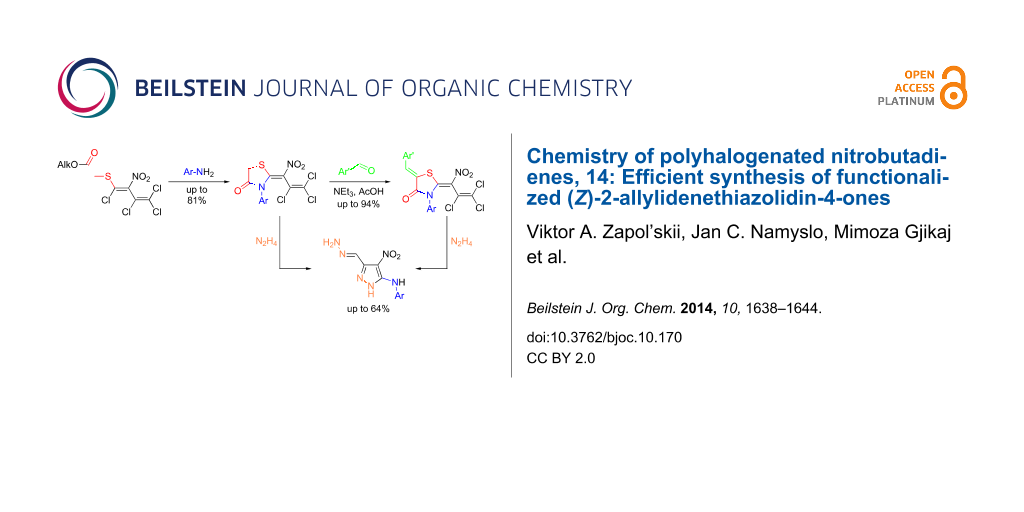
Abstract
The reaction of mercaptoacetic acid esters with pentachloro-2-nitro-1,3-butadiene (1) provides an appropriate precursor for the synthesis of special thiazolidin-4-ones. Applying different anilines as the second constituent for the requisite cyclization step, a series of (Z)-2-allylidenethiazolidin-4-ones was obtained in yields up to 81%. Some subsequent reactions have been examined too, such as the formation of perfunctionalized 1H-pyrazoles upon treatment with hydrazine. Thiazolidinones are as well known for their physiological activities as for their application in optoelectronics.

Graphical Abstract
Introduction
Preliminary studies in the field of polyhalogenated nitrobutadienes have already shown the enormous potential of pentachloro-2-nitro-1,3-butadiene (1) as a precursor for the “click synthesis” of highly functionalized (hetero)cyclic as well as acyclic compounds [1-15]. The corresponding syntheses that we have developed up to now [2-13], always start with the attack of an appropriate nucleophile at the activated terminal carbon atom of the nitrodichlorovinyl group within 1 to undergo a vinylic substitution. Thus, in case of, e.g., sulfur nucleophiles, the corresponding thioperchlorobutadiene derivatives are easily accessible [2,5,16,17]. In this paper, we describe the formation of uniquely substituted thiazolidin-4-ones, a class of compounds which has proven to exhibit distinctive bioactivity, e.g., antifungal, antibacterial, antitubercular, and anticonvulsant properties [18-28].
Additionally, more recent studies often focus on their anticancer [29-31] and anti-HIV potential [30,32]. Several 4-thiazolidinone derivatives such as ralitoline (anticonvulsant), etozoline (antihypertensive), pioglitazone (hypoglycemic) and thiazolidomycin (activity against streptomyces species) are already in the market [33]. Beyond that, among industrial applications thiazolidinones serve as electron donors, e.g., as ligands in coordination compounds or as vulcanizing agents [31]. Furthermore, it is well known that these heterocycles play a major role in corresponding organic electronics, e.g., they lead to high quantum efficiency as donor compound in solar cells [34]. Very recently, thiazolidinones became a target in the field of photonics and optoelectronics, potentially applicable for reversible optical data storage, photo switching of optical elements, photochromic polymers, and similar applications [35].
The background of thiazolidinone chemistry including classical synthetic strategies has been reviewed comprehensively [18-28,36-39]. Even though among possible synthetic pathways the use of mercaptoacetic acid and its derivatives is well-established [40-44], our approach, starting from a mercaptoacetic acid derivative of perchloro-2-nitro-1,3-butadiene and an aniline, is unprecedented up to now. In this context, it is very interesting that the reaction of a 2-nitro-1-thioperchlorobuta-1,3-diene with N-nucleophiles usually yields a 1-amino-2-nitro-1-thioperchlorobuta-1,3-diene [16,45]. However, our novel ring closing reaction incorporates an arylamine as source for the nitrogen of the thiazolidin-4-one heterocycle.
