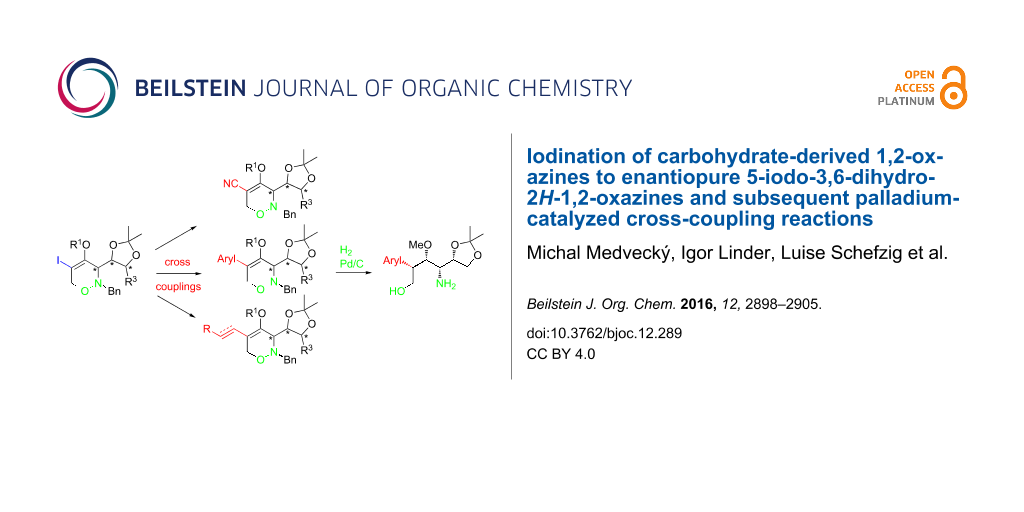
Abstract
Iodination of carbohydrate-derived 3,6-dihydro-2H-1,2-oxazines of type 3 using iodine and pyridine in DMF furnished 5-iodo-substituted 1,2-oxazine derivatives 4 with high efficacy. The alkenyl iodide moiety of 1,2-oxazine derivatives syn-4 and anti-4 was subsequently exploited for the introduction of new functionalities at the C-5 position by applying palladium-catalyzed carbon–carbon bond-forming reactions such as Sonogashira, Heck, or Suzuki coupling reactions as well as a cyanation reaction. These cross-coupling reactions led to a series of 5-alkynyl-, 5-alkenyl-, 5-aryl- and 5-cyano-substituted 1,2-oxazine derivatives being of considerable interest for further synthetic elaborations. This was exemplarily demonstrated by the hydrogenation of syn-21 and anti-24 and by a click reaction of a 5-alkynyl-substituted precursor.

Graphical Abstract
Introduction
Over the last decade, we have intensively studied syntheses and applications of 3,6-dihydro-2H-1,2-oxazines of type 3 [1,2]. These N,O-heterocycles are easily prepared in enantiopure form by a stereodivergent [3 + 3] cyclization of carbohydrate-derived nitrones 2 and lithiated alkoxyallenes 1 (Scheme 1) [3,4]. Subsequently we investigated modifications and synthetic applications of 1,2-oxazines 3 including the preparation of seven-membered N,O-heterocycles by ring enlargement [5], functionalization of the enol ether unit [6-11], and N,O-cleavage reactions leading to amino alcohols [8,10,12], pyrroles [13] or α,β-unsaturated β-alkoxy-γ-amino-aldehydes and ketones [14]. In this context, a series of publications of our group reported on syntheses of carbohydrate mimetics [15-20] that are based on aminopyrans, aminooxepanes or other aminopolyols and that were examined for example as ligands of L- and P-selectin [21,22]. We previously reported the synthesis of enantiopure 1,2-oxazin-4-yl nonaflates and phosphates starting from precursors of type 3 and their conversion into differently C-4-substituted products employing palladium-catalyzed cross-coupling reactions [23]. In a related study, we have also investigated the synthesis of 4-halogen- and 4,5-bis(halogen)-substituted 6H-1,2-oxazines by halogenation of 6H-1,2-oxazines and subsequent palladium-catalyzed coupling reactions such as Sonogashira or Suzuki–Miyaura reactions [24] leading to aryl- and alkynyl-functionalized products. The synthetic potential of the mono- and bisalkynyl-substituted 6H-1,2-oxazines was additionally demonstrated by Lewis-acid-mediated conversion into highly substituted pyridine derivatives [25] by cycloaddition of in situ generated azapyrylium intermediates [26] and alkynes. Inspired by these previously reported results, we focused our interest on the so far unknown functionalization at the C-5 position of the synthetically useful enantiopure 3,6-dihydro-1,2-oxazines 3. Herein, we now disclose our results on the iodination of 3 to provide the previously undescribed 5-iodo-1,2-oxazine derivatives 4. These new intermediates allow subsequent C–C functionalization at the C-5 position employing various palladium-catalyzed cross-coupling reactions thus expanding the library of available enantiopure 3,6-dihydro-2H-1,2-oxazines.
![[1860-5397-12-289-i1]](/bjoc/content/inline/1860-5397-12-289-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Access to enantiopure 3,6-dihydro-1,2-oxazines 3 via lithiated alkoxyallenes 1 and carbohydrate-derived nitrones 2.
Scheme 1: Access to enantiopure 3,6-dihydro-1,2-oxazines 3 via lithiated alkoxyallenes 1 and carbohydrate-der...
Results and Discussion
Numerous procedures exist for the synthesis of β-iodo enol ethers [27], although the direct β-iodination of enol ethers using a suitable electrophilic iodine reagent is relatively underdeveloped. For the iodination of 4-alkoxy-3,6-dihydro-1,2-oxazines 3, we selected molecular iodine as the most simple iodination reagent in the presence of a base [28-30]. A clean reaction occurred upon treatment of D-glyceraldehyde-derived syn-configured 3,6-dihydro-1,2-oxazines syn-3a–c with three equivalents of iodine and one equivalent of pyridine as base in DMF at room temperature giving the desired 5-iodo-substituted 1,2-oxazines syn-4a–c in 55–87% yield after purification by column chromatography (Scheme 2). This operationally simple iodination protocol was also successfully applied to the anti-configured 1,2-oxazine anti-3a and the D-arabinose-derived starting material anti-3d furnishing the expected iodinated products anti-4a and anti-4d in good yields.
![[1860-5397-12-289-i2]](/bjoc/content/inline/1860-5397-12-289-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Iodination of 1,2-oxazines syn-3a–c and anti-3a,d leading to 5-iodo-substituted 1,2-oxazines syn-4a–c and anti-4a,d [TMSE = (2-trimethylsilyl)ethyl].
Scheme 2: Iodination of 1,2-oxazines syn-3a–c and anti-3a,d leading to 5-iodo-substituted 1,2-oxazines syn-4a...
Having attained an access to 5-iodo-substituted 1,2-oxazines 4, we turned our attention to their conversion into subsequent products by taking advantage of the alkenyl iodide functionality for various palladium-catalyzed cross-coupling reactions. As a first approach to form a new C–C bond at C-5 we envisioned the Sonogashira coupling. For this purpose, we selected (trimethylsilyl)acetylene as alkyne component. The Sonogashira couplings of 5-iodo-1,2-oxazines syn-4a and anti-4a were carried out under standard conditions using a catalytic system consisting of PdCl2(PPh3)2, CuI and triethylamine in toluene at room temperature to furnish the corresponding 5-(trimethylsilyl)ethynyl-substituted 1,2-oxazines syn-5 and anti-5 in good yields (Scheme 3). When these reaction conditions were applied to the D-arabinose-derived 5-iodo-1,2-oxazine anti-4d, the desired coupling product anti-6 was formed merely in 28% yield. Gratifyingly, this transformation was considerably improved when an alternative coupling protocol (Pd(OAc)2, PPh3, CuI in NEt3/DMF) was employed, the yield was significantly enhanced and the coupling product anti-6 was obtained in 59% yield.
![[1860-5397-12-289-i3]](/bjoc/content/inline/1860-5397-12-289-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Sonogashira reactions of 4-methoxy-1,2-oxazines syn-4a, anti-4a and anti-4d leading to 5-alkynyl-substituted 1,2-oxazines syn-5, anti-5 and anti-6.
Scheme 3: Sonogashira reactions of 4-methoxy-1,2-oxazines syn-4a, anti-4a and anti-4d leading to 5-alkynyl-su...
Sonogashira couplings of 4-benzyloxy- and 4-(2-trimethylsilyl)ethoxy-substituted 1,2-oxazines syn-4b and syn-4c with phenylacetylene worked equally well and provided under the standard conditions the desired phenylethynyl-substituted products syn-7 and syn-8, respectively, in comparable yields (Scheme 4). Even the unprotected propargyl alcohol could be applied under these coupling conditions affording the corresponding 5-alkynyl-substituted 1,2-oxazine syn-9, albeit in moderate yield (52%). In the last example of Scheme 4, syn-4a was coupled with an imidazolyl-substituted alkyne 10, that also derives from lithiated methoxyallene [31] and that was already successfully applied as alkyne component in other Sonogashira couplings presented in former publications [23,32]. The reaction of syn-4a and alkyne 10 using palladium acetate, triphenylphosphine, and copper(I) iodide in a solvent mixture of diisopropylamine and DMF at room temperature gave the 5-(imidazolylethynyl)-substituted 1,2-oxazine syn-11 in 41% yield.
![[1860-5397-12-289-i4]](/bjoc/content/inline/1860-5397-12-289-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Sonogashira reactions of D-glyceraldehyde-derived 1,2-oxazines syn-4a–c leading to 5-alkynyl-substituted 1,2-oxazines syn-7, syn-8, syn-9 and syn-11.
Scheme 4: Sonogashira reactions of D-glyceraldehyde-derived 1,2-oxazines syn-4a–c leading to 5-alkynyl-substi...
Next, we briefly studied 5-iodo-1,2-oxazine syn-4a as substrate in Heck reactions. The substrate was reacted with the alkyl acrylates 12a (R1 = Me) and 12b (R1 = t-Bu) under phosphine-free conditions [33,34] using 6 mol % of palladium(II) acetate, triethylamine as base and lithium chloride [35] leading to the expected coupling products syn-13 and syn-14 in 39% and 82% yield, respectively (Scheme 5). In both cases, only the E-configured 2-substituted alkyl acrylates were isolated. The moderate yield in the Heck reaction with methyl acrylate 12a is very likely caused by the tendency of this olefin to polymerize under the conditions applied. Consequently, the change of the olefin component from the methyl to the tert-butyl ester allowed the preparation of the corresponding coupling product syn-14 in significantly better yield. The use of dehydroamino acids such as olefin 16 [36] in Heck reactions [37-42] is also of interest, since these coupling products are useful intermediates for the synthesis of non-proteinogenic amino acids [43]. To our delight, the Heck coupling of syn-4a and 16 could be efficiently achieved employing Jeffery´s conditions [44]. When the coupling was performed using palladium(II) acetate, solid NaHCO3 and tetra-n-butylammonium bromide (TBAB) at 130 °C [37,38,42] the desired β-1,2-oxazinyl-substituted dehydroamino acid derivative syn-17 was obtained in 69% yield. We did not prove the Z-configuration of the external C=C bond, since it is well documented that in related cross-coupling reactions of 16 exclusively the Z-isomers are formed [40].
![[1860-5397-12-289-i5]](/bjoc/content/inline/1860-5397-12-289-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Heck reactions of 1,2-oxazine syn-4a leading to 5-alkenyl-substituted 1,2-oxazines syn-13, syn-14 and to dehydroamino acid syn-17.
Scheme 5: Heck reactions of 1,2-oxazine syn-4a leading to 5-alkenyl-substituted 1,2-oxazines syn-13, syn-14 a...
The 5-iodo-1,2-oxazines syn-4a,b reacted smoothly under standard conditions of Suzuki–Miyaura coupling reactions [45] with vinylboronic acid 18 and 3-methoxyphenylboronic acid 20 to furnish the expected 5-styryl-substituted derivative syn-19 and the 5-aryl-substituted derivatives syn-21 and syn-22, respectively, in moderate to good yield (Scheme 6). Notably, the D-arabinose-derived 5-iodo-1,2-oxazine anti-4d was also efficiently converted into the 5-phenyl-substituted compound anti-24 employing phenylboronic acid 23 under the same reaction conditions in excellent yield.
![[1860-5397-12-289-i6]](/bjoc/content/inline/1860-5397-12-289-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Suzuki–Miyaura reactions of 1,2-oxazines syn-4a, syn-4b and anti-4d leading to 5-styryl-substituted 1,2-oxazine syn-19 and 5-aryl-substituted 1,2-oxazines syn-21, syn-22 and anti-24.
Scheme 6: Suzuki–Miyaura reactions of 1,2-oxazines syn-4a, syn-4b and anti-4d leading to 5-styryl-substituted...
In the last example of palladium-catalyzed reactions, we set out to prove a 5-iodo-1,2-oxazine 4 as substrate in a cyanation reaction that would lead to a 5-cyano-substituted derivative. The installation of a cyano group at the 5-position would lead to a push–pull system that could open new synthetic opportunities including the attack of nucleophiles at C-4. Metal cyanides such as KCN represent synthetically valuable C-1 building blocks that could efficiently be coupled by palladium catalysis to alkenyl triflates [46,47] or alkenyl halides [48,49] forming α,β-unsaturated nitriles. As shown in Scheme 7, we adopted a protocol described by Yamamura and Murahashi [48] and found that the palladium-catalyzed coupling of anti-4d with potassium cyanide in the presence of 18-crown-6 at 80 °C in toluene afforded the desired 5-cyano-substituted product anti-25 in moderate yield (not optimized).
![[1860-5397-12-289-i7]](/bjoc/content/inline/1860-5397-12-289-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Cross-coupling reaction of 1,2-oxazine anti-4d leading to 5-cyano-substituted 1,2-oxazine anti-25.
Scheme 7: Cross-coupling reaction of 1,2-oxazine anti-4d leading to 5-cyano-substituted 1,2-oxazine anti-25.
The synthetic usefulness of the obtained 5-alkynyl-, 5-alkenyl- and 5-aryl-substituted 1,2-oxazine derivatives depends on their ability to undergo subsequent transformations. Due to their high degree of functionalization, the C-5-substituted 1,2-oxazine derivatives prepared by the coupling reactions described above should be versatile precursors for a variety of subsequent reactions, e.g., acid-catalyzed hydrolysis, hydrogenations or cyclization reactions. For example, we envisioned that 3,6-dihydro-2H-1,2-oxazines bearing the newly installed alkynyl group at C-5 are ideal candidates for efficient subsequent transformations. A very popular and widely applied reaction of terminal alkynes is the copper-catalyzed azide–alkyne cycloaddition, also termed as click reaction, efficiently leading to 1,4-disubstituted 1,2,3-triazoles [50]. After the desilylation of syn-5 using potassium fluoride in methanol (Scheme 8) the resulting mono-substituted alkyne was subjected to an established protocol using benzyl azide, copper(I) iodide, triethylamine and TBTA [51] (for a recent review see [52]). The cycloaddition proceeded well and afforded the expected 5-(1,2,3-triazolyl)-substituted 1,2-oxazine syn-26 in moderate yield (54%).
![[1860-5397-12-289-i8]](/bjoc/content/inline/1860-5397-12-289-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Desilylation of 1,2-oxazine syn-5 and subsequent click reaction with benzyl azide leading to 5-(1,2,3-triazolyl)-substituted 1,2-oxazine syn-26.
Scheme 8: Desilylation of 1,2-oxazine syn-5 and subsequent click reaction with benzyl azide leading to 5-(1,2...
Hydrogenolysis belongs to the well-established transformations of 1,2-oxazines, often successfully leading to valuable compounds including 1,4-amino alcohols or pyrrolidine derivatives. We therefore briefly examined the reaction of 5-aryl-substituted 1,2-oxazines syn-21 and anti-24 under previously established conditions [53]. The hydrogenolysis of 1,2-oxazine derivative syn-21 in methanol using palladium on charcoal as catalyst afforded the expected α-(3-methoxyphenyl)-substituted γ-amino alcohol derivative 27 in 51% yield as 96:4 mixture of two diastereomers (Scheme 9). Subsequent ring closure of γ-amino alcohol 27 by treatment with mesyl chloride in the presence of triethylamine [53] furnished the N-mesylated pyrrolidine derivative 28 in 65% yield with excellent diastereoselectivity (dr 95:5).
![[1860-5397-12-289-i9]](/bjoc/content/inline/1860-5397-12-289-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Hydrogenation of 1,2-oxazine syn-21 leading to γ-amino alcohols 27a,b and subsequent ring closure to pyrrolidine derivatives 28a,b.
Scheme 9: Hydrogenation of 1,2-oxazine syn-21 leading to γ-amino alcohols 27a,b and subsequent ring closure t...
In contrast, when these hydrogenolysis conditions were applied to the D-arabinose-derived 5-phenyl-1,2-oxazine anti-24 merely an inseparable complex mixture of products was obtained. After shortening of the reaction time from one day to one hour, we were able to isolate two products, the N-debenzylated 3,6-dihydro-2H-1,2-oxazine anti-29 in 37% yield and the 3,4,5,6-tetrahydro-2H-1,2-oxazine anti-30 in 36% yield as a 93:7 mixture of diastereomers (Scheme 10). The dependence of product distribution, especially in the latter case, revealed that a specific reaction sequence is operating during the hydrogenolysis processes. As already discussed in previous publications [23,53], the hydrogenolysis of 1,2-oxazines of type 3 with palladium on charcoal very likely starts with a fast N-debenzylation, followed by the reduction of the C-4/C-5 double bond forming the corresponding 3,4,5,6-tetrahydro-2H-1,2-oxazines that after cleavage of the N–O bond provide the corresponding amino alcohols. In the second reaction step, the hydrogen attacks the C=C bond mainly from the less hindered side (here trans to the fairly bulky 3-dioxolanyl group) leading to the preferred configuration of anti-30a as depicted in Scheme 10.
![[1860-5397-12-289-i10]](/bjoc/content/inline/1860-5397-12-289-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Hydrogenation of 1,2-oxazine anti-24 to products anti-29 and anti-30.
Scheme 10: Hydrogenation of 1,2-oxazine anti-24 to products anti-29 and anti-30.
The three successful transformations demonstrate the potential of 1,2-oxazines with substituents at C-5 for further elaborations, they also show that careful optimizations are required in each individual case, in particular for the hydrogenolysis reactions.
Conclusion
We have demonstrated that the enol ether unit of 3,6-dihydro-2H-1,2-oxazines 3 can efficiently be converted into the corresponding 5-iodo-substituted compounds 4 under mild reaction conditions using molecular iodine in the presence of pyridine as base. The obtained alkenyl iodides 4 are ideal candidates for further transformations. As shown in this report, the subsequent cross-coupling reactions at C-5 position considerably broaden the scope of available 3,6-dihydro-2H-1,2-oxazines that are highly functionalized and have high potential for further synthetic elaborations, in particular for the preparation of enantiopure acyclic and cyclic amino alcohols.
Supporting Information
| Supporting Information File 1: General information, all experimental procedures and analytical data. | ||
| Format: PDF | Size: 532.5 KB | Download |
| Supporting Information File 2: Copies of 1H and 13C NMR spectra of compounds 4–9, 11, 14, 19, 21, 24–27, 29 and 30. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U. Chem. Soc. Rev. 2014, 43, 2888–2903. doi:10.1039/c3cs60429b
Return to citation in text: [1] -
Schade, W.; Reissig, H.-U. Synlett 1999, 632–634. doi:10.1055/s-1999-2662
Return to citation in text: [1] -
Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627
Return to citation in text: [1] -
Al-Harrasi, A.; Fischer, S.; Zimmer, R.; Reissig, H.-U. Synthesis 2010, 304–314. doi:10.1055/s-0029-1217126
Return to citation in text: [1] -
Pulz, R.; Al-Harrasi, A.; Reissig, H.-U. Org. Lett. 2002, 4, 2353–2355. doi:10.1021/ol0260573
Return to citation in text: [1] -
Dekaris, V.; Reissig, H.-U. Synlett 2010, 42–46. doi:10.1055/s-0029-1218531
Return to citation in text: [1] -
Dekaris, V.; Pulz, R.; Al-Harrasi, A.; Lentz, D.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 3210–3219. doi:10.1002/ejoc.201100230
Return to citation in text: [1] [2] -
Jasiński, M.; Lentz, D.; Reissig, H.-U. Beilstein J. Org. Chem. 2012, 8, 662–674. doi:10.3762/bjoc.8.74
Return to citation in text: [1] -
Jasiński, M.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 442–454. doi:10.1002/ejoc.201301406
Return to citation in text: [1] [2] -
Jasiński, M.; Utecht, G.; Fruziński, A.; Reissig, H.-U. Synthesis 2016, 48, 893–905. doi:10.1055/s-0035-1560398
Return to citation in text: [1] -
Parmeggiani, C.; Cardona, F.; Giusti, L.; Reissig, H.-U.; Goti, A. Chem. – Eur. J. 2013, 19, 10595–10604. doi:10.1002/chem.201301320
Return to citation in text: [1] -
Jasiński, M.; Watanabe, T.; Reissig, H.-U. Eur. J. Org. Chem. 2013, 605–610. doi:10.1002/ejoc.201201210
Return to citation in text: [1] -
Al-Harrasi, A.; Bouché, L.; Zimmer, R.; Reissig, H.-U. Synthesis 2011, 109–118. doi:10.1055/s-0030-1258326
Return to citation in text: [1] -
Bouché, L.; Reissig, H.-U. Pure Appl. Chem. 2012, 84, 23–36. doi:10.1351/PAC-CON-11-09-20
Return to citation in text: [1] -
Bouché, L.; Kandziora, M.; Reissig, H.-U. Beilstein J. Org. Chem. 2014, 10, 213–223. doi:10.3762/bjoc.10.17
Return to citation in text: [1] -
Bouché, L.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 3697–3703. doi:10.1002/ejoc.201402191
Return to citation in text: [1] -
Kandziora, M.; Reissig, H.-U. Beilstein J. Org. Chem. 2014, 10, 1749–1758. doi:10.3762/bjoc.10.182
Return to citation in text: [1] -
Kandziora, M.; Reissig, H.-U. Eur. J. Org. Chem. 2015, 370–377. doi:10.1002/ejoc.201403186
Return to citation in text: [1] -
Kandziora, M.; Mucha, E.; Zucker, S. P.; Reissig, H.-U. Synlett 2015, 26, 367–374. doi:10.1055/s-0034-1379503
Return to citation in text: [1] -
Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a
Return to citation in text: [1] -
Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/c1ob05583f
Return to citation in text: [1] -
Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Synthesis 2013, 45, 2752–2762. doi:10.1055/s-0033-1339509
Return to citation in text: [1] [2] [3] -
Zimmer, R.; Schmidt, E.; Andrä, M.; Duhs, M.-A.; Linder, I.; Reissig, H.-U. Beilstein J. Org. Chem. 2009, 5, No. 44. doi:10.3762/bjoc.5.44
Return to citation in text: [1] -
Linder, I.; Gerhard, M.; Schefzig, L.; Andrä, M.; Bentz, C.; Reissig, H.-U.; Zimmer, R. Eur. J. Org. Chem. 2011, 6070–6077. doi:10.1002/ejoc.201100765
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U.; Homann, K. J. Prakt. Chem. 1995, 337, 521–528. doi:10.1002/prac.199533701112
Return to citation in text: [1] -
Nubbemeyer, U. Sci. Synth. 2008, 32, 169–200.
Return to citation in text: [1] -
Sha, C.-K.; Shen, C.-Y.; Jean, T.-S.; Chiu, R.-T.; Tseng, W.-H. Tetrahedron Lett. 1993, 34, 7641–7644. doi:10.1016/S0040-4039(00)60422-7
Return to citation in text: [1] -
Paintner, F. F.; Allmendinger, L.; Bauschke, C. Synlett 2005, 2735–2738. doi:10.1055/s-2005-918915
Return to citation in text: [1] -
Hanessian, S.; Focken, T.; Oza, R. Org. Lett. 2010, 12, 3172–3175. doi:10.1021/ol101103q
Return to citation in text: [1] -
Gwiazda, M.; Reissig, H.-U. Synthesis 2008, 990–994. doi:10.1055/s-2007-990933
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398
Return to citation in text: [1] -
Voigt, K.; von Zezschwitz, P.; Rosauer, K.; Lansky, A.; Adams, A.; Reiser, O.; de Meijere, A. Eur. J. Org. Chem. 1998, 1521–1534. doi:10.1002/(SICI)1099-0690(199808)1998:8<1521::AID-EJOC1521>3.0.CO;2-E
Return to citation in text: [1] -
Lyapkalo, I. M.; Webel, M.; Reissig, H.-U. Eur. J. Org. Chem. 2001, 4189–4194. doi:10.1002/1099-0690(200111)2001:22<4189::AID-EJOC4189>3.0.CO;2-I
Return to citation in text: [1] -
Merlic, C. A.; Semmelhack, M. F. J. Organomet. Chem. 1990, 391, C23–C27. doi:10.1016/0022-328X(90)80183-Z
Return to citation in text: [1] -
Crestey, F.; Collot, V.; Stiebing, S.; Rault, S. Synthesis 2006, 3506–3514. doi:10.1055/s-2006-950242
Return to citation in text: [1] -
Carlström, A.-S.; Frejd, T. Synthesis 1989, 414–418. doi:10.1055/s-1989-27270
Return to citation in text: [1] [2] -
Carlström, A.-S.; Frejd, T. Acta Chem. Scand. 1992, 46, 163–171. doi:10.3891/acta.chem.scand.46-0163
Return to citation in text: [1] [2] -
Arcadi, A.; Cacchi, S.; Marinelli, F.; Morera, E.; Ortar, G. Tetrahedron 1990, 46, 7151–7164. doi:10.1016/S0040-4020(01)87897-2
Return to citation in text: [1] -
Yokoyama, Y.; Takahashi, M.; Takashima, M.; Kohno, Y.; Kobayashi, H.; Kataoka, K.; Shidori, K.; Murakami, Y. Chem. Pharm. Bull. 1994, 42, 832–838. doi:10.1248/cpb.42.832
Return to citation in text: [1] [2] -
de Azambuja, F.; Duarte Correia, C. R. Tetrahedron Lett. 2011, 52, 42–45. doi:10.1016/j.tetlet.2010.10.132
Return to citation in text: [1] -
Bozell, J. J.; Vogt, C. E.; Gozum, J. J. Org. Chem. 1991, 56, 2584–2587. doi:10.1021/jo00007a060
Return to citation in text: [1] [2] -
Taszarek, M.; Reissig, H.-U. ChemistrySelect 2016, 1, 5712–5714. doi:10.1002/slct.201601241
Return to citation in text: [1] -
Ajana, W.; Feliu, L.; Alvarez, M.; Joule, J. A. Tetrahedron 1998, 54, 4405–4412. doi:10.1016/S0040-4020(98)00154-9
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Quirante, J.; Escolano, C.; Massot, M.; Bonjoch, J. Tetrahedron 1997, 53, 1391–1402. doi:10.1016/S0040-4020(96)01051-4
Return to citation in text: [1] -
Hübner, H.; Haubmann, C.; Utz, W.; Gmeiner, P. J. Med. Chem. 2000, 43, 756–762. doi:10.1021/jm991098z
Return to citation in text: [1] -
Yamamura, K.; Murahashi, S.-I. Tetrahedron Lett. 1977, 18, 4429–4430. doi:10.1016/S0040-4039(01)83527-9
Return to citation in text: [1] [2] -
Piers, E.; Fleming, F. F. Can. J. Chem. 1993, 71, 1867–1872. doi:10.1139/v93-234
Return to citation in text: [1] -
Singh, M. S.; Chowdhury, S.; Koley, S. Tetrahedron 2016, 72, 5257–5283. doi:10.1016/j.tet.2016.07.044
Return to citation in text: [1] -
Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e
Return to citation in text: [1] -
Haldón, E.; Nicasio, M. C.; Pérez, P. J. Org. Biomol. Chem. 2015, 13, 9528–9550. doi:10.1039/C5OB01457C
Return to citation in text: [1] -
Pulz, R.; Watanabe, T.; Schade, W.; Reissig, H.-U. Synlett 2000, 983–986. doi:10.1055/s-2000-6654
Return to citation in text: [1] [2] [3]
| 48. | Yamamura, K.; Murahashi, S.-I. Tetrahedron Lett. 1977, 18, 4429–4430. doi:10.1016/S0040-4039(01)83527-9 |
| 49. | Piers, E.; Fleming, F. F. Can. J. Chem. 1993, 71, 1867–1872. doi:10.1139/v93-234 |
| 48. | Yamamura, K.; Murahashi, S.-I. Tetrahedron Lett. 1977, 18, 4429–4430. doi:10.1016/S0040-4039(01)83527-9 |
| 50. | Singh, M. S.; Chowdhury, S.; Koley, S. Tetrahedron 2016, 72, 5257–5283. doi:10.1016/j.tet.2016.07.044 |
| 1. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h |
| 2. | Zimmer, R.; Reissig, H.-U. Chem. Soc. Rev. 2014, 43, 2888–2903. doi:10.1039/c3cs60429b |
| 8. | Dekaris, V.; Pulz, R.; Al-Harrasi, A.; Lentz, D.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 3210–3219. doi:10.1002/ejoc.201100230 |
| 10. | Jasiński, M.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 442–454. doi:10.1002/ejoc.201301406 |
| 12. | Parmeggiani, C.; Cardona, F.; Giusti, L.; Reissig, H.-U.; Goti, A. Chem. – Eur. J. 2013, 19, 10595–10604. doi:10.1002/chem.201301320 |
| 28. | Sha, C.-K.; Shen, C.-Y.; Jean, T.-S.; Chiu, R.-T.; Tseng, W.-H. Tetrahedron Lett. 1993, 34, 7641–7644. doi:10.1016/S0040-4039(00)60422-7 |
| 29. | Paintner, F. F.; Allmendinger, L.; Bauschke, C. Synlett 2005, 2735–2738. doi:10.1055/s-2005-918915 |
| 30. | Hanessian, S.; Focken, T.; Oza, R. Org. Lett. 2010, 12, 3172–3175. doi:10.1021/ol101103q |
| 6. | Pulz, R.; Al-Harrasi, A.; Reissig, H.-U. Org. Lett. 2002, 4, 2353–2355. doi:10.1021/ol0260573 |
| 7. | Dekaris, V.; Reissig, H.-U. Synlett 2010, 42–46. doi:10.1055/s-0029-1218531 |
| 8. | Dekaris, V.; Pulz, R.; Al-Harrasi, A.; Lentz, D.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 3210–3219. doi:10.1002/ejoc.201100230 |
| 9. | Jasiński, M.; Lentz, D.; Reissig, H.-U. Beilstein J. Org. Chem. 2012, 8, 662–674. doi:10.3762/bjoc.8.74 |
| 10. | Jasiński, M.; Moreno-Clavijo, E.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 442–454. doi:10.1002/ejoc.201301406 |
| 11. | Jasiński, M.; Utecht, G.; Fruziński, A.; Reissig, H.-U. Synthesis 2016, 48, 893–905. doi:10.1055/s-0035-1560398 |
| 31. | Gwiazda, M.; Reissig, H.-U. Synthesis 2008, 990–994. doi:10.1055/s-2007-990933 |
| 5. | Al-Harrasi, A.; Fischer, S.; Zimmer, R.; Reissig, H.-U. Synthesis 2010, 304–314. doi:10.1055/s-0029-1217126 |
| 26. | Zimmer, R.; Reissig, H.-U.; Homann, K. J. Prakt. Chem. 1995, 337, 521–528. doi:10.1002/prac.199533701112 |
| 23. | Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Synthesis 2013, 45, 2752–2762. doi:10.1055/s-0033-1339509 |
| 53. | Pulz, R.; Watanabe, T.; Schade, W.; Reissig, H.-U. Synlett 2000, 983–986. doi:10.1055/s-2000-6654 |
| 3. | Schade, W.; Reissig, H.-U. Synlett 1999, 632–634. doi:10.1055/s-1999-2662 |
| 4. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fišera, L.; Hlobilová, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 21. | Dernedde, J.; Enders, S.; Reissig, H.-U.; Roskamp, M.; Schlecht, S.; Yekta, S. Chem. Commun. 2009, 932–934. doi:10.1039/b818263a |
| 22. | Roskamp, M.; Enders, S.; Pfrengle, F.; Yekta, S.; Dekaris, V.; Dernedde, J.; Reissig, H.-U.; Schlecht, S. Org. Biomol. Chem. 2011, 9, 7448–7456. doi:10.1039/c1ob05583f |
| 24. | Zimmer, R.; Schmidt, E.; Andrä, M.; Duhs, M.-A.; Linder, I.; Reissig, H.-U. Beilstein J. Org. Chem. 2009, 5, No. 44. doi:10.3762/bjoc.5.44 |
| 53. | Pulz, R.; Watanabe, T.; Schade, W.; Reissig, H.-U. Synlett 2000, 983–986. doi:10.1055/s-2000-6654 |
| 15. | Bouché, L.; Reissig, H.-U. Pure Appl. Chem. 2012, 84, 23–36. doi:10.1351/PAC-CON-11-09-20 |
| 16. | Bouché, L.; Kandziora, M.; Reissig, H.-U. Beilstein J. Org. Chem. 2014, 10, 213–223. doi:10.3762/bjoc.10.17 |
| 17. | Bouché, L.; Reissig, H.-U. Eur. J. Org. Chem. 2014, 3697–3703. doi:10.1002/ejoc.201402191 |
| 18. | Kandziora, M.; Reissig, H.-U. Beilstein J. Org. Chem. 2014, 10, 1749–1758. doi:10.3762/bjoc.10.182 |
| 19. | Kandziora, M.; Reissig, H.-U. Eur. J. Org. Chem. 2015, 370–377. doi:10.1002/ejoc.201403186 |
| 20. | Kandziora, M.; Mucha, E.; Zucker, S. P.; Reissig, H.-U. Synlett 2015, 26, 367–374. doi:10.1055/s-0034-1379503 |
| 25. | Linder, I.; Gerhard, M.; Schefzig, L.; Andrä, M.; Bentz, C.; Reissig, H.-U.; Zimmer, R. Eur. J. Org. Chem. 2011, 6070–6077. doi:10.1002/ejoc.201100765 |
| 53. | Pulz, R.; Watanabe, T.; Schade, W.; Reissig, H.-U. Synlett 2000, 983–986. doi:10.1055/s-2000-6654 |
| 14. | Al-Harrasi, A.; Bouché, L.; Zimmer, R.; Reissig, H.-U. Synthesis 2011, 109–118. doi:10.1055/s-0030-1258326 |
| 51. | Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e |
| 13. | Jasiński, M.; Watanabe, T.; Reissig, H.-U. Eur. J. Org. Chem. 2013, 605–610. doi:10.1002/ejoc.201201210 |
| 23. | Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Synthesis 2013, 45, 2752–2762. doi:10.1055/s-0033-1339509 |
| 52. | Haldón, E.; Nicasio, M. C.; Pérez, P. J. Org. Biomol. Chem. 2015, 13, 9528–9550. doi:10.1039/C5OB01457C |
| 35. | Merlic, C. A.; Semmelhack, M. F. J. Organomet. Chem. 1990, 391, C23–C27. doi:10.1016/0022-328X(90)80183-Z |
| 23. | Moinizadeh, N.; Klemme, R.; Kansy, M.; Zimmer, R.; Reissig, H.-U. Synthesis 2013, 45, 2752–2762. doi:10.1055/s-0033-1339509 |
| 32. | Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398 |
| 33. | Voigt, K.; von Zezschwitz, P.; Rosauer, K.; Lansky, A.; Adams, A.; Reiser, O.; de Meijere, A. Eur. J. Org. Chem. 1998, 1521–1534. doi:10.1002/(SICI)1099-0690(199808)1998:8<1521::AID-EJOC1521>3.0.CO;2-E |
| 34. | Lyapkalo, I. M.; Webel, M.; Reissig, H.-U. Eur. J. Org. Chem. 2001, 4189–4194. doi:10.1002/1099-0690(200111)2001:22<4189::AID-EJOC4189>3.0.CO;2-I |
| 45. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 46. | Quirante, J.; Escolano, C.; Massot, M.; Bonjoch, J. Tetrahedron 1997, 53, 1391–1402. doi:10.1016/S0040-4020(96)01051-4 |
| 47. | Hübner, H.; Haubmann, C.; Utz, W.; Gmeiner, P. J. Med. Chem. 2000, 43, 756–762. doi:10.1021/jm991098z |
| 37. | Carlström, A.-S.; Frejd, T. Synthesis 1989, 414–418. doi:10.1055/s-1989-27270 |
| 38. | Carlström, A.-S.; Frejd, T. Acta Chem. Scand. 1992, 46, 163–171. doi:10.3891/acta.chem.scand.46-0163 |
| 42. | Bozell, J. J.; Vogt, C. E.; Gozum, J. J. Org. Chem. 1991, 56, 2584–2587. doi:10.1021/jo00007a060 |
| 40. | Yokoyama, Y.; Takahashi, M.; Takashima, M.; Kohno, Y.; Kobayashi, H.; Kataoka, K.; Shidori, K.; Murakami, Y. Chem. Pharm. Bull. 1994, 42, 832–838. doi:10.1248/cpb.42.832 |
| 43. | Taszarek, M.; Reissig, H.-U. ChemistrySelect 2016, 1, 5712–5714. doi:10.1002/slct.201601241 |
| 44. | Ajana, W.; Feliu, L.; Alvarez, M.; Joule, J. A. Tetrahedron 1998, 54, 4405–4412. doi:10.1016/S0040-4020(98)00154-9 |
| 36. | Crestey, F.; Collot, V.; Stiebing, S.; Rault, S. Synthesis 2006, 3506–3514. doi:10.1055/s-2006-950242 |
| 37. | Carlström, A.-S.; Frejd, T. Synthesis 1989, 414–418. doi:10.1055/s-1989-27270 |
| 38. | Carlström, A.-S.; Frejd, T. Acta Chem. Scand. 1992, 46, 163–171. doi:10.3891/acta.chem.scand.46-0163 |
| 39. | Arcadi, A.; Cacchi, S.; Marinelli, F.; Morera, E.; Ortar, G. Tetrahedron 1990, 46, 7151–7164. doi:10.1016/S0040-4020(01)87897-2 |
| 40. | Yokoyama, Y.; Takahashi, M.; Takashima, M.; Kohno, Y.; Kobayashi, H.; Kataoka, K.; Shidori, K.; Murakami, Y. Chem. Pharm. Bull. 1994, 42, 832–838. doi:10.1248/cpb.42.832 |
| 41. | de Azambuja, F.; Duarte Correia, C. R. Tetrahedron Lett. 2011, 52, 42–45. doi:10.1016/j.tetlet.2010.10.132 |
| 42. | Bozell, J. J.; Vogt, C. E.; Gozum, J. J. Org. Chem. 1991, 56, 2584–2587. doi:10.1021/jo00007a060 |
© 2016 Medvecký et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)