Abstract
Strained azirinium ylides derived from 2H-azirines and α-diazoketones under Rh(II)-catalysis can undergo either irreversible ring opening across the N–C2 bond to 2-azabuta-1,3-dienes that further cyclize to 2H-1,4-oxazines or reversibly undergo a 1,5-cyclization to dihydroazireno[2,1-b]oxazoles. Dihydroazireno[2,1-b]oxazoles derived from 3-aryl-2H-azirines and 3-diazoacetylacetone or ethyl diazoacetoacetate are able to cycloadd to acetyl(methyl)ketene generated from 3-diazoacetylacetone under Rh(II) catalysis to give 4,6-dioxa-1-azabicyclo[3.2.1]oct-2-ene and/or 5,7-dioxa-1-azabicyclo[4.3.1]deca-3,8-diene-2-one derivatives. According to DFT calculations (B3LYP/6-31+G(d,p)), the cycloaddition can involve two modes of nucleophilic attack of the dihydroazireno[2,1-b]oxazole intermediate on acetyl(methyl)ketene followed by aziridine ring opening into atropoisomeric oxazolium betaines and cyclization. Azirinium ylides generated from 2,3-di- and 2,2,3-triaryl-substituted azirines give rise to only 2-azabuta-1,3-dienes and/or 2H-1,4-oxazines.

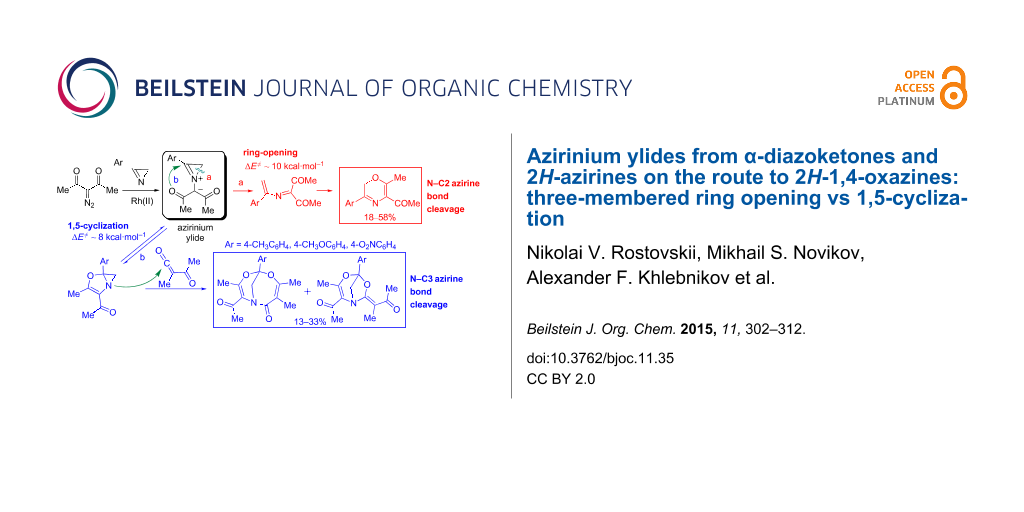
Graphical Abstract
Introduction
2H-Azirines are unique strained compounds which have found various applications in organic synthesis due to their ability to react both with retention and opening of the three-membered ring. Even though each of the three bonds in the azirine ring can be cleaved under certain conditions, the majority of synthetic applications of these compounds imply cleavage of either the N–C2 or N–C3 bond. In these syntheses 2H-azirines serve as C–C–N three-atomic building blocks for the construction of both acyclic compounds, such as nonproteinogenic amino acids and peptides [1-3], and various 4–7-membered heterocycles [4-11]. The reactions of azirines with acylketenes [12,13], carboxylic acids [14] and amino acids [1] proceed via N–C3 bond cleavage to afford 5,7-dioxa-1-azabicyclo[4.4.1]undeca-3,8-diene derivatives, ketamides, and aminoamides, respectively. On the other hand, rhodium carbenoids derived from α-diazocarbonyl compounds transform 2H-azirines 1 to azirinium ylides 5 (Scheme 1) which undergo facile N–C2 bond cleavage to give 2-azabuta-1,3-dienes 3. Recently we showed that the use of α-diazo-β-ketoesters 2 in these reactions, which are finished by 1,6-cyclization of azadienes onto the keto group, provides a rapid access to non-fused 2H-1,4-oxazine-5-carboxylates 4, a new type of non-spirocyclic 1,4-oxazine photochromes [15].
![[1860-5397-11-35-i1]](/bjoc/content/inline/1860-5397-11-35-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Rh(II)-catalyzed synthesis of photochromic 2H-1,4-oxazines from 2H-azirines and α-diazo-β-ketoesters.
Scheme 1: Rh(II)-catalyzed synthesis of photochromic 2H-1,4-oxazines from 2H-azirines and α-diazo-β-ketoester...
In the search for new compounds with useful photochromic characteristics and a high fatigue resistance we focused on 5-unsubstituted, 5-aryl-, and 5-acyl-substituted 2H-1,4-oxazines. They could, in principle, be prepared from azirines and α-diazo ketones or 2-diazo-1,3-diketones via ring opening across the N–C2 bond in the intermediate azirinium ylides to form 2-azabuta-1,3-dienes and subsequent cyclization of the latter into 2H-1,4-oxazines. In the work reported herein we studied Rh(II)-catalyzed reactions of alkyl-, aryl-, and hetaryl-substituted 2Н-azirines with α-diazo-α-phenylacetone (2a), α-diazo-4-chloroacetophenone (2b), and 3-diazoacetylacetone (2c). A new type of transformation of azirinium ylides, specifically 1,5-cyclization into dihydroazireno[2,1-b]oxazoles capable of cycloadding to acyl ketenes, was discovered in the study on the reactivity of 3-monosubstituted azirines toward 3-diazoacetylacetone (2c) under catalytic conditions. The competition between ring opening and 1,5-cyclization in azirinium ylides, as well as the mechanism of trapping of dihydroazireno[2,1-b]oxazole intermediates by acetyl(methyl)ketene were investigated by the DFT method.