Results and Discussion
The reaction of pentachloro-2-nitro-1,3-butadiene (1) with 1.1 equivalents of ethyl 2-mercaptoacetate (2) at room temperature without a solvent for 10 days furnished ethyl 2-((1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl)thio)acetate (3) as a single isomer in 80% yield (for further details see Supporting Information File 1). In contrast, the conversion of 1 (neat) with 2.1 equivalents of methyl 2-mercaptoacetate (4) results in a mixture of methyl 2-((1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl)thio)acetate (5) and 1,1-bis(methoxycarbonylmethylthio)-2-nitro-3,4,4-trichlorobuta-1,3-diene (6) after 10 days at room temperature with 67% and 29% yields, respectively. These nitrodienes 5 and 6 were separated by flash column chromatography and then characterized. Interestingly, the dithio compound 6 was also accessible by treatment of the aforementioned monothio derivative 5 with one equivalent of mercaptoacetate 4. Following this pathway, the disubstituted diene 6 was obtained in 80% yield (Scheme 1).
![[1860-5397-10-170-i1]](/bjoc/content/inline/1860-5397-10-170-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: SNVin reactions of pentachloro-2-nitro-1,3-butadiene (1).
Scheme 1: SNVin reactions of pentachloro-2-nitro-1,3-butadiene (1).
The subsequent reaction of the mercaptoacetates 3 and 5 with 2.1 equivalents of various arylamines is the key step of our thiazolidin-4-one synthesis, that led to the corresponding (Z)-3-aryl-2-(2,3,3-trichloro-1-nitroallylidene)thiazolidin-4-ones 7–18 with up to 81% yield. As an example for the sequential reactivity of the persubstituted allylidene side chain, the thiazolidin-4-one 15 was reacted with p-tolylthiolate. Thereby, the C(2)–Cl position of the allylidene group was selectively substituted by the reactive thiolate nucleophile, again in a SNVin-type process, to give (Z)-3-(4-tolyl)-2-(3,3-dichloro-1-nitro-2-[4-tolylthio]allylidene)thiazolidin-4-one (19) in 70% yield (Scheme 2).
![[1860-5397-10-170-i2]](/bjoc/content/inline/1860-5397-10-170-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of thiazolidin-4-ones 7–19.
Scheme 2: Formation of thiazolidin-4-ones 7–19.
According to proton NMR, the ring-closing step with the parent aniline or with one of its para-substituted derivatives in each case led to a single N-arylthiazolidin-4-one isomer 7–15. In the case of the ortho- or meta-substituted anilines 16–18 two atropisomers were generated. This phenomenon is due to a less hindered rotation of the C–N bond of the former anilines (Figure 1).
![[1860-5397-10-170-1]](/bjoc/content/figures/1860-5397-10-170-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Hindered rotation in the case of ortho- or meta-substituted aniline precursors.
Figure 1: Hindered rotation in the case of ortho- or meta-substituted aniline precursors.
The steric hindrance is additionally forcing by the s-cis conformation of the nitrobutadiene moiety (cf. Figure 2). A DFT calculation of the energy barrier for rotation of the aromatic substituent resulted in extraordinary high values of 169 and 191 kJ/mol for the ortho-methyl compound 18 and the larger, but less rigid ortho-methoxy derivative 16, respectively. As expected, the meta-anisyl derivative 17 showed a lower barrier of 73 kJ/mol (see Supporting Information File 1). For comparison, the well-known atropisomer 1,1’-binaphthalene-2,2’-diol (BINOL) shows a rotational barrier of 158 kJ/mol with calculated data in good accordance with the experimental value [46].
In addition to substantial NMR, IR, and mass spectral characterization of the thiazolidinones 7–18, as an example the 4-iodophenyl substituted thiazolidinone 11 was subjected to X-ray analysis (see Supporting Information File 1). This confirmed the Z-configuration of the inner double bond which bears the sulfur atom of the hetero ring as well as the nitro group. The structural plot also gives an idea of the abovementioned bulkiness, even in the case of the depicted para-substitution (Figure 2).
![[1860-5397-10-170-2]](/bjoc/content/figures/1860-5397-10-170-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray analysis of thiazolidin-4-one 11.
Figure 2: X-ray analysis of thiazolidin-4-one 11.
The formation of the thiazolidin-4-ones 7–18 is assumed to consist of three individual steps. In a SNVin type reaction with the aniline derivative intermediate A (Scheme 3) is formed. Subsequently, the thus introduced amino group attacks the carbonyl carbon of the mercaptoacetate moiety. This ring closure affords the temporary imide hemiacetal B which is then stabilized upon elimination of the corresponding alcohol to give the desired thiazolidin-4-ones 7–18 (Scheme 3).
![[1860-5397-10-170-i3]](/bjoc/content/inline/1860-5397-10-170-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Assumed mechanism for the formation of thiazolidin-4-ones 7–18.
Scheme 3: Assumed mechanism for the formation of thiazolidin-4-ones 7–18.
Furthermore, in addition to the anilines described above (Scheme 2), we applied 1-naphthylamine as a bulkier aromatic representative and morpholine as a secondary, more basic aliphatic amine. Conversion of 3 and 5 with morpholine and 1-naphthylamine afforded the open chain products 20 and 21 in 93% and 80% yield, respectively (Scheme 4), without a subsequent cyclization under one-pot conditions. Unexpectedly, even under forcing conditions cyclization of isolated 21 did not take place. Compound 21 was exclusively obtained as E-isomer. This was indicated by a proton NMR signal shifted downfield to about 12 ppm due to a strong hydrogen bond between the amino hydrogen and the nitro group. In contrast, lacking the opportunity to build such a hydrogen bridge, the regiochemistry of compound 20 (also formed as one single isomer) remained unclear.
