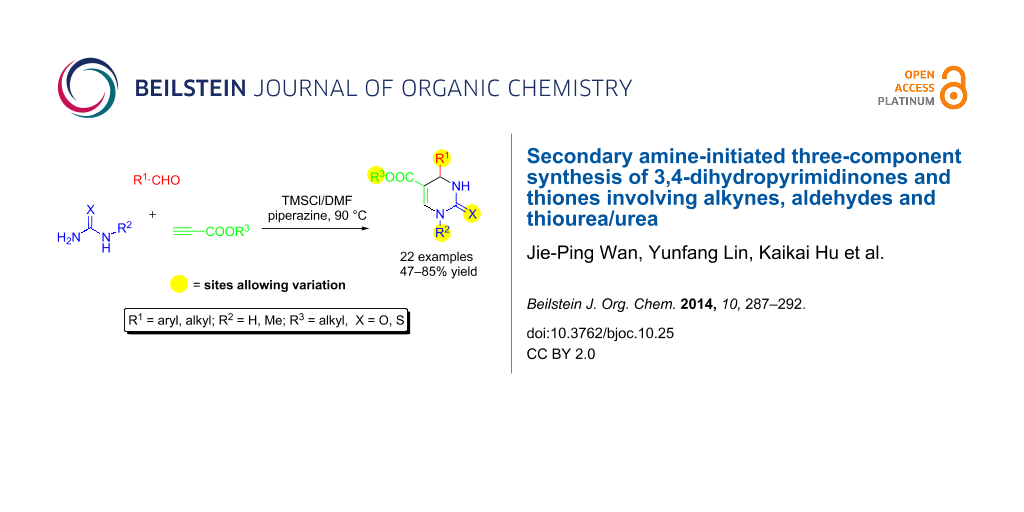
Abstract
The three-component reactions of aldehydes, electron deficient alkynes and ureas/thioureas have been smoothly performed to yield a class of unprecedented 3,4-dihydropyrimidinones and thiones (DHPMs). The reactions are initiated by the key transformation of an enamine-type activation involving the addition of a secondary amine to an alkyne, which enables the subsequent incorporation of aldehydes and ureas/thioureas. This protocol tolerates a broad range of aryl- or alkylaldehydes, N-substituted and unsubstituted ureas/thioureas and alkynes to yield the corresponding DHPMs with specific regioselectivity.

Graphical Abstract
Introduction
DHPMs are well-known heterocyclic scaffolds with abundant biological relevance [1-3]. The DHPM backbone has been found in a class of marine natural products possessing anti-HIV activity [4]. What’s more, diversified other biological activities have been discovered in many synthesized small DHPMs. For example, monastrol (A) [5], (R)-SQ 32926 (B) [6] and (+)-SNAP-7941(C) [7] are lead compounds possessing outstanding antitumor, antihypertensive and melanin-concentrating hormone receptor antagonism activities, respectively (Figure 1).
![[1860-5397-10-25-1]](/bjoc/content/figures/1860-5397-10-25-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some DHPMs-based lead compounds.
Figure 1: Some DHPMs-based lead compounds.
More recently, it was shown that DHPMs display many new bioactivities such as antioxidation [8], antibacterial [9], antimalaria [10], antimicrobial [11] and sodium iodide symporter inhibition [12], suggesting the great potential of DHPMs in discovering new lead compounds and medicines. Besides their attractiveness in biological and medicinal researches, DHPMs have also been demonstrated as quite flexible precursors for the synthesis of many other derived heterocyclic scaffolds [13].
