based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1. |
| 2. |
Sears, P.; Wong, C.-H. Angew. Chem. 1999, 111, 2446–2471. doi:10.1002/(SICI)1521-3757(19990816)111:16<2446::AID-ANGE2446>3.0.CO;2-4
Angew. Chem., Int. Ed. 1999, 38, 2300-2324. doi:10.1002/(SICI)1521-3773(19990816)38:16<2300::AID-ANIE2300>3.0.CO;2-6 |
| 3. | Koester, D. C.; Holkenbrink, A.; Werz, D. B. Synthesis 2010, 3217–3242. doi:10.1055/s-0030-1258228 |
| 9. | Sahabuddin, S.; Roy, A.; Drew, M. G. B.; Roy, B. G.; Achari, B.; Mandal, S. B. J. Org. Chem. 2006, 71, 5980–5992. doi:10.1021/jo0606554 |
| 10. | Tripathi, S.; Roy, B. G.; Drew, M. G. B.; Achari, B.; Mandal, S. B. J. Org. Chem. 2007, 72, 7427–7430. doi:10.1021/jo070846m |
| 11. | Bhattacharjee, A.; Datta, S.; Chattopadhyay, P.; Ghoshal, N.; Kundu, A. P.; Pal, A.; Mukhopadhyay, R.; Chowdhury, S.; Bhattacharjya, A.; Patra, A. Tetrahedron 2003, 59, 4623–4639. doi:10.1016/S0040-4020(03)00634-3 |
| 27. | Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d |
| 28. | Bouché, L.; Reissig, H.-U. Pure Appl. Chem. 2012, 84, 23–36. doi:10.1351/PAC-CON-11-09-20 |
| 54. | Su, J.-K.; Jia, Y.-M.; He, R.; Rui, P.-X.; Han, N.; He, X.; Xiang, J.; Chen, X.; Zhu, J.; Yu, C.-Y. Synlett 2010, 1609–1616. doi:10.1055/s-0029-1258085 |
| 8. | Saha, J.; Peczuh, M. W. Synthesis and properties of septanose carbohydrates. In Advances in Carbohydrate Chemistry and Biochemistry; Derek, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2011; Vol. 66, pp 121–186. doi:10.1016/B978-0-12-385518-3.00003-1 |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 33. | Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a |
| 34. | Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/C1OB05583F |
| 6. | Kakinuma, K.; Ikekawa, N.; Nakagawa, A.; Omura, S. J. Am. Chem. Soc. 1979, 101, 3402–3404. doi:10.1021/ja00506a056 |
| 7. | Funel-Le Bon, C.; Berrué, F.; Thomas, O. P.; Reyes, F.; Amade, P. J. Nat. Prod. 2005, 68, 1284–1287. doi:10.1021/np050100o |
| 32. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h |
| 49. |
Al-Harrasi, A.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Z. Kristallogr. NCS 2006, 221, 131–132.
See for an X-ray analysis of the corresponding TBS-protected oxepane of 27. |
| 4. | Lee, Y. C.; Lee, R. T. Acc. Chem. Res. 1995, 28, 321–327. doi:10.1021/ar00056a001 |
| 5. | Lundquist, J. J.; Toone, E. J. Chem. Rev. 2002, 102, 555–578. doi:10.1021/cr000418f |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 50. | Smith, G. V.; Notheisz, F. Hydrogenolysis of Benzyl-Nitrogen Bonds. Heterogeneous Catalysis in Organic Chemistry; Academic Press, 1999; pp 171–172. |
| 51. | Fu, X.; Cook, J. M. J. Am. Chem. Soc. 1992, 114, 6910–6912. doi:10.1021/ja00043a043 |
| 52. | Fu, X.; Cook, J. M. J. Org. Chem. 1993, 58, 661–672. doi:10.1021/jo00055a019 |
| 53. | The same type of side products was also obtained in our group: Zimmer, R.; Kandziora, M.; Salta, J. unpublished results. |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 22. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2009, 74, 739–746. doi:10.1021/jo801967s |
| 29. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T. Synth. Commun. 1994, 24, 2537–2550. doi:10.1080/00397919408010565 |
| 30. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804 |
| 46. | Lal, B.; Pramanik, B. N.; Manhas, M. S.; Bose, A. K. Tetrahedron Lett. 1977, 18, 1977–1980. doi:10.1016/S0040-4039(01)83657-1 |
| 19. | Fyvie, W. S.; Morton, M.; Peczuh, M. W. Carbohydr. Res. 2004, 339, 2363–2370. doi:10.1016/j.carres.2004.07.009 |
| 20. | Wong, J. C. Y.; Lacombe, P.; Sturino, C. F. Tetrahedron Lett. 1999, 40, 8751–8754. doi:10.1016/S0040-4039(99)01881-X |
| 31. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 47. | Bressel, B.; Egart, B.; Al-Harrasi, A.; Pulz, R.; Reissig, H.-U.; Brüdgam, I. Eur. J. Org. Chem. 2008, 467–474. doi:10.1002/ejoc.200700792 |
| 48. | Jasiński, M.; Lentz, D.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2012, 3304–3316. doi:10.1002/ejoc.201200158 |