![[1860-5397-10-170-i4]](/bjoc/content/inline/1860-5397-10-170-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Substitution reactions of the precursors 3 and 5 with additional amines.
Scheme 4: Substitution reactions of the precursors 3 and 5 with additional amines.
With the thiazolidinones in hand, in the case of 7, 8, 10, and 12 we carried out a Knoevenagel-type condensation with benzaldehyde and 3,4-dichlorobenzaldehyde in acetic acid at 118 °C in the presence of triethylamine. Thus, the 5-arylmethylidenethiazolidin-4-ones 22–26 were obtained in 68–94% yield. The presence of only one signal for the benzylidene proton at 7.83–7.98 ppm in the 1H NMR spectra of 22–26 suggested the formation of a single isomer, which was assigned to the Z-configuration according to the literature for similar compounds [47,48]. Interestingly, close analogues of these structures, i.e., the 5-arylmethylidene rhodanines, possess photosynthesis-inhibiting and antialgal properties [49], show anticancer activity [50,51], and are inhibitors of bacterial enzyme synthetase MurD with E. coli [52].
Additional treatment of thiazolidin-4-one 8 with a fivefold excess of hydrazine hydrate resulted in the formation of a 1H-pyrazole: A total of two hydrazine molecules were incorporated into the final product, whereas two additional equivalents were consumed capturing hydrochloric acid. In this way, the 1H-pyrazole 27 with an exceptional substitution pattern was obtained in 64% yield. The same reaction was applied to 5-arylmethylidene-thiazolidin-4-one 25 which furnished the corresponding 1H-pyrazole 28 in 52% yield (Scheme 5).
![[1860-5397-10-170-i5]](/bjoc/content/inline/1860-5397-10-170-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 5-arylmethylidenethiazolidin-4-ones 22–26 and 1H-pyrazoles 27, 28.
Scheme 5: Synthesis of 5-arylmethylidenethiazolidin-4-ones 22–26 and 1H-pyrazoles 27, 28.
A plausible multistep mechanism of this conversion is shown in Scheme 6: Initially, a first molecule of the strong nucleophile hydrazine is assumed to substitute the single C–Cl group within the trichlorovinyl subunit of 8 to give the intermediate I (SNVin reaction). Thereby, a second equivalent of hydrazine captures hydrochloric acid. The amino group of the hydrazone then attacks the electrophilic C-2 position of the thiazolidinone. This way a threefold substituted 1H-pyrazole is formed (intermediate II) under ring opening of the thiazolidinone. In the course of the reaction, probably at this stage, the N-aryl amide is hydrazinolysed to give the intermediate III, which is converted into the monohydrazino compound IV upon consumption of two further equivalents of hydrazine. Finally, 27 is obtained upon elimination of hydrochloric acid, caught by a fifth equivalent of hydrazine (Scheme 6). An analogous mechanism is also plausible for the formation of pyrazole 28 from thiazolidinone 25. However, instead of the elimination of 2-mercaptoacetic acid from intermediate II, in the latter case 3-(3,4-dichlorophenyl)-2-mercaptoacrylic acid is released.
![[1860-5397-10-170-i6]](/bjoc/content/inline/1860-5397-10-170-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Assumed mechanism for the formation of 1H-pyrazole 27.
Scheme 6: Assumed mechanism for the formation of 1H-pyrazole 27.
Moreover, nitrodiene 1 was subjected to a vinylic substitution with ethyl 2-mercaptopropanoate to give ethyl 2-[(1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl)thio]propanoate (29) as an inseparable mixture of Z- and E-isomers in a 4:1 ratio with 78% total yield. Treatment of dienes (Z,E)-29 with different anilines in ethanol at 0 °C to rt furnished (Z,E)-3-aryl-5-methylthiazolidinones 30–32. As an example, thiazolidinone 32 was reacted with 4-chlorothiophenol in the presence of sodium ethoxide to give 4-chlorophenylthio compound 33 (60%), again as a mixture of Z- and E-isomers, but this time in a 2:1 ratio (Scheme 7).
![[1860-5397-10-170-i7]](/bjoc/content/inline/1860-5397-10-170-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Formation of ethyl propanoate 29 and subsequent reactions.
Scheme 7: Formation of ethyl propanoate 29 and subsequent reactions.