For a rather long period, the Biginelli reaction involving the condensation of aldehydes, β-ketoesters and ureas (thioureas) [14] has been dominantly employed for DHPMs synthesis in both racemic [15-18] and asymmetric versions [19-23]. Despite of many recognized advantages of the Biginelli reaction, the product diversity suffered from limitations because β-ketoesters or 1,3-diketones are intrinsically required as donors of the C5–C6 fragment in this reaction, which predetermined the presence of a C6 substitution in the produced DHPMs. On the other hand, DHPMs without a substituent at the C6 site were hardly accessible by the classical Biginelli reaction, probably because of either the rare availability of the corresponding β-formylketone/ester substrates or the intolerance of β-formylketones/esters in the Biginelli reaction. In regard to the daily increasing requirements on molecular diversity, developing powerful methods for the rapid synthesis of DHPMs with diverse and unprecedented substitution patterns has become an issue of central importance. During the last decade, tremendous endeavours have been made to devise efficient synthetic routes to access structurally diverse DHPMs by employing multicomponent reactions (MCRs) [24-26]. Interestingly, in the process of designing new MCRs yielding DHPMs, the utilization of new C5–C6 fragment donors constituted the major strategy. Representative new C5–C6 building blocks used in the multicomponent synthesis of DHPMs are 2-oxosuccinic acid [27], acetylaldehydes [28], cyclic and acyclic ketones [29-31], β-oxo dithioesters [32], diketenes [33] and enaminones [34]. On the other hand, as frequently utilized building blocks in organic synthesis, alkynes have been known to possess versatile reactivity in the synthesis of small molecules. For example, a previous protocol employing aryl alkynes, aldehydes and urea/thiourea has been found to selectively provide various 1,3-thiazine derivatives 4 [35]. Amazingly, a generally applicable alkyne-based method regioselectively yielding DHPMs has not yet been achieved [36]. Herein, we report the regioselective three-component synthesis of DHPMs employing alkynes, aldehydes and ureas/thioureas by making use of the activation effect of a secondary amine to alkynes (Scheme 1) [37].
![[1860-5397-10-25-i1]](/bjoc/content/inline/1860-5397-10-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Regioselective 1,3-thiazines and DHPMs via aldehydes, ureas/thioureas and alkynes.
Scheme 1: Regioselective 1,3-thiazines and DHPMs via aldehydes, ureas/thioureas and alkynes.
Results and Discussion
The work began from the three-component model reaction of p-chlorobenzaldehyde (1a), thiourea (2a) and ethyl propiolate (3a). The optimization results are outlined in Table 1. Firstly, parallel studies respectively employing TMSCl, morpholine and mixed TMSCl/morpholine as catalysts have been conducted. It was found that the target product could be formed only when both morpholine and TMSCl were present (Table 1, entries 1–3). Extended experiments using different amounts and types of amine catalysts demonstrated that 0.5 equiv of piperazine was favorable (Table 1, entries 4–6). Reducing the amount of TMSCl led to a decrease in product yield (Table 1, entry 7). Other Lewis acid or Brønsted acids such as FeCl3 and p-TSA gave no better result for the same reaction (Table 1, entries 8 and 9). In addition, the non-polar solvent toluene was not able to mediate the reaction, while a lower yield of product was observed when MeCN was used as solvent (Table 1, entries 10 and 11). Altering the reaction temperature also failed to enhance the yield (Table 1, entries 12 and 13). Finally, employing additional 0.5 equiv of p-TSA has been found to significantly improve the yield (Table 1, entry 14). This result may be attributed to the double activation effect involving both Lewis and Brønsted acid (see Scheme 3).