| 15. | Snyder, N. L.; Haines, H. M.; Peczuh, M. W. Tetrahedron 2006, 62, 9301–9320. doi:10.1016/j.tet.2006.07.021 |
| 16. | Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 44, 4057–4061. doi:10.1016/S0040-4039(03)00849-9 |
| 17. | Castro, S.; Johnson, C. S.; Surana, B.; Peczuh, M. W. Tetrahedron 2009, 65, 7921–7926. doi:10.1016/j.tet.2009.07.041 |
| 18. |
Riley, D. L.; van Otterlo, W. A. L. Oxepines and Azepines. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 535–568. doi:10.1002/9783527634880
See for a recent review about oxepines in natural product synthesis. |
| 12. | Hoberg, J. O. Tetrahedron 1998, 54, 12631–12670. doi:10.1016/S0040-4020(98)00596-1 |
| 13. | Kotkar, D.; Ghosh, P. K. J. Chem. Soc., Chem. Commun. 1986, 650–651. doi:10.1039/C39860000650 |
| 14. | Pavlik, C.; Onorato, A.; Castro, S.; Morton, M.; Peczuh, M. W.; Smith, M. B. Org. Lett. 2009, 11, 3722–3725. doi:10.1021/ol9013427 |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 24. | Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 25. | Pfrengle, F.; Reissig, H.-U. Chem.–Eur. J. 2010, 16, 11915–11925. doi:10.1002/chem.201001060 |
| 26. |
Pfrengle, F.; Lentz, D.; Reissig, H.-U. Angew. Chem. 2009, 121, 3211–3215. doi:10.1002/ange.200805724
Angew. Chem., Int. Ed. 2009, 48, 3165-3169 doi:10.1002/anie.200805724 |
| 27. | Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev. 2010, 39, 549–557. doi:10.1039/b914356d |
| 28. | Bouché, L.; Reissig, H.-U. Pure Appl. Chem. 2012, 84, 23–36. doi:10.1351/PAC-CON-11-09-20 |
| 45. |
Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Synthesis 2013, 45, 2752–2762. doi:10.1055/s-0033-1339509
See for Click reactions using 1,2-oxazines which were recently described by our group. |
| 35. | Abushanab, E.; Vemishetti, P.; Leiby, R. W.; Singh, H. K.; Mikkilineni, A. B.; Wu, D. C.-J.; Saibaba, R.; Panzica, R. P. J. Org. Chem. 1988, 53, 2598–2602. doi:10.1021/jo00246a037 |
| 24. | Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 31. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 33. | Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a |
| 34. | Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/C1OB05583F |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 38. | Dondoni, A.; Junquera, F.; Merchan, F. L.; Merino, P.; Tejero, T. Synthesis 1994, 1450–1456. doi:10.1055/s-1994-25712 |
| 39. | Merino, P.; Lanaspa, A.; Merchan, F. L.; Tejero, T. J. Org. Chem. 1996, 61, 9028–9032. doi:10.1021/jo961293a |
| 30. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 49. |
Al-Harrasi, A.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Z. Kristallogr. NCS 2006, 221, 131–132.
See for an X-ray analysis of the corresponding TBS-protected oxepane of 27. |
| 30. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T.; Bertolasi, V. Chem.–Eur. J. 1995, 1, 505–520. doi:10.1002/chem.19950010804 |
| 31. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 24. | Al-Harrasi, A.; Pfrengle, F.; Prisyazhnyuk, V.; Yekta, S.; Koóš, P.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 11632–11641. doi:10.1002/chem.200900996 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 31. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 36. | The ethyl ester was obtained as mixture of diastereomers (52:48), see Supporting Information. |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
| 37. | Al-Harrasi, A. New Transformations of Enantiopure 3,6-Dihydro-2H-1,2-oxazines: Ring Cleavages, Ring Enlargements and a Novel Approach to Carbohydrate Mimetics. Ph.D. Thesis, Freie Universität Berlin, Germany, 2005. |
| 29. | Dondoni, A.; Franco, S.; Junquera, F.; Merchán, F. L.; Merino, P.; Tejero, T. Synth. Commun. 1994, 24, 2537–2550. doi:10.1080/00397919408010565 |
| 23. |
Al-Harrasi, A.; Reissig, H.-U. Angew. Chem. 2005, 117, 6383–6387. doi:10.1002/ange.200501127
Angew. Chem., Int. Ed. 2005, 44, 6227-6231. doi:10.1002/anie.200501127 |
© 2014 Bouché et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)