Conclusion
A two-step synthesis of 2-allylidene-N-arylthiazolidinones 7–18 has been developed, starting from our building block 2-nitroperchlorobutadiene (1), 2-mercaptoacetates 2 and 4, and anilines. Inclusion of o- or m-substituted N-aryl groups leads to the formation of stable thiazolidinone atropisomers, due to hindered rotation between the trichlorovinyl and the arylamino groups. X-ray analysis proved the Z-configuration of the nitrovinylidene group of 11. Subsequent reactions of the thiazolidinones, such as the SNVin thiolation in 2-position of the allylidene backbone, Knoevenagel condensation of the heterocyclic rings with aromatic aldehydes, and an unusual formation of persubstituted pyrazoles demonstrate the chemical versatility of these heterocyclic systems. The newly formed thiazolidinones deserve additional synthetic interest as starting materials, also due to their trichlorovinyl group. Tests on their physiological activities and potential applications as optoelectronic materials are on the way.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 355.5 KB | Download |
References
-
Kaberdin, R. V.; Potkin, V. I.; Zapol’skii, V. A. Russ. Chem. Rev. 1997, 66, 827–842. doi:10.1070/RC1997v066n10ABEH000310
Return to citation in text: [1] -
Zapol’skii, V. A.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Heterocycles 2004, 63, 1281–1298. doi:10.3987/COM-04-10020
Return to citation in text: [1] [2] [3] -
Zapol’skii, V. A.; Nutz, E.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Synthesis 2006, 2927–2933. doi:10.1055/s-2006-950187
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Namyslo, J. C.; Blaschkowski, B.; Kaufmann, D. E. Synlett 2006, 3464–3468. doi:10.1055/s-2006-956490
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. ARKIVOC 2007, No. i, 76–93.
Return to citation in text: [1] [2] [3] -
Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. Synlett 2007, 1507–1512. doi:10.1055/s-2007-982554
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Namyslo, J. C.; Altug, C.; Gjikaj, M.; Kaufmann, D. E. Synthesis 2008, 304–310. doi:10.1055/s-2007-990948
Return to citation in text: [1] [2] -
Nutz, E.; Zapol’skii, V. A.; Kaufmann, D. E. Synthesis 2009, 2719–2724. doi:10.1055/s-0029-1216906
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Fischer, R.; Namyslo, J. C.; Kaufmann, D. E. Bioorg. Med. Chem. 2009, 17, 4206–4215. doi:10.1016/j.bmc.2009.01.001
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. Z. Naturforsch., B 2010, 65b, 843–860.
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Namyslo, J. C.; de Meijere, A.; Kaufmann, D. E. Beilstein J. Org. Chem. 2012, 8, 621–628. doi:10.3762/bjoc.8.69
Return to citation in text: [1] [2] -
Vogt, E.-J.; Zapol’skii, V. A.; Nutz, E.; Kaufmann, D. E. Z. Naturforsch., B 2012, 67, 285–294.
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Vogt, E.-J.; Gjikaj, M.; Kaufmann, D. E. Heterocycles 2012, 86, 1431–1447. doi:10.3987/COM-12-S(N)108
Return to citation in text: [1] [2] -
Ibis, C. Bull. Soc. Chim. Belg. 1996, 105, 317–320.
Return to citation in text: [1] -
Ibis, C.; Sayil, M. C.; Deniz, N. G. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o800–o801. doi:10.1107/S1600536806001838
Return to citation in text: [1] -
Ol'dekop, Y. A.; Kaberdin, R. V.; Potkin, V. I. J. Org. Chem. USSR 1980, 16, 469–472.
Return to citation in text: [1] [2] -
Ibiş, C.; Kirbaşlar, F. G.; Aydinli, G. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 365–374. doi:10.1080/104265090508433
Return to citation in text: [1] -
Brown, F. C. Chem. Rev. 1961, 61, 463–521. doi:10.1021/cr60213a002
Return to citation in text: [1] [2] -
Singh, S. P.; Parmar, S. S.; Raman, K.; Stenberg, V. I. Chem. Rev. 1981, 81, 175–203. doi:10.1021/cr00042a003
Return to citation in text: [1] [2] -
Chandrasekhar, B. J. Sulfur Chem. 2012, 33, 439–503. doi:10.1080/17415993.2012.693490
Return to citation in text: [1] [2] -
Tian, Y.; Zhan, P.; Rai, D.; Zhang, J.; De Clercq, J.; Liu, X. Curr. Med. Chem. 2012, 19, 2026–2037. doi:10.2174/092986712800167383
Return to citation in text: [1] [2] -
Jain, A. K.; Vaidya, A.; Ravichandran, V.; Kashaw, S. K.; Agrawal, R. K. Bioorg. Med. Chem. 2012, 20, 3378–3395. doi:10.1016/j.bmc.2012.03.069
Return to citation in text: [1] [2] -
Verma, A.; Saraf, S. K. Eur. J. Med. Chem. 2008, 43, 897–905. doi:10.1016/j.ejmech.2007.07.017
Return to citation in text: [1] [2] -
Zhang, Q.; Zhou, H.; Zhai, S.; Yan, B. Curr. Pharm. Des. 2010, 16, 1826–1842. doi:10.2174/138161210791208983
Return to citation in text: [1] [2] -
Mehta, D. P.; Sengar, N. P. S.; Pathak, A. K. Orient. J. Chem. 2008, 24, 441–454.
Return to citation in text: [1] [2] -
Edwards, P. J. Drug Discovery Today 2008, 13, 1107–1108. doi:10.1016/j.drudis.2008.10.004
Return to citation in text: [1] [2] -
Cunico, W.; Gomes, C. R. B.; Vellasco, W. T., Jr. Mini-Rev. Org. Chem. 2008, 5, 336–344. doi:10.2174/157019308786242232
Return to citation in text: [1] [2] -
Newkome, G. R.; Nayak, A. Adv. Heterocycl. Chem. 1980, 25, 83–112. doi:10.1016/S0065-2725(08)60690-X
Return to citation in text: [1] [2] -
Kawakami, M.; Koya, K.; Ukai, T.; Tatsuta, N.; Ikegawa, A.; Ogawa, K.; Shishido, T.; Chen, L. B. J. Med. Chem. 1998, 41, 130–142. doi:10.1021/jm970590k
Return to citation in text: [1] -
Rida, S. M.; Ashour, F. A.; El-Hawash, S. A. M.; ElSemary, M. M.; Badr, M. H.; Shalaby, M. A. Eur. J. Med. Chem. 2005, 40, 949–959. doi:10.1016/j.ejmech.2005.03.023
Return to citation in text: [1] [2] -
Hamama, W. S.; Ismail, M. A.; Shaaban, S.; Zoorob, H. H. J. Heterocycl. Chem. 2008, 45, 939–956. doi:10.1002/jhet.5570450401
Return to citation in text: [1] [2] -
Graciet, J. C.; Faury, P.; Camplo, M.; Charvet, A. S.; Mourier, N.; Trabaud, C.; Niddam, V.; Simon, V.; Kraus, J. L. Nucleosides Nucleotides 1995, 14, 1379–1392. doi:10.1080/15257779508010698
Return to citation in text: [1] -
Tripathi, A. C.; Gupta, S. J.; Fatima, G. N.; Sonar, P. K.; Verma, A.; Saraf, S. K. Eur. J. Med. Chem. 2014, 72, 52–77. doi:10.1016/j.ejmech.2013.11.017
Return to citation in text: [1] -
Takahashi, K.; Nakatani, S.; Matsuda, T.; Nanbu, H.; Komura, T.; Murata, K. Chem. Lett. 1994, 23, 2001–2004.
Return to citation in text: [1] -
Smokal, V.; Derkowska, B.; Czaplicki, R.; Krupka, O.; Kolendo, A.; Sahraoui, B. Opt. Mater. 2009, 31, 554–557. doi:10.1016/j.optmat.2007.10.019
Return to citation in text: [1] -
Singh, V. P.; Upadhyay, G. S.; Singh, H. Asian J. Chem. Rev. 1992, 3, 12–21.
Return to citation in text: [1] -
Abdel-Rahman, R. M. Boll. Chim. Farm. 2001, 140, 401–410.
Return to citation in text: [1] -
Lesyk, R. B.; Zimenkovsky, B. S. Curr. Org. Chem. 2004, 8, 1547–1577. doi:10.2174/1385272043369773
Return to citation in text: [1] -
Dudhe, R.; Imran, M.; Maurya, N. P.; Sharma, P. K. Orient. J. Chem. 2007, 23, 167–176.
Return to citation in text: [1] -
Fahmy, H. T. Y. Boll. Chim. Farm. 2001, 140, 422–427.
Return to citation in text: [1] -
Vovk, M. V.; Dorokhov, V. I. Russ. J. Org. Chem. 1993, 29, 1471–1473.
Return to citation in text: [1] -
Matsubara, Y.; Nakamura, T.; Yoshihara, M.; Maeshima, T. Chem. Pharm. Bull. 1985, 33, 3009–3011.
Return to citation in text: [1] -
Satzinger, G. Justus Liebigs Ann. Chem. 1963, 665, 150–165. doi:10.1002/jlac.19636650118
Return to citation in text: [1] -
Marković, R.; Vitnik, Ž.; Baranac, M.; Juranic, I. J. Chem. Res., Synop. 2002, 485–489. doi:10.3184/030823402103170673
Return to citation in text: [1] -
Ibiş, C.; Gökmen, Z. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 2537–2542. doi:10.1080/10426500490485615
Return to citation in text: [1] -
Meca, L.; Řeha, D.; Havlas, Z. J. Org. Chem. 2003, 68, 5677–5680. doi:10.1021/jo034344u
Return to citation in text: [1] -
Ishida, T.; In, Y.; Inoue, M.; Ueno, Y.; Tanaka, C. Tetrahedron Lett. 1989, 30, 959–962. doi:10.1016/S0040-4039(00)95290-0
Return to citation in text: [1] -
Delgado, P.; Quiroga, J.; Cobo, J.; Low, J. N.; Glidewell, C. Acta Crystallogr., Sect. C 2005, 61, o477–o482. doi:10.1107/S0108270105021888
Return to citation in text: [1] -
Opletalova, V.; Dolezel, J.; Kralova, K.; Pesko, M.; Kunes, J.; Jampilek, J. Molecules 2011, 16, 5207–5227. doi:10.3390/molecules16065207
Return to citation in text: [1] -
Cutshall, N. S.; O’Day, C.; Prezhdo, M. Bioorg. Med. Chem. Lett. 2005, 15, 3374–3379. doi:10.1016/j.bmcl.2005.05.034
Return to citation in text: [1] -
Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Zaprutko, L.; Gzella, A.; Lesyk, R. Eur. J. Med. Chem. 2009, 44, 1396–1404. doi:10.1016/j.ejmech.2008.09.032
Return to citation in text: [1] -
Zidar, N.; Tomašić, T.; Šink, R.; Rupnik, V.; Kovač, A.; Turk, S.; Patin, D.; Blanot, D.; Martel, C. C.; Dessen, A.; Müller Premru, M.; Zega, A.; Gobec, S.; Mašič, L. P.; Kikelj, D. J. Med. Chem. 2010, 53, 6584–6594. doi:10.1021/jm100285g
Return to citation in text: [1]
| 50. | Cutshall, N. S.; O’Day, C.; Prezhdo, M. Bioorg. Med. Chem. Lett. 2005, 15, 3374–3379. doi:10.1016/j.bmcl.2005.05.034 |
| 51. | Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Zaprutko, L.; Gzella, A.; Lesyk, R. Eur. J. Med. Chem. 2009, 44, 1396–1404. doi:10.1016/j.ejmech.2008.09.032 |
| 52. | Zidar, N.; Tomašić, T.; Šink, R.; Rupnik, V.; Kovač, A.; Turk, S.; Patin, D.; Blanot, D.; Martel, C. C.; Dessen, A.; Müller Premru, M.; Zega, A.; Gobec, S.; Mašič, L. P.; Kikelj, D. J. Med. Chem. 2010, 53, 6584–6594. doi:10.1021/jm100285g |
| 1. | Kaberdin, R. V.; Potkin, V. I.; Zapol’skii, V. A. Russ. Chem. Rev. 1997, 66, 827–842. doi:10.1070/RC1997v066n10ABEH000310 |
| 2. | Zapol’skii, V. A.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Heterocycles 2004, 63, 1281–1298. doi:10.3987/COM-04-10020 |
| 3. | Zapol’skii, V. A.; Nutz, E.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Synthesis 2006, 2927–2933. doi:10.1055/s-2006-950187 |
| 4. | Zapol’skii, V. A.; Namyslo, J. C.; Blaschkowski, B.; Kaufmann, D. E. Synlett 2006, 3464–3468. doi:10.1055/s-2006-956490 |
| 5. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. ARKIVOC 2007, No. i, 76–93. |
| 6. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. Synlett 2007, 1507–1512. doi:10.1055/s-2007-982554 |
| 7. | Zapol’skii, V. A.; Namyslo, J. C.; Altug, C.; Gjikaj, M.; Kaufmann, D. E. Synthesis 2008, 304–310. doi:10.1055/s-2007-990948 |
| 8. | Nutz, E.; Zapol’skii, V. A.; Kaufmann, D. E. Synthesis 2009, 2719–2724. doi:10.1055/s-0029-1216906 |
| 9. | Zapol’skii, V. A.; Fischer, R.; Namyslo, J. C.; Kaufmann, D. E. Bioorg. Med. Chem. 2009, 17, 4206–4215. doi:10.1016/j.bmc.2009.01.001 |
| 10. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. Z. Naturforsch., B 2010, 65b, 843–860. |
| 11. | Zapol’skii, V. A.; Namyslo, J. C.; de Meijere, A.; Kaufmann, D. E. Beilstein J. Org. Chem. 2012, 8, 621–628. doi:10.3762/bjoc.8.69 |
| 12. | Vogt, E.-J.; Zapol’skii, V. A.; Nutz, E.; Kaufmann, D. E. Z. Naturforsch., B 2012, 67, 285–294. |
| 13. | Zapol’skii, V. A.; Vogt, E.-J.; Gjikaj, M.; Kaufmann, D. E. Heterocycles 2012, 86, 1431–1447. doi:10.3987/COM-12-S(N)108 |
| 14. | Ibis, C. Bull. Soc. Chim. Belg. 1996, 105, 317–320. |
| 15. | Ibis, C.; Sayil, M. C.; Deniz, N. G. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o800–o801. doi:10.1107/S1600536806001838 |
| 29. | Kawakami, M.; Koya, K.; Ukai, T.; Tatsuta, N.; Ikegawa, A.; Ogawa, K.; Shishido, T.; Chen, L. B. J. Med. Chem. 1998, 41, 130–142. doi:10.1021/jm970590k |
| 30. | Rida, S. M.; Ashour, F. A.; El-Hawash, S. A. M.; ElSemary, M. M.; Badr, M. H.; Shalaby, M. A. Eur. J. Med. Chem. 2005, 40, 949–959. doi:10.1016/j.ejmech.2005.03.023 |
| 31. | Hamama, W. S.; Ismail, M. A.; Shaaban, S.; Zoorob, H. H. J. Heterocycl. Chem. 2008, 45, 939–956. doi:10.1002/jhet.5570450401 |
| 47. | Ishida, T.; In, Y.; Inoue, M.; Ueno, Y.; Tanaka, C. Tetrahedron Lett. 1989, 30, 959–962. doi:10.1016/S0040-4039(00)95290-0 |
| 48. | Delgado, P.; Quiroga, J.; Cobo, J.; Low, J. N.; Glidewell, C. Acta Crystallogr., Sect. C 2005, 61, o477–o482. doi:10.1107/S0108270105021888 |
| 18. | Brown, F. C. Chem. Rev. 1961, 61, 463–521. doi:10.1021/cr60213a002 |
| 19. | Singh, S. P.; Parmar, S. S.; Raman, K.; Stenberg, V. I. Chem. Rev. 1981, 81, 175–203. doi:10.1021/cr00042a003 |
| 20. | Chandrasekhar, B. J. Sulfur Chem. 2012, 33, 439–503. doi:10.1080/17415993.2012.693490 |
| 21. | Tian, Y.; Zhan, P.; Rai, D.; Zhang, J.; De Clercq, J.; Liu, X. Curr. Med. Chem. 2012, 19, 2026–2037. doi:10.2174/092986712800167383 |
| 22. | Jain, A. K.; Vaidya, A.; Ravichandran, V.; Kashaw, S. K.; Agrawal, R. K. Bioorg. Med. Chem. 2012, 20, 3378–3395. doi:10.1016/j.bmc.2012.03.069 |
| 23. | Verma, A.; Saraf, S. K. Eur. J. Med. Chem. 2008, 43, 897–905. doi:10.1016/j.ejmech.2007.07.017 |
| 24. | Zhang, Q.; Zhou, H.; Zhai, S.; Yan, B. Curr. Pharm. Des. 2010, 16, 1826–1842. doi:10.2174/138161210791208983 |
| 25. | Mehta, D. P.; Sengar, N. P. S.; Pathak, A. K. Orient. J. Chem. 2008, 24, 441–454. |
| 26. | Edwards, P. J. Drug Discovery Today 2008, 13, 1107–1108. doi:10.1016/j.drudis.2008.10.004 |
| 27. | Cunico, W.; Gomes, C. R. B.; Vellasco, W. T., Jr. Mini-Rev. Org. Chem. 2008, 5, 336–344. doi:10.2174/157019308786242232 |
| 28. | Newkome, G. R.; Nayak, A. Adv. Heterocycl. Chem. 1980, 25, 83–112. doi:10.1016/S0065-2725(08)60690-X |
| 49. | Opletalova, V.; Dolezel, J.; Kralova, K.; Pesko, M.; Kunes, J.; Jampilek, J. Molecules 2011, 16, 5207–5227. doi:10.3390/molecules16065207 |
| 2. | Zapol’skii, V. A.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Heterocycles 2004, 63, 1281–1298. doi:10.3987/COM-04-10020 |
| 5. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. ARKIVOC 2007, No. i, 76–93. |
| 16. | Ol'dekop, Y. A.; Kaberdin, R. V.; Potkin, V. I. J. Org. Chem. USSR 1980, 16, 469–472. |
| 17. | Ibiş, C.; Kirbaşlar, F. G.; Aydinli, G. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 365–374. doi:10.1080/104265090508433 |
| 16. | Ol'dekop, Y. A.; Kaberdin, R. V.; Potkin, V. I. J. Org. Chem. USSR 1980, 16, 469–472. |
| 45. | Ibiş, C.; Gökmen, Z. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 2537–2542. doi:10.1080/10426500490485615 |
| 2. | Zapol’skii, V. A.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Heterocycles 2004, 63, 1281–1298. doi:10.3987/COM-04-10020 |
| 3. | Zapol’skii, V. A.; Nutz, E.; Namyslo, J. C.; Adam, A. E.; Kaufmann, D. E. Synthesis 2006, 2927–2933. doi:10.1055/s-2006-950187 |
| 4. | Zapol’skii, V. A.; Namyslo, J. C.; Blaschkowski, B.; Kaufmann, D. E. Synlett 2006, 3464–3468. doi:10.1055/s-2006-956490 |
| 5. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. ARKIVOC 2007, No. i, 76–93. |
| 6. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. Synlett 2007, 1507–1512. doi:10.1055/s-2007-982554 |
| 7. | Zapol’skii, V. A.; Namyslo, J. C.; Altug, C.; Gjikaj, M.; Kaufmann, D. E. Synthesis 2008, 304–310. doi:10.1055/s-2007-990948 |
| 8. | Nutz, E.; Zapol’skii, V. A.; Kaufmann, D. E. Synthesis 2009, 2719–2724. doi:10.1055/s-0029-1216906 |
| 9. | Zapol’skii, V. A.; Fischer, R.; Namyslo, J. C.; Kaufmann, D. E. Bioorg. Med. Chem. 2009, 17, 4206–4215. doi:10.1016/j.bmc.2009.01.001 |
| 10. | Zapol’skii, V. A.; Namyslo, J. C.; Gjikaj, M.; Kaufmann, D. E. Z. Naturforsch., B 2010, 65b, 843–860. |
| 11. | Zapol’skii, V. A.; Namyslo, J. C.; de Meijere, A.; Kaufmann, D. E. Beilstein J. Org. Chem. 2012, 8, 621–628. doi:10.3762/bjoc.8.69 |
| 12. | Vogt, E.-J.; Zapol’skii, V. A.; Nutz, E.; Kaufmann, D. E. Z. Naturforsch., B 2012, 67, 285–294. |
| 13. | Zapol’skii, V. A.; Vogt, E.-J.; Gjikaj, M.; Kaufmann, D. E. Heterocycles 2012, 86, 1431–1447. doi:10.3987/COM-12-S(N)108 |
| 46. | Meca, L.; Řeha, D.; Havlas, Z. J. Org. Chem. 2003, 68, 5677–5680. doi:10.1021/jo034344u |
| 34. | Takahashi, K.; Nakatani, S.; Matsuda, T.; Nanbu, H.; Komura, T.; Murata, K. Chem. Lett. 1994, 23, 2001–2004. |
| 18. | Brown, F. C. Chem. Rev. 1961, 61, 463–521. doi:10.1021/cr60213a002 |
| 19. | Singh, S. P.; Parmar, S. S.; Raman, K.; Stenberg, V. I. Chem. Rev. 1981, 81, 175–203. doi:10.1021/cr00042a003 |
| 20. | Chandrasekhar, B. J. Sulfur Chem. 2012, 33, 439–503. doi:10.1080/17415993.2012.693490 |
| 21. | Tian, Y.; Zhan, P.; Rai, D.; Zhang, J.; De Clercq, J.; Liu, X. Curr. Med. Chem. 2012, 19, 2026–2037. doi:10.2174/092986712800167383 |
| 22. | Jain, A. K.; Vaidya, A.; Ravichandran, V.; Kashaw, S. K.; Agrawal, R. K. Bioorg. Med. Chem. 2012, 20, 3378–3395. doi:10.1016/j.bmc.2012.03.069 |
| 23. | Verma, A.; Saraf, S. K. Eur. J. Med. Chem. 2008, 43, 897–905. doi:10.1016/j.ejmech.2007.07.017 |
| 24. | Zhang, Q.; Zhou, H.; Zhai, S.; Yan, B. Curr. Pharm. Des. 2010, 16, 1826–1842. doi:10.2174/138161210791208983 |
| 25. | Mehta, D. P.; Sengar, N. P. S.; Pathak, A. K. Orient. J. Chem. 2008, 24, 441–454. |
| 26. | Edwards, P. J. Drug Discovery Today 2008, 13, 1107–1108. doi:10.1016/j.drudis.2008.10.004 |
| 27. | Cunico, W.; Gomes, C. R. B.; Vellasco, W. T., Jr. Mini-Rev. Org. Chem. 2008, 5, 336–344. doi:10.2174/157019308786242232 |
| 28. | Newkome, G. R.; Nayak, A. Adv. Heterocycl. Chem. 1980, 25, 83–112. doi:10.1016/S0065-2725(08)60690-X |
| 36. | Singh, V. P.; Upadhyay, G. S.; Singh, H. Asian J. Chem. Rev. 1992, 3, 12–21. |
| 37. | Abdel-Rahman, R. M. Boll. Chim. Farm. 2001, 140, 401–410. |
| 38. | Lesyk, R. B.; Zimenkovsky, B. S. Curr. Org. Chem. 2004, 8, 1547–1577. doi:10.2174/1385272043369773 |
| 39. | Dudhe, R.; Imran, M.; Maurya, N. P.; Sharma, P. K. Orient. J. Chem. 2007, 23, 167–176. |
| 31. | Hamama, W. S.; Ismail, M. A.; Shaaban, S.; Zoorob, H. H. J. Heterocycl. Chem. 2008, 45, 939–956. doi:10.1002/jhet.5570450401 |
| 40. | Fahmy, H. T. Y. Boll. Chim. Farm. 2001, 140, 422–427. |
| 41. | Vovk, M. V.; Dorokhov, V. I. Russ. J. Org. Chem. 1993, 29, 1471–1473. |
| 42. | Matsubara, Y.; Nakamura, T.; Yoshihara, M.; Maeshima, T. Chem. Pharm. Bull. 1985, 33, 3009–3011. |
| 43. | Satzinger, G. Justus Liebigs Ann. Chem. 1963, 665, 150–165. doi:10.1002/jlac.19636650118 |
| 44. | Marković, R.; Vitnik, Ž.; Baranac, M.; Juranic, I. J. Chem. Res., Synop. 2002, 485–489. doi:10.3184/030823402103170673 |
| 33. | Tripathi, A. C.; Gupta, S. J.; Fatima, G. N.; Sonar, P. K.; Verma, A.; Saraf, S. K. Eur. J. Med. Chem. 2014, 72, 52–77. doi:10.1016/j.ejmech.2013.11.017 |
| 30. | Rida, S. M.; Ashour, F. A.; El-Hawash, S. A. M.; ElSemary, M. M.; Badr, M. H.; Shalaby, M. A. Eur. J. Med. Chem. 2005, 40, 949–959. doi:10.1016/j.ejmech.2005.03.023 |
| 32. | Graciet, J. C.; Faury, P.; Camplo, M.; Charvet, A. S.; Mourier, N.; Trabaud, C.; Niddam, V.; Simon, V.; Kraus, J. L. Nucleosides Nucleotides 1995, 14, 1379–1392. doi:10.1080/15257779508010698 |
| 35. | Smokal, V.; Derkowska, B.; Czaplicki, R.; Krupka, O.; Kolendo, A.; Sahraoui, B. Opt. Mater. 2009, 31, 554–557. doi:10.1016/j.optmat.2007.10.019 |
© 2014 Zapol’skii et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)