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-25-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalysts | Solvent | T (°C) | Yield (%)b |
|---|---|---|---|---|
| 1 | morpholine/TMSCl | DMF | 90 | 45 |
| 2c | TMSCl | DMF | 90 | nr |
| 3c | morpholine | DMF | 90 | nr |
| 4d | morpholine/TMSCl | DMF | 90 | 25 |
| 5 | pyrrolidine/TMSCl | DMF | 90 | 13 |
| 6 | piperazine/TMSCl | DMF | 90 | 59 |
| 7e | piperazine/TMSCl | DMF | 90 | 40 |
| 8 | piperazine/FeCl3 | DMF | 90 | nr |
| 9 | piperazine/p-TSA | DMF | 90 | 15 |
| 10 | piperazine/TMSCl | CH3CN | 90 | 39 |
| 11 | piperazine/TMSCl | toluene | 90 | nr |
| 12 | piperazine/TMSCl | DMF | 80 | 27 |
| 13 | piperazine/TMSCl | DMF | 100 | 39 |
| 14f | piperazine/TMSCl | DMF | 90 | 81 |
aGeneral conditions: 1a (0.3 mmol), 2a (0.4 mmol), 3a (0.3 mmol), secondary amine (0.15 mmol) and acid (0.6 mmol) in 4 mL solvent, stirred for 12 h. bYields of isolated product. cNo reaction. d0.09 mmol (30 mol %) morpholine was used. e0.45 mmol TMSCl was used. fAdditional 0.5 equiv of p-TSA was used.
With the optimal conditions in hand, we conducted the investigation on examining the application scope. Various aldehydes of different properties have been subjected to react with thioureas/N-substituted thioureas/urea as well as different propiolates. Typical results were listed in Table 2. It can be seen from these reactions that aldehydes containing various functional groups tolerate the protocol of the corresponding DHPMs synthesis. For reactions involving aromatic aldehydes, the electronic properties of the substituent exhibited evident impact on the product yield. Aldehydes containing an electron withdrawing group (EWG) facilitated the reactions to give related DHPMs with evidently higher yields than those containing an electron donating group (EDG) (Table 2, products 5a–5e, 5i–5k). A similar tendency occurred in the experiments using N-methyl thiourea (Table 2, products 5f–5h). Attempts on employing EDG-substituted aldehydes such as p-tolylaldehyde to react with N-substituted thiourea and alkyne were not successful. On the other hand, benzaldehydes with ortho- and meta-substitution could also react with thioureas and propiolates to give the corresponding DHPMs 5l–5p. However, compared with thiourea, urea has been found to undergo a similar transformation more toughly, and DHPMs 5q–5s from urea reactions have been obtained with only moderate yields under the conditions of refluxing THF (Table 2, products 5q–5s). Notably, this synthetic methodology displayed also good tolerance to aliphatic aldehydes to provide 4-alkyl DHPMs 5t–5v with good to excellent yield (Table 2).
Table 2: Multicomponent synthesis of different DHPMs.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-25-i5.svg?max-width=637&scale=1.0)
|
|||||
| R1 | R2 | R3 | X | Product | Yield (%)b |
|---|---|---|---|---|---|
| 4-ClC6H4 | H | Et | S | 5a | 81 |
| 4-BrC6H4 | H | Et | S | 5b | 70 |
| 4-CF3C6H4 | H | Et | S | 5c | 72 |
| 4-NO2C6H4 | H | Et | S | 5d | 85 |
| 4-MeC6H4 | H | Et | S | 5e | 58 |
| 4-ClC6H4 | Me | Et | S | 5f | 78 |
| 4-BrC6H4 | Me | Et | S | 5g | 63 |
| 4-CF3C6H4 | Me | Et | S | 5h | 83 |
| 4-ClC6H4 | H | Me | S | 5i | 72 |
| 4-CF3C6H4 | H | Me | S | 5j | 81 |
| 4-MeC6H4 | H | Me | S | 5k | 66 |
| 3-OHCC6H4 | H | Et | S | 5l | 68 |
| 3-MeOC6H4 | H | Et | S | 5m | 61 |
| 2,4-Cl2C6H3 | H | Et | S | 5n | 64 |
| 2-ClC6H4 | Me | Et | S | 5o | 75 |
| 2-ClC6H4 | H | Me | S | 5p | 60 |
| 4-ClC6H4 | H | Et | O | 5qc | 43 |
| 4-BrC6H4 | H | Et | O | 5rc | 55 |
| 4-NO2C6H4 | H | Et | O | 5sc | 47 |
| Et | H | Et | S | 5t | 82 |
| Pr | H | Et | S | 5u | 68 |
| PhCH2 | H | Et | S | 5v | 81 |
aGeneral conditions: 1 (0.3 mmol), 2 (0.4 mmol), 3 (0.3 mmol), piperazine (0.15 mmol), TMSCl (0.6 mmol), p-TSA (0.15 mmol) in 4 mL DMF, stirred at 90 °C for 12 h. bYield of isolated product. cReactions in refluxing THF, piperazine (0.15 mmol), TMSCl (0.9 mmol) and p-TSA (0.3 mmol).
Following these obtained results, especially the key function of the secondary amine to activate electron deficient alkynes [34] we conducted the control experiments on both the synthesis of the possible enamino ester and its transformation to the corresponding DHPM product. The results proved that enamino ester 6a could be easily generated and efficiently transformed to target product 5a under standard conditions (without using secondary amine, Scheme 2).
![[1860-5397-10-25-i2]](/bjoc/content/inline/1860-5397-10-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of enamino ester intermediate and its transformation to DHPM.
Scheme 2: Synthesis of enamino ester intermediate and its transformation to DHPM.
Based on the results from the control experiments, we postulate the reaction mechanism: At first, the addition of the secondary amine to the propiolate gives enamino ester intermediate 6. On the other hand, ureas/thioureas were known to be readily activated by TMSCl to give intermediate 10 [38,39]. Intermediate 10 consequently condenses with the aldehyde which was activated by p-TSA to generate imine 7. The combination of 6 and 7 allows the production of iminium ion 8. Finally, an intramolecular cyclization of 8 leads to the formation of 9 which subsequently undergoes deaminative elimination to result product 5 by releasing the amine catalyst for further recycling (Scheme 3).
![[1860-5397-10-25-i3]](/bjoc/content/inline/1860-5397-10-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have established an unprecedented amine-initiated three-component protocol for the synthesis of new DHPMs wherein readily available alkynes served as C5–C6 building blocks. This methodology displayed general applicability for aryl- and alkylaldehydes, urea, thiourea, N-substituted thiourea and different alkyl propiolates. The method is useful for the synthesis of diverse new DHPMs which were hardly accessible through known methods such as the Biginelli reaction.
Experimental
General information
All reagents were obtained from commercial sources and used directly without further purification, solvents have been treated following standard processes prior to use. 1H and 13C NMR spectra were recorded on a 400 MHz or 600 MHz apparatus. The frequencies for 1H NMR and 13C NMR experiments are 400 MHz/600 MHz and 100 MHz/150 MHz, respectively. The chemical shifts were reported in ppm employing TMS as internal standard. Melting points were measured with an X-4A instrument without correcting the temperature, IR spectra were measured in KBr on a Spectrum One apparatus and the HRMS were obtained under ESI mode in a Bruker 7-tesla FT-ICR MS instrument.
General procedure for the three-component synthesis of DHPMs 5
Aldehyde 1 (0.3 mmol), urea/thiourea 2 (0.4 mmol) and alkyl propionate 3 (0.3 mmol) piperazine (0.15 mmol), and p-tolylsulfonic acid (0.15 mmol, 0.3 mmol for the reaction of urea) were charged in a 25 mL round bottom flask equipped with a stirring bar. DMF (THF for the reaction of urea) (4 mL) and TMSCl (0.6 mmol, 0.9 mmol for the reaction of urea) were added and the mixture was stirred at 90 °C for 12 h (TLC). After cooling down to room temperature, 5 mL water was added, and the resulting mixture was extracted with ethyl acetate (3 × 8 mL). The organic layers were combined and dried overnight with anhydrous MgSO4. After filtration and removing of the solvent under reduced pressure, the residue was subjected to flash column chromatography to provide pure products.
Synthesis of intermediate 6a. Into a 25 mL round bottom flask was added ethyl propiolate (0.6 mmol) and piperazine (0.3 mmol). 1.5 mL DMF was added and the mixture was stirred at rt for 8 h (TLC). Upon completion, 10 mL water was added and the resulting mixture was extracted with EtOAc (3 × 10 mL). The combined organic layer was dried with anhydrous Na2SO4. After removing of the solid by filtration and evaporation of the solvent the product 6a was isolated as white solid.
Supporting Information
| Supporting Information File 1: Experimental details on the synthesis of all DHPMs 5 and intermediate 6a, full characterization data as well as 1H and 13C NMR spectra of all products 5 and 6a. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Kappe, C. O. Eur. J. Med. Chem. 2000, 35, 1043. doi:10.1016/S0223-5234(00)01189-2
Return to citation in text: [1] -
Singh, K.; Arora, D.; Singh, S. Mini-Rev. Med. Chem. 2009, 9, 95. doi:10.2174/138955709787001686
Return to citation in text: [1] -
Wan, J.-P.; Pan, Y. Mini-Rev. Med. Chem. 2012, 12, 337. doi:10.2174/138955712799829267
Return to citation in text: [1] -
Heys, L.; Moore, C. G.; Murphy, P. J. Chem. Soc. Rev. 2000, 29, 57. doi:10.1039/a903712h
Return to citation in text: [1] -
Mayer, T. U.; Kapoor, T. M.; Haggarty, S. J.; King, R. W.; Schreiber, S. L.; Mitchison, T. J. Science 1999, 286, 971. doi:10.1126/science.286.5441.971
Return to citation in text: [1] -
Schnell, B.; Strauss, U. T.; Verdino, P.; Faber, K.; Kappe, C. O. Tetrahedron: Asymmetry 2000, 11, 1449. doi:10.1016/S0957-4166(00)00081-1
Return to citation in text: [1] -
Borowsky, B.; Durkin, M. M.; Ogozalek, K.; Marzabadi, M. R.; DeLeon, J.; Heurich, R.; Lichtblau, H.; Shaposhnik, Z.; Daniewska, I.; Blackburn, T. P.; Branchek, T. A.; Gerald, C.; Vaysse, P. J.; Forray, C. Nat. Med. 2002, 8, 825. doi:10.1038/nm0902-1039b
Return to citation in text: [1] -
Stefani, H. A.; Oliveira, C. B.; Almeida, R. B.; Pereira, C. M. P.; Braga, R. C.; Cella, R.; Borges, V. C.; Savegnago, L.; Nogueira, C. W. Eur. J. Med. Chem. 2006, 41, 513. doi:10.1016/j.ejmech.2006.01.007
Return to citation in text: [1] -
Ashok, M.; Holla, B. S.; Kumari, N. S. Eur. J. Med. Chem. 2007, 42, 380. doi:10.1016/j.ejmech.2006.09.003
Return to citation in text: [1] -
October, N.; Watermeyer, N. D.; Yardley, V.; Egan, T. J.; Ncokazi, K.; Chibale, K. ChemMedChem 2008, 3, 1649. doi:10.1002/cmdc.200800172
Return to citation in text: [1] -
Chitra, S.; Devanathan, D.; Pandiarajan, K. Eur. J. Med. Chem. 2010, 45, 367. doi:10.1016/j.ejmech.2009.09.018
Return to citation in text: [1] -
Lacotte, P.; Puente, C.; Ambroise, Y. ChemMedChem 2013, 8, 104. doi:10.1002/cmdc.201200417
Return to citation in text: [1] -
Matache, M.; Dobrota, C.; Bogdan, N. D.; Funeriu, D. P. Curr. Org. Synth. 2011, 3, 356. doi:10.2174/157017911795529218
Return to citation in text: [1] -
Biginelli, P. Gazz. Chim. Ital. 1893, 23, 360.
Return to citation in text: [1] -
Kappe, C. O. Tetrahedron 1993, 49, 6937. doi:10.1016/S0040-4020(01)87971-0
Return to citation in text: [1] -
Kappe, C. O. Acc. Chem. Res. 2000, 33, 879. doi:10.1021/ar000048h
Return to citation in text: [1] -
Kappe, C. O.; Stadler, A. Org. React. 2004, 63, 1. doi:10.1002/0471264180.or063.01
Return to citation in text: [1] -
Suresh; Sandhu, J. S. ARKIVOC 2012, No. i, 66.
Return to citation in text: [1] -
Huang, Y.; Yang, F.; Zhu, C. J. Am. Chem. Soc. 2005, 127, 16386. doi:10.1021/ja056092f
Return to citation in text: [1] -
Chen, X.-H.; Xu, X.-Y.; Liu, H.; Cun, L.-F.; Gong, L.-Z. J. Am. Chem. Soc. 2006, 128, 14802. doi:10.1021/ja065267y
Return to citation in text: [1] -
Gong, L.-Z.; Chen, X.-H.; Xu, X.-Y. Chem.–Eur. J. 2007, 13, 8920. doi:10.1002/chem.200700840
Return to citation in text: [1] -
Xin, J.; Chang, L.; Hou, Z.; Shang, D.; Liu, X.; Feng, X. Chem.–Eur. J. 2008, 14, 3177. doi:10.1002/chem.200701581
Return to citation in text: [1] -
Saha, S.; Moorthy, J. N. J. Org. Chem. 2011, 76, 396. doi:10.1021/jo101717m
Return to citation in text: [1] -
Wan, J.-P.; Liu, Y. Synthesis 2010, 3943. doi:10.1055/s-0030-1258290
See for a review on new MCRs for DHPMs synthesis.
Return to citation in text: [1] -
Vugts, D. J.; Jansen, H.; Schmitz, R. F.; de Kanter, F. J. J.; Orru, R. V. A. Chem. Commun. 2003, 2594. doi:10.1039/b308243a
Return to citation in text: [1] -
Vugts, D. J.; Koningstein, M. M.; Schmitz, R. F.; de Kanter, F. J. J.; Groen, M. B.; Orru, R. V. A. Chem.–Eur. J. 2006, 12, 7178. doi:10.1002/chem.200600168
Return to citation in text: [1] -
Bussolari, J. C.; McDonnell, P. A. J. Org. Chem. 2000, 65, 6777. doi:10.1021/jo005512a
Return to citation in text: [1] -
Bailey, C. D.; Houlden, C. E.; Bar, G. L. J.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. Chem. Commun. 2007, 2932. doi:10.1039/b707361e
Return to citation in text: [1] -
Albelman, M. M.; Smith, S. C.; James, D. R. Tetrahedron Lett. 2003, 44, 4559. doi:10.1016/S0040-4039(03)00985-7
Return to citation in text: [1] -
Wang, Z.-T.; Xu, L.-W.; Xia, C.-G.; Wang, H.-Q. Tetrahedron Lett. 2004, 45, 7951. doi:10.1016/j.tetlet.2004.08.107
Return to citation in text: [1] -
Shen, Z.-L.; Xu, X.-P.; Ji, S.-J. J. Org. Chem. 2010, 75, 1162. doi:10.1021/jo902394y
Return to citation in text: [1] -
Nandi, G. C.; Samai, S.; Singh, M. S. J. Org. Chem. 2010, 75, 7785. doi:10.1021/jo101572c
Return to citation in text: [1] -
Shaabani, A.; Seyyedhamzeh, M.; Maleki, A.; Hajishaabanha, F. Tetrahedron 2010, 66, 4040. doi:10.1016/j.tet.2010.04.028
Return to citation in text: [1] -
Wan, J.-P.; Pan, Y.-J. Chem. Commun. 2009, 2768. doi:10.1039/b901112a
Return to citation in text: [1] [2] -
Huang, S.; Pan, Y.; Zhu, Y.; Wu, A. Org. Lett. 2005, 7, 3797. doi:10.1021/ol051458e
Return to citation in text: [1] -
Khanina, E. L.; Mutsenietse, D. K.; Dubur, G. Y. Khim. Geterotsikl. Soedin. 1984, 4, 529.
Return to citation in text: [1] -
Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471. doi:10.1021/cr0684016
See for a comprehensive review on enamine activation.
Return to citation in text: [1] -
Ryabukhin, S. V.; Plaskon, A. S.; Volochnyuk, D. M.; Shivanyuk, A. N.; Tolmachev, A. A. J. Org. Chem. 2007, 72, 7417. doi:10.1021/jo0712087
Return to citation in text: [1] -
Ryabukhin, S. V.; Plaskon, A. S.; Ostapchuk, E. N.; Volochnyuk, D. M.; Tolmachev, A. A. Synthesis 2007, 417. doi:10.1055/s-2007-965881
Return to citation in text: [1]
| 1. | Kappe, C. O. Eur. J. Med. Chem. 2000, 35, 1043. doi:10.1016/S0223-5234(00)01189-2 |
| 2. | Singh, K.; Arora, D.; Singh, S. Mini-Rev. Med. Chem. 2009, 9, 95. doi:10.2174/138955709787001686 |
| 3. | Wan, J.-P.; Pan, Y. Mini-Rev. Med. Chem. 2012, 12, 337. doi:10.2174/138955712799829267 |
| 7. | Borowsky, B.; Durkin, M. M.; Ogozalek, K.; Marzabadi, M. R.; DeLeon, J.; Heurich, R.; Lichtblau, H.; Shaposhnik, Z.; Daniewska, I.; Blackburn, T. P.; Branchek, T. A.; Gerald, C.; Vaysse, P. J.; Forray, C. Nat. Med. 2002, 8, 825. doi:10.1038/nm0902-1039b |
| 24. |
Wan, J.-P.; Liu, Y. Synthesis 2010, 3943. doi:10.1055/s-0030-1258290
See for a review on new MCRs for DHPMs synthesis. |
| 25. | Vugts, D. J.; Jansen, H.; Schmitz, R. F.; de Kanter, F. J. J.; Orru, R. V. A. Chem. Commun. 2003, 2594. doi:10.1039/b308243a |
| 26. | Vugts, D. J.; Koningstein, M. M.; Schmitz, R. F.; de Kanter, F. J. J.; Groen, M. B.; Orru, R. V. A. Chem.–Eur. J. 2006, 12, 7178. doi:10.1002/chem.200600168 |
| 6. | Schnell, B.; Strauss, U. T.; Verdino, P.; Faber, K.; Kappe, C. O. Tetrahedron: Asymmetry 2000, 11, 1449. doi:10.1016/S0957-4166(00)00081-1 |
| 27. | Bussolari, J. C.; McDonnell, P. A. J. Org. Chem. 2000, 65, 6777. doi:10.1021/jo005512a |
| 5. | Mayer, T. U.; Kapoor, T. M.; Haggarty, S. J.; King, R. W.; Schreiber, S. L.; Mitchison, T. J. Science 1999, 286, 971. doi:10.1126/science.286.5441.971 |
| 15. | Kappe, C. O. Tetrahedron 1993, 49, 6937. doi:10.1016/S0040-4020(01)87971-0 |
| 16. | Kappe, C. O. Acc. Chem. Res. 2000, 33, 879. doi:10.1021/ar000048h |
| 17. | Kappe, C. O.; Stadler, A. Org. React. 2004, 63, 1. doi:10.1002/0471264180.or063.01 |
| 18. | Suresh; Sandhu, J. S. ARKIVOC 2012, No. i, 66. |
| 4. | Heys, L.; Moore, C. G.; Murphy, P. J. Chem. Soc. Rev. 2000, 29, 57. doi:10.1039/a903712h |
| 19. | Huang, Y.; Yang, F.; Zhu, C. J. Am. Chem. Soc. 2005, 127, 16386. doi:10.1021/ja056092f |
| 20. | Chen, X.-H.; Xu, X.-Y.; Liu, H.; Cun, L.-F.; Gong, L.-Z. J. Am. Chem. Soc. 2006, 128, 14802. doi:10.1021/ja065267y |
| 21. | Gong, L.-Z.; Chen, X.-H.; Xu, X.-Y. Chem.–Eur. J. 2007, 13, 8920. doi:10.1002/chem.200700840 |
| 22. | Xin, J.; Chang, L.; Hou, Z.; Shang, D.; Liu, X.; Feng, X. Chem.–Eur. J. 2008, 14, 3177. doi:10.1002/chem.200701581 |
| 23. | Saha, S.; Moorthy, J. N. J. Org. Chem. 2011, 76, 396. doi:10.1021/jo101717m |
| 11. | Chitra, S.; Devanathan, D.; Pandiarajan, K. Eur. J. Med. Chem. 2010, 45, 367. doi:10.1016/j.ejmech.2009.09.018 |
| 13. | Matache, M.; Dobrota, C.; Bogdan, N. D.; Funeriu, D. P. Curr. Org. Synth. 2011, 3, 356. doi:10.2174/157017911795529218 |
| 10. | October, N.; Watermeyer, N. D.; Yardley, V.; Egan, T. J.; Ncokazi, K.; Chibale, K. ChemMedChem 2008, 3, 1649. doi:10.1002/cmdc.200800172 |
| 9. | Ashok, M.; Holla, B. S.; Kumari, N. S. Eur. J. Med. Chem. 2007, 42, 380. doi:10.1016/j.ejmech.2006.09.003 |
| 8. | Stefani, H. A.; Oliveira, C. B.; Almeida, R. B.; Pereira, C. M. P.; Braga, R. C.; Cella, R.; Borges, V. C.; Savegnago, L.; Nogueira, C. W. Eur. J. Med. Chem. 2006, 41, 513. doi:10.1016/j.ejmech.2006.01.007 |
| 12. | Lacotte, P.; Puente, C.; Ambroise, Y. ChemMedChem 2013, 8, 104. doi:10.1002/cmdc.201200417 |
| 32. | Nandi, G. C.; Samai, S.; Singh, M. S. J. Org. Chem. 2010, 75, 7785. doi:10.1021/jo101572c |
| 28. | Bailey, C. D.; Houlden, C. E.; Bar, G. L. J.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. Chem. Commun. 2007, 2932. doi:10.1039/b707361e |
| 29. | Albelman, M. M.; Smith, S. C.; James, D. R. Tetrahedron Lett. 2003, 44, 4559. doi:10.1016/S0040-4039(03)00985-7 |
| 30. | Wang, Z.-T.; Xu, L.-W.; Xia, C.-G.; Wang, H.-Q. Tetrahedron Lett. 2004, 45, 7951. doi:10.1016/j.tetlet.2004.08.107 |
| 31. | Shen, Z.-L.; Xu, X.-P.; Ji, S.-J. J. Org. Chem. 2010, 75, 1162. doi:10.1021/jo902394y |
| 38. | Ryabukhin, S. V.; Plaskon, A. S.; Volochnyuk, D. M.; Shivanyuk, A. N.; Tolmachev, A. A. J. Org. Chem. 2007, 72, 7417. doi:10.1021/jo0712087 |
| 39. | Ryabukhin, S. V.; Plaskon, A. S.; Ostapchuk, E. N.; Volochnyuk, D. M.; Tolmachev, A. A. Synthesis 2007, 417. doi:10.1055/s-2007-965881 |
| 37. |
Mukherjee, S.; Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471. doi:10.1021/cr0684016
See for a comprehensive review on enamine activation. |
| 35. | Huang, S.; Pan, Y.; Zhu, Y.; Wu, A. Org. Lett. 2005, 7, 3797. doi:10.1021/ol051458e |
| 36. | Khanina, E. L.; Mutsenietse, D. K.; Dubur, G. Y. Khim. Geterotsikl. Soedin