Results and Discussion
Rhodium(II) carbenoids generated from α-diazoketones or 2-diazo-1,3-diketones react with various nitrogen-containing compounds, such as amines [16], amides [17-19], and nitriles [20,21], to give N–H insertion products or N- or N,O-heterocyclic systems. The reactivity of acyl- or diacyl-substituted Rh(II) carbenoids toward an sp2-hybridized nitrogen [22-24] is much less studied, while examples of their reactions with 2H-azirines are unknown at all. Our study of the chemical behavior of 2H-azirines under Rh(II)-catalyzed decomposition of diazoketones was started with searching for optimal conditions for the catalytic reaction of 2,3-diphenyl-2H-azirine (1a) with 1-diazo-1-phenylpropan-2-one (2a), leading to oxazine 4a. The most successful procedure involving addition of Rh2(OAc)4 to a boiling 1:1.25 mixture of azirine 1a and diazo compound 2a in 1,2-dichloroethane (DCE) (procedure A) gave rise to oxazine 4a and azadiene Z-3a in 60 and 7% yields, respectively (Scheme 2, Table 1, entry 1). It was also shown that azadiene Z-3a did not cyclize into oxazine 4a under the reaction conditions. According to our previous computational and experimental results for transformations of 3,4-diphenyl-substituted 2-azabuta-1,3-dienes derived from α-diazo-β-ketoesters, oxazine 4a formed via rapid cyclization of azadiene E-3a [15], whereas 2-azabuta-1,3-dienes with the C=C bond in Z-configuration have a sufficiently high activation barrier for cyclization and are usually stable up to 90–100 °C.
![[1860-5397-11-35-i2]](/bjoc/content/inline/1860-5397-11-35-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Rh(II)-catalyzed reaction of azirines 1a−g with diazo compounds 2a–c.
Scheme 2: Rh(II)-catalyzed reaction of azirines 1a−g with diazo compounds 2a–c.
Table 1: Rh(II)-catalyzed reaction of azirines 1a–g with diazo compounds 2a–c (method A, DCE, 84 °C).
| Entry | 1 | Ar | R1 | R2 | R3 | R4 | 2 | Catalyst | Isolated yields, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 6 | 7 | 8 | |||||||||
| 1 | 1a | Ph | Me | Ph | H | Ph | 2a | Rh2(OAc)4 | 7 (Z-a)a | 60 (a) | |||
| 2 | 1b | Ph | Me | Ph | H | 1-Btb | 2a | Rh2(OAc)4 | 55 (b) | ||||
| 3 | 1c | Ph | Me | Ph | Me | Me | 2a | Rh2(OAc)4 | 58 (c) | ||||
| 4 | 1d | Ph | Me | Ph | Ph | Ph | 2a | Rh2(OAc)4 | 75 (d)c | 7 (d) | |||
| 5 | 1c | Ph | 4-ClC6H4 | H | Me | Me | 2b | Rh2(OAc)4 | 43 (e)d | 67 (e)e | |||
| 6 | 1e | 4-MeC6H4 | Me | MeCO | H | H | 2c | Rh2(OAc)4 | 17 (f) | –f | –f | –f | |
| 7 | 1e | 4-MeC6H4 | Me | MeCO | H | H | 2c | Rh2(Oct)4 | 22 (f)g | 23 (f) | 7 (f) | 4 (f) | |
| 8 | 1f | 4-MeOC6H4 | Me | MeCO | H | H | 2c | Rh2(Oct)4 | 18 (g) | 26 (g)h | 6 (g) | 4 (g)h | |
| 9 | 1g | 4-NO2C6H4 | Me | MeCO | H | H | 2c | Rh2(Oct)4 | 58 (h) | 13 (h) | |||
| 10 | 1a | Ph | Me | MeCO | H | Ph | 2c | Rh2(OAc)4 | 47 (i)i | ||||
aCompound Z-3a is a 1.1:1 mixture of isomers across C=N bond. b1H-Benzotriazol-1-yl. cCompound 3d is a 1.2:1 mixture of isomers across C=N bond. dReaction temperature 60 °C. eObtained from azadiene 3e at 84 °C. fNot isolated. gEt3N-doped eluent for chromatography was used. hThe yield was determined by 1H NMR spectroscopy with acenaphtene as internal standard. The compound decomposes on silica gel. iThe reaction was carried out according to method B.
Oxazine 4b was synthesized in a similar way from azirine 1b and diazo compound 2a (Table 1, entry 2), but in this case no traces of azadiene Z-3b were detected. 4,4-Dimethyl- and 4,4-diphenyl-2-azabuta-1,3-dienes 3c,d derived from azirines 1c,d (Table 1, entries 3 and 4) showed a different behavior. The former rapidly cyclized into oxazine 4c, isolated in 58% yield. On the contrary, oxazine 4d was obtained in as low as 7% yield, while the main reaction product was azadiene 3d. Moreover, when dissolved separately in CDCl3, compounds 3d and 4d both converted into 4.5:1 3d + 4d equilibrium mixtures after a week at room temperature.
To prepare a C5-unsubstituted 1,4-oxazine derivative, we synthesized α-diazo-4-chloroacetophenone (2b) according to the published procedure [25]. Diazo compound 2b slowly decomposes in the presence of Rh2(OAc)4 and 2,2-dimethyl-3-phenylazirine (1c) even at room temperature, but no conversion of the latter was observed according to 1H NMR spectroscopy. A satisfactory result was obtained, when the reaction was carried out at 60 °С: azirine 1c was completely consumed to give azadiene 3e isolated by column chromatography in 43% yield (Table 1, entry 5). The structure of azadiene 3e was confirmed by X-ray diffraction analysis (Figure 1). It should be noted that azadiene 3e in crystal exists in the s-trans-conformation across the single С–N bond (the С2–N1–С3–C4 dihedral angle is 4.3°, Figure 1), unlike its C4-aryl-substituted analogs with the angle of 73–75° [26]. The Е-configuration of the С=N bond, unfavorable for cyclization into 1,4-oxazine, explains the enhanced thermal stability of compound 3e.
![[1860-5397-11-35-1]](/bjoc/content/figures/1860-5397-11-35-1.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of azadiene 3e.
Figure 1: X-ray crystal structure of azadiene 3e.
Heating azadiene 3e in DCE under reflux (84 °C) for 3.5 h gave a 6:1 equilibrium mixture of 1,4-oxazine 4e and azadiene 3e (according to 1Н NMR spectroscopy). Oxazine 4e is stable enough at room temperature to be isolated by column chromatography (Table 1, entry 5).
When 3-(p-tolyl)azirine 1e was reacted with diazoacetylacetone 2c under similar conditions (Rh2(OAc)4, 60 °C, DCE), four products were detected by 1H NMR spectroscopy (Scheme 2, Table 1, entry 6). However, chromatography of the reaction mixture on silica gel gave only oxazine 4f in 17% yield, whereas other compounds completely decomposed. The use of dirhodium tetraoctanoate Rh2(Oct)4 as a catalyst in refluxing DCE, as well as an Et3N-doped eluent gave a slightly higher yield of 4f (Table 1, entry 7) and allowed isolation of three byproducts formed via cleavage of the azirine N–C3 bond: bicycle 6f (hereinafter referred to as the [3.2.1] adduct), bicycle 7f (hereinafter referred to as the [4.3.1] adduct), and 5,7-dioxa-1-azabicyclo[4.4.1]undeca-3,8-diene derivative 8f. The same catalyst and the same reaction and purification conditions were used in further experiments. Analogous reaction of 4-methoxyphenyl-substituted azirine 1f yielded the same set of products (Scheme 2, Table 1, entry 8). Unfortunately, [3.2.1] adduct 6g and imide 8g were too unstable to be isolated by chromatography on silica gel, even using Et3N-doped eluents. Their presence in the reaction mixture was unambiguously confirmed by 1H NMR spectroscopy (Table 1, entry 8). At the same time, oxazine 4g and adduct 7g are stable on silica and were isolated in a pure form. The reaction of azirine 1g containing a 4-nitrophenyl substituent on C3 produces only oxazine 4h and [4.3.1] adduct 7h, which were isolated in 58 and 13% yields, respectively (Table 1, entry 9). Compounds 6–8 were characterized by standard spectral methods and the structures of adducts 6f and 7h were additionally confirmed by X-ray diffraction analysis (Figure 2). The reaction of diazoacetylacetone 2c with 2,3-diphenyl-2H-azirine (1a) provides oxazine 4i as a sole product (Table 1, entry 10). It was isolated with the highest yield of 47% by slow addition of a solution of the diazo compound to a solution of the azirine and Rh2(OAc)4 in DCE at 60 °С (procedure B).
![[1860-5397-11-35-2]](/bjoc/content/figures/1860-5397-11-35-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structures of compounds 6f and 7h.
Figure 2: X-ray crystal structures of compounds 6f and 7h.
To the best of our knowledge, neither heterocyclic systems with a 4,6-dioxa-1-azabicyclo[3.2.1]oct-2-ene backbone (compounds 6) nor systems with a 5,7-dioxa-1-azabicylo[4.4.1]undec-8-ene backbone (compounds 7) have ever been reported. On the contrary, bicyclic compounds 8f,g are representatives of a known class of 5,7-dioxa-1-azabicylo[4.4.1]undeca-3,8-diene-2,10-dione derivatives formed by the recently reported reaction of 3-arylazirines with acyl ketenes generated by thermolysis of 2-diazo-1,3-diketones [12] or 5-arylfuran-2,3-diones [13]. Therefore, the presence of compounds 8f,g among the reaction products provides evidence for the formation of some amounts of acetyl(methyl)ketene (12) under the reaction conditions, which, in turn, gives us insight to the mechanism of formation of adducts 6 and 7 (Scheme 3). A separate experiment was performed to show that these compounds are formed via independent pathways, as they do not interconvert under the reaction conditions (Rh2(Oct)4, 84 °C, DCE).
We assumed that the reaction sequence leading to bicycles 6 and 7 involves the 1,5-cyclization of azirinium ylide 9f–h to dihydroazireno[2,1-b]oxazole 10f–h followed by cycloaddition of the latter to ketene 12 to give two regioisomeric adducts 6f,g and 7f–h (Scheme 3).
![[1860-5397-11-35-i3]](/bjoc/content/inline/1860-5397-11-35-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: General scheme for the formation of compounds 4,6 and 7.
Scheme 3: General scheme for the formation of compounds 4,6 and 7.
Several examples of the 1,5-cyclization of azomethine ylides bearing an α-keto group into oxazole derivatives were reported [27-29]. As also known, the azomethine ylide derived from N-benzylideneanisidine and diazoacetylacetone under Rh2(OAc)4-catalysis undergoes 1,3-cyclization to an aziridine derivative in high yield, rather than 1,5-cyclization [22]. However, no cyclizations of azirinium ylides, cyclic analogs of azomethine ylides, are known. This is not surprising in view of the high strain of the azirinium system, and until now ring opening in these systems seemed much more preferable than annelation of a new cycle. Nevertheless, we decided to study two competing pathways for isomerization of the model azirinium ylide 9j: ring opening into azadiene 3j and 1,5-cyclization into azirenooxazole 10j (Scheme 4), by means of DFT calculations (B3LYP/6-31+G(d,p)). In addition, two reasonable pathways for the formation of adducts 6j and 7j formed from azirenooxazole 10j and ketene 12 were studied at the same level of theory (Scheme 4).
![[1860-5397-11-35-i4]](/bjoc/content/inline/1860-5397-11-35-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Possible pathways for the formation of 6j and 7j from azirenooxazole 10j and ketene 12.
Scheme 4: Possible pathways for the formation of 6j and 7j from azirenooxazole 10j and ketene 12.
According to the calculations, the barrier to the 1,5-cyclization of ylide 9j to compound 10j was found to be even slightly lower (7.9 kcal/mol) than the barrier to the ring opening across the N–С2 bond to azadiene 3j (10.1 kcal/mol) (Figure 3). Azirenooxazole 10j is thermodynamically more stable than ylide 9j, by ca. 15 kcal/mol, but the barrier to the reverse reaction 10j→9j is not too high (22.6 kcal/mol). The ring opening in azirinium ylide 9j into azadiene 3j, too, has a low activation barrier but occurs irreversibly. Therefore, azirenooxazole 10j might form in this reaction, and, moreover, its formation from ylide 9j is kinetically preferred over the formation of azadiene 3j. However, in view of the reversibility of the 1,5-cyclization 9j →10j and in the absence of an active trap for azirenooxazole 10j in the reaction mixture, it isomerizes via ylide 9j to a much more thermodynamically stable open-chain form 3j.
![[1860-5397-11-35-3]](/bjoc/content/figures/1860-5397-11-35-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Energy profiles [DFT B3LYP/6-31+G(d,p), 357 K, 1,2-dichloroethane (PCM)] for the transformation of ylide 9j into azadiene 3j and dihydroazireno[2,1-b]oxazole 10j and for the transformation of 10j to adducts 6j,7j.
Figure 3: Energy profiles [DFT B3LYP/6-31+G(d,p), 357 K, 1,2-dichloroethane (PCM)] for the transformation of ...
Curiously, the same energy profile (see Supporting Information File 1, Scheme S1) was obtained for isomerization of azirinium ylide 5 (Scheme 1, R1 = Me, R3 = R4 = H, R5 = Ph, Alk = Me) which contains one acyl group and one ester group at the carbanion center. From these results it follows that 1,5-cyclization of azirinium ylides is a general stabilization route for ylides derived from diazo compounds containing an α-keto group. However, azirenooxazoles 10 reversibly formed in these reactions could not be detected in the absence of an active electrophilic trap in the reaction mixture. 3-Diazoacetylacetone under Rh(II)-catalysis gives, along with a Rh(II) carbenoid, a highly electrophilic acetyl(methyl)ketene (12) via Wolff rearrangement (Scheme 3).
The nucleophilic attack of the azirenooxazole 10j nitrogen on the ketene sp-carbon followed by cyclization provides two isomeric adducts 6j and 7j. There are two reasonable pathways to these unexpected products. The first pathway involves the addition of bicycle 10j to ketene 12 to form atropoisomeric azirenooxazolium betains 13j,13′j (Scheme 4) which can further undergo aziridine ring opening to give atropoisomeric oxazinium betaines 14j,14′j and their cyclization into adducts 6j and 7j, respectively. Besides, betaines 14j,14′j can result from aziridine ring opening in azirenooxazole 10j into carbonyl ylide 15j and its addition to ketene 12 (Scheme 4).
The energy profiles for both the cycloaddition pathways of 10j to 12 are represented in Figure 3 with the use of relative scale for total energy ΔΔE. According to the calculations, the two attack modes of azirenooxazole 10j to ketene 12 (red and blue lines on the plot) give rise to atropoisomeric azirenooxazolium betains 13j,13′j (not shown in the plot) which undergo a virtually barrierless aziridine ring opening to give oxazinium betaines 14j,14′j. The activation barriers to both the transformation pathways of 10j to 13 are close to each other (10.5 and 11.6 kcal/mol, respectively) but much lower than that to the ring opening of azirenooxazole 10j to azirinium ylide 9j. The cyclizations of betaines 14j,14′j into bicycles 6j,7j proceed with extremely low activation barriers (1.1 and 4.7 kcal/mol). The alternative pathway to compounds 6j,7j, involving reversible formation of carbonyl ylide 15j, can result in exclusive formation of [3.2.1] adduct 6j, because the transition state TS9 leading to betaine 14′j, a precursor of [4.3.1] adduct 7j, has a higher energy than the transition state TS5 for the competitive pathway. In spite of the fact that the transition states TS3 and TS8 for alternative betaine 14j formation pathways are very close in energy, the reaction sequence 10j→13j→14j leading to [3.2.1] adduct 6j seems to be more reasonable than 10j→15j→14j due to the much higher concentrations of the reacting species. Actually, the activation barrier to the bicyclic C–N bond cleavage in 10j is very low, but the equilibrium between 10j and 15j is strongly shifted toward bicyclic isomer 10j, and, therefore, carbonyl ylide 15j should be formed in an extremely low concentration.
Thus, the computations predict: a) two competing pathways for transformation of azirinium ylides derived both from 2-diazo-1,3-diketons and α-diazo-β-ketoesters; b) a high reactivity of azirenooxazole 10j toward acetyl(methyl)ketene (12); c) two competing modes of the attack of 10j on ketene 12, leading to adducts 6j,7j via two short-lived betaine intermediates 13j,13′j and 14j,14′j.
It is worthy to notice that the distribution of products 6,7 strongly depends on the electronic effects of the para substituents in the aryl group of 3-aryl-2H-azirine 1. The [3.2.1] adduct is preferably formed from azirines 1e,f with electron-donating substituents (Table 1, entries 7 and 8), while 3-(4-nitrophenyl)-2H-azirine (1g) provides the [4.3.1] adduct only (Table 1, entry 9).
It is known that the Rh(II)-catalyzed reaction of 3-aryl-2H-azirines with ethyl diazoacetoacetate (2d) gives rise to 2H-1,4-oxazines as a single product [15]. The absence of products like 6 or 7 may be caused by a decreased propensity of the carbenoid derived from 2d to undergo a Wolff rearrangement into a ketene derivative. To obtain experimental evidence for the formation of azirenooxazole intermediates 10 in the reactions of azirines 1 with α-diazo-β-ketoesters, we reacted azirine 1g with a mixture of two diazo compounds, ethyl diazoacetoacetate (2d) and diazoacetylacetone (2c), in the presence of Rh2(Oct)4 (Scheme 5). We suggested that the azirenooxazole formed from the azirine and diazo compound 2d will be trapped by ketene 12 generated from diazoacetylacetone 2c via Wolff rearrangement. The 1H NMR and TLC analysis of the reaction mixture revealed, along with oxazines 4k, 4h and adduct 7h, one compound. The latter was not detected among the products of the reactions of azirine 1g separately with each of the diazo compounds. This product was isolated by column chromatography on silica gel, and its structure corresponding to the [4.3.1] adduct 7k formed by cycloaddition of ketene 12 to azirenoxazole 10k was assigned on the basis of the 1H and 13C NMR and mass spectra. According to the 1H NMR spectrum of the reaction mixture, the 4k:4h:7k:7h ratio was 16:7:1:2. In the analogous reaction of 3-(p-tolyl)azirine (1e) with a mixture of diazo compounds 2c,d, no other products than [3.2.1] adduct 6l (see Supporting Information File 1, Scheme S1) formed from the ethoxycarbonyl-substituted azirenooxazole derivative and ketene 12 were detected.
![[1860-5397-11-35-i5]](/bjoc/content/inline/1860-5397-11-35-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Rh2(Oct)4-catalyzed reaction of azirine 1g with diazo compound 2d in the presence of diazo compound 2c.
Scheme 5: Rh2(Oct)4-catalyzed reaction of azirine 1g with diazo compound 2d in the presence of diazo compound ...
Obviously, the bridgehead nitrogen in azirenooxazoles 10 must be sterically accessible for smooth addition of 10 to ketene 12. For example, methyl or phenyl substitution in the one-atom bridge in the related 5-oxa-1-azabicyclo[4.1.0]hept-3-ene system completely suppresses the reactivity of the latter toward acylketenes [12]. This is a possible reason for the formation of neither 6 nor 7 adduct in the reaction of diazoacetylacetone (2c) with 2,3-diphenyl-2H-azirine (1a, Table 1, entry 10). The moderate yield of compound 4i is explained by the formation of an unstable by-product of the transformation of the acetyl group in target oxazine 4i under the reaction conditions. We succeeded in isolating a by-product of this type in the analogous reaction of spiroazirine 1h with diazo compound 2c (Scheme 6). Along with oxazine 16 (57%), small amount of 1,4-oxazine 17 with a modified acetyl group at С5 was isolated by column chromatography. Compound 17 obviously resulted from cycloaddition of acyl ketene 12 derived from 2c under the reaction conditions to the carbonyl group of oxazine 16. Actually, compound 17 formed in the reaction of a pure oxazine 16 with diazo compound 2c in the presence of Rh2(OAc)4. Its structure was assigned by standard spectral methods and confirmed by X-ray diffraction analysis (Figure 4). It was found that, when kept for a week in a CDCl3 solution in the dark at room temperature, oxazine 17 undergoes reversible ring opening to form a 1:2.3 equilibrium mixture of 17 and 18. Azadiene 18 was isolated by chromatography and characterized by standard spectral methods.
![[1860-5397-11-35-i6]](/bjoc/content/inline/1860-5397-11-35-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Rh2(OAc)4-catalyzed reaction of azirine 1h with diazo compound 2c.
Scheme 6: Rh2(OAc)4-catalyzed reaction of azirine 1h with diazo compound 2c.
![[1860-5397-11-35-4]](/bjoc/content/figures/1860-5397-11-35-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structure of compound 17.
Figure 4: X-ray crystal structure of compound 17.
The synthesized 5-phenyl- (4a–d) and 5-unsubstituted (4e) 2H-1,4-oxazines proved to be stable in the dark but undergo an irreversible ring opening to azadienes 3 under UV irradiation. Thus, irradiation of oxazine 4a for 1.5 h gives quantitatively azadiene Z-3a which was also obtained by the reaction of azirine 1a and α-diazo-α-phenylacetone (2a, Table 1, entry 1). By contrast, 5-acetyl-substituted oxazines 4f–i are photochromic compounds. Being pale yellow colored, under UV irradiation in a CDCl3 solution at room temperature they undergo a ring opening to yellow–orange azadienes 3f–i which cyclize back to oxazines 4f–i in the dark. The effect of the C5-substituent in 2H-1,4-oxazine on its photochromic activity can be tracked by changes in the half-life times of open-chain isomers Z-3a,Z-3i and 19 (Scheme 7). It was found that the azadiene cyclization rate strongly depends on the electron-withdrawing ability of substituent R and increases as it changes from Ph or H to CO2Et or COMe.
![[1860-5397-11-35-i7]](/bjoc/content/inline/1860-5397-11-35-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Effect of the C5-substituent in the 2H-1,4-oxazine system on its photochromic activity.
Scheme 7: Effect of the C5-substituent in the 2H-1,4-oxazine system on its photochromic activity.
Conclusion
5-Unsubstituted and 5-aryl-substituted 2H-1,4-oxazines can be readily prepared by the Rh(II)-catalyzed reaction of 2H-azirines with α-diazo ketones. The reaction involves intermediate formation of azirinium ylides and their irreversible ring opening across the N–C2 bond to 2-azabuta-1,3-diene followed by 1,6-cyclization. Along with ring opening, transient azirinium ylides more readily undergo reversible 1,5-cyclization to dihydroazireno[2,1-b]oxazoles which can be trapped with such active electrophiles as acylketenes to give 4,6-dioxa-1-azabicyclo[3.2.1]oct-2-ene and/or 5,7-dioxa-1-azabicyclo[4.3.1]deca-3,8-diene-2-one derivatives. This reaction sequence partly occurs, when 3-aryl-2H-azirines and 3-diazoacetylacetone, which is able to produce acetyl(methyl)ketenes via Rh(II)-catalyzed Wolff rearrangement, are used as starting materials. According to DFT B3LYP/6-31+G(d,p) calculations, the cycloaddition of dihydroazireno[2,1-b]oxazoles to acetyl(methyl)ketene, leading to the final bicyclic products, proceeds via consecutive low barrier formation of two pairs of atropoisomeric betaine intermediates followed by their cyclization.
The reaction of 2,3-di- and 2,2,3-triaryl-2H-azirines with both α-diazo-α-phenylacetone and 3-diazoacetylacetone gives rise to 2-azabuta-1,3-dienes and/or 2H-1,4-oxazines. The alternative reaction pathway via N–C3 azirine bond cleavage does not take place in this case because of the steric shielding of the nitrogen atom in the dihydroazireno[2,1-b]oxazole intermediate. Under UV irradiation 5-unsubstituted and 5-aryl-substituted 2H-1,4-oxazines produce stable 2-azabuta-1,3-dienes. In contrast, 5-acyl-substituted 2H-1,4-oxazines are photochromic compounds, i.e., under UV irradiation they undergo ring opening to 2-azabuta-1,3-dienes which transform back to the oxazines in the dark at room temperature. The rate of the reverse cyclization reaction increases with increasing electron-withdrawing ability of C1-substituent in 2-azabuta-1,3-diene.
Supporting Information
| Supporting Information File 1: Experimental part, computational details and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 4.5 MB | Download |
Acknowledgements
We gratefully acknowledge the financial support of the Russian Foundation for Basic Research (grant no. 14-03-00187, 14-03-31117) and Saint Petersburg State University (grant no. 12.38.239.2014, 12.38.217.2015). This research used resources of the resource center ‘Computer Center’, ‘Research resource center for Magnetic Resonance’, ‘Center for Chemical Analysis and Material Research’, and ‘Research resource Centre for X-ray Diffraction Studies’ of Saint Petersburg State University.
References
-
Heimgartner, H. Angew. Chem., Int. Ed. Engl. 1991, 30, 238–264. doi:10.1002/anie.199102381
Return to citation in text: [1] [2] -
Koch, K. N.; Linden, A.; Heimgartner, H. Tetrahedron 2001, 57, 2311–2326. doi:10.1016/S0040-4020(01)00091-6
Return to citation in text: [1] -
Brun, K. A.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2002, 85, 3422–3443. doi:10.1002/1522-2675(200210)85:10<3422::AID-HLCA3422>3.0.CO;2-N
Return to citation in text: [1] -
Huang, C.-Y.; Doyle, A. G. Chem. Rev. 2014, 114, 8153–8198. doi:10.1021/cr500036t
Return to citation in text: [1] -
Khlebnikov, A. F.; Novikov, M. S. Tetrahedron 2013, 69, 3363–3401. doi:10.1016/j.tet.2013.02.020
Return to citation in text: [1] -
Rostovskii, N. V.; Novikov, M. S.; Khlebnikov, A. F.; Korneev, S. M.; Yufit, D. S. Org. Biomol. Chem. 2013, 11, 5535–5545. doi:10.1039/c3ob40708j
Return to citation in text: [1] -
Teixeira Costa, F. In Heterocyclic Targets in Advanced Organic Synthesis; do Carmo Carreiras, M.; Marco-Contelles, J., Eds.; Research Signpost: Trivandrum, India, 2011; p 145.
Return to citation in text: [1] -
Padwa, A. Adv. Heterocycl. Chem. 2010, 99, 1–31. doi:10.1016/S0065-2725(10)09901-0
Return to citation in text: [1] -
Lemos, A. Molecules 2009, 14, 4098–4119. doi:10.3390/molecules14104098
Return to citation in text: [1] -
Palacios, F.; Ochoa de Retana, A. M.; Martínez de Marigorta, E.; de los Santos, J. M. In Recent Developments in Heterocyclic Chemistry; Pinho e Melo, T. M. V. D.; d'A Rocha-Gonsalves, A. M., Eds.; Research Signpost: Trivandrum, India, 2007; p 27.
Return to citation in text: [1] -
Palacios, F.; Ochoa de Retana, A. M.; Martínez de Marigorta, E.; de los Santos, J. M. Eur. J. Org. Chem. 2001, 2401–2414. doi:10.1002/1099-0690(200107)2001:13<2401::AID-EJOC2401>3.0.CO;2-U
Return to citation in text: [1] -
Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Yufit, D. S. J. Org. Chem. 2011, 76, 9344–9352. doi:10.1021/jo201563b
Return to citation in text: [1] [2] [3] -
Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Iakovenko, R. O.; Yufit, D. S. Beilstein J. Org. Chem. 2014, 10, 784–793. doi:10.3762/bjoc.10.74
Return to citation in text: [1] [2] -
Palacios, F.; Aparicio, D.; Ochoa de Retana, A. M.; de los Santos, J. M.; Gil, J. I.; Alonso, J. M. J. Org. Chem. 2002, 67, 7283–7288. doi:10.1021/jo025995d
Return to citation in text: [1] -
Rostovskii, N. V.; Novikov, M. S.; Khlebnikov, A. F.; Khlebnikov, V. A.; Korneev, S. M. Tetrahedron 2013, 69, 4292–4301. doi:10.1016/j.tet.2013.03.106
Return to citation in text: [1] [2] [3] -
Livant, P.; Jie, Y.; Wang, X. Tetrahedron Lett. 2005, 46, 2113–2116. doi:10.1016/j.tetlet.2005.01.131
Return to citation in text: [1] -
Bréhu, L.; Fernandes, A.-C.; Lavergne, O. Tetrahedron Lett. 2005, 46, 1437–1440. doi:10.1016/j.tetlet.2005.01.045
Return to citation in text: [1] -
Lee, S.-H.; Yoshida, K.; Matsushita, H.; Clapham, B.; Koch, G.; Zimmermann, J.; Janda, K. D. J. Org. Chem. 2004, 69, 8829–8835. doi:10.1021/jo048353u
Return to citation in text: [1] -
Zhang, X.; Sui, Z. Tetrahedron Lett. 2006, 47, 5953–5955. doi:10.1016/j.tetlet.2006.06.053
Return to citation in text: [1] -
Lee, Y. R.; Suk, J. Y. Tetrahedron Lett. 2000, 41, 4795–4799. doi:10.1016/S0040-4039(00)00716-4
Return to citation in text: [1] -
Lee, Y. R.; Suk, J. Y. Heterocycles 1998, 48, 875–883. doi:10.3987/COM-97-7881
Return to citation in text: [1] -
Zhang, X.-J.; Yan, M.; Huang, D. Org. Biomol. Chem. 2009, 7, 187–192. doi:10.1039/b813763c
Return to citation in text: [1] [2] -
Dong, C.; Deng, G.; Wang, J. J. Org. Chem. 2006, 71, 5560–5564. doi:10.1021/jo0605039
Return to citation in text: [1] -
Padwa, A.; Dean, D. C. J. Org. Chem. 1990, 55, 405–406. doi:10.1021/jo00289a001
Return to citation in text: [1] -
Wilds, A. L.; Mearder, A. L., Jr. J. Org. Chem. 1948, 13, 763–769. doi:10.1021/jo01163a024
Return to citation in text: [1] -
Khlebnikov, A. F.; Novikov, M. S.; Amer, A. A.; Kostikov, R. R.; Magull, J.; Vidovic, D. Russ. J. Org. Chem. 2006, 42, 515–526. doi:10.1134/S1070428006040075
Return to citation in text: [1] -
Shimoharada, H.; Ikeda, S.; Kajigaeshi, S.; Kanemasa, S. Chem. Lett. 1977, 6, 1237–1238. doi:10.1246/cl.1977.1237
Return to citation in text: [1] -
Beletskii, E. V.; Kuznetsov, M. A. Russ. J. Org. Chem. 2009, 45, 1229–1240. doi:10.1134/S107042800908020X
Return to citation in text: [1] -
Kuznetsov, M. A.; Voronin, V. V. Chem. Heterocycl. Compd. 2011, 47, 173–181. doi:10.1007/s10593-011-0738-8
Return to citation in text: [1]
| 12. | Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Yufit, D. S. J. Org. Chem. 2011, 76, 9344–9352. doi:10.1021/jo201563b |
| 22. | Zhang, X.-J.; Yan, M.; Huang, D. Org. Biomol. Chem. 2009, 7, 187–192. doi:10.1039/b813763c |
| 15. | Rostovskii, N. V.; Novikov, M. S.; Khlebnikov, A. F.; Khlebnikov, V. A.; Korneev, S. M. Tetrahedron 2013, 69, 4292–4301. doi:10.1016/j.tet.2013.03.106 |
| 1. | Heimgartner, H. Angew. Chem., Int. Ed. Engl. 1991, 30, 238–264. doi:10.1002/anie.199102381 |
| 2. | Koch, K. N.; Linden, A.; Heimgartner, H. Tetrahedron 2001, 57, 2311–2326. doi:10.1016/S0040-4020(01)00091-6 |
| 3. | Brun, K. A.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2002, 85, 3422–3443. doi:10.1002/1522-2675(200210)85:10<3422::AID-HLCA3422>3.0.CO;2-N |
| 1. | Heimgartner, H. Angew. Chem., Int. Ed. Engl. 1991, 30, 238–264. doi:10.1002/anie.199102381 |
| 13. | Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Iakovenko, R. O.; Yufit, D. S. Beilstein J. Org. Chem. 2014, 10, 784–793. doi:10.3762/bjoc.10.74 |
| 14. | Palacios, F.; Aparicio, D.; Ochoa de Retana, A. M.; de los Santos, J. M.; Gil, J. I.; Alonso, J. M. J. Org. Chem. 2002, 67, 7283–7288. doi:10.1021/jo025995d |
| 27. | Shimoharada, H.; Ikeda, S.; Kajigaeshi, S.; Kanemasa, S. Chem. Lett. 1977, 6, 1237–1238. doi:10.1246/cl.1977.1237 |
| 28. | Beletskii, E. V.; Kuznetsov, M. A. Russ. J. Org. Chem. 2009, 45, 1229–1240. doi:10.1134/S107042800908020X |
| 29. | Kuznetsov, M. A.; Voronin, V. V. Chem. Heterocycl. Compd. 2011, 47, 173–181. doi:10.1007/s10593-011-0738-8 |
| 12. | Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Yufit, D. S. J. Org. Chem. 2011, 76, 9344–9352. doi:10.1021/jo201563b |
| 13. | Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Iakovenko, R. O.; Yufit, D. S. Beilstein J. Org. Chem. 2014, 10, 784–793. doi:10.3762/bjoc.10.74 |
| 26. | Khlebnikov, A. F.; Novikov, M. S.; Amer, A. A.; Kostikov, R. R.; Magull, J.; Vidovic, D. Russ. J. Org. Chem. 2006, 42, 515–526. doi:10.1134/S1070428006040075 |
| 4. | Huang, C.-Y.; Doyle, A. G. Chem. Rev. 2014, 114, 8153–8198. doi:10.1021/cr500036t |
| 5. | Khlebnikov, A. F.; Novikov, M. S. Tetrahedron 2013, 69, 3363–3401. doi:10.1016/j.tet.2013.02.020 |
| 6. | Rostovskii, N. V.; Novikov, M. S.; Khlebnikov, A. F.; Korneev, S. M.; Yufit, D. S. Org. Biomol. Chem. 2013, 11, 5535–5545. doi:10.1039/c3ob40708j |
| 7. | Teixeira Costa, F. In Heterocyclic Targets in Advanced Organic Synthesis; do Carmo Carreiras, M.; Marco-Contelles, J., Eds.; Research Signpost: Trivandrum, India, 2011; p 145. |
| 8. | Padwa, A. Adv. Heterocycl. Chem. 2010, 99, 1–31. doi:10.1016/S0065-2725(10)09901-0 |
| 9. | Lemos, A. Molecules 2009, 14, 4098–4119. doi:10.3390/molecules14104098 |
| 10. | Palacios, F.; Ochoa de Retana, A. M.; Martínez de Marigorta, E.; de los Santos, J. M. In Recent Developments in Heterocyclic Chemistry; Pinho e Melo, T. M. V. D.; d'A Rocha-Gonsalves, A. M., Eds.; Research Signpost: Trivandrum, India, 2007; p 27. |
| 11. | Palacios, F.; Ochoa de Retana, A. M.; Martínez de Marigorta, E.; de los Santos, J. M. Eur. J. Org. Chem. 2001, 2401–2414. doi:10.1002/1099-0690(200107)2001:13<2401::AID-EJOC2401>3.0.CO;2-U |
| 12. | Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Yufit, D. S. J. Org. Chem. 2011, 76, 9344–9352. doi:10.1021/jo201563b |
| 20. | Lee, Y. R.; Suk, J. Y. Tetrahedron Lett. 2000, 41, 4795–4799. doi:10.1016/S0040-4039(00)00716-4 |
| 21. | Lee, Y. R.; Suk, J. Y. Heterocycles 1998, 48, 875–883. doi:10.3987/COM-97-7881 |
| 15. | Rostovskii, N. V.; Novikov, M. S.; Khlebnikov, A. F.; Khlebnikov, V. A.; Korneev, S. M. Tetrahedron 2013, 69, 4292–4301. doi:10.1016/j.tet.2013.03.106 |
| 17. | Bréhu, L.; Fernandes, A.-C.; Lavergne, O. Tetrahedron Lett. 2005, 46, 1437–1440. doi:10.1016/j.tetlet.2005.01.045 |
| 18. | Lee, S.-H.; Yoshida, K.; Matsushita, H.; Clapham, B.; Koch, G.; Zimmermann, J.; Janda, K. D. J. Org. Chem. 2004, 69, 8829–8835. doi:10.1021/jo048353u |
| 19. | Zhang, X.; Sui, Z. Tetrahedron Lett. 2006, 47, 5953–5955. doi:10.1016/j.tetlet.2006.06.053 |
| 25. | Wilds, A. L.; Mearder, A. L., Jr. J. Org. Chem. 1948, 13, 763–769. doi:10.1021/jo01163a024 |
| 16. | Livant, P.; Jie, Y.; Wang, X. Tetrahedron Lett. 2005, 46, 2113–2116. doi:10.1016/j.tetlet.2005.01.131 |
| 15. | Rostovskii, N. V.; Novikov, M. S.; Khlebnikov, A. F.; Khlebnikov, V. A.; Korneev, S. M. Tetrahedron 2013, 69, 4292–4301. doi:10.1016/j.tet.2013.03.106 |
| 22. | Zhang, X.-J.; Yan, M.; Huang, D. Org. Biomol. Chem. 2009, 7, 187–192. doi:10.1039/b813763c |
| 23. | Dong, C.; Deng, G.; Wang, J. J. Org. Chem. 2006, 71, 5560–5564. doi:10.1021/jo0605039 |
| 24. | Padwa, A.; Dean, D. C. J. Org. Chem. 1990, 55, 405–406. doi:10.1021/jo00289a001 |
© 2015 Rostovskii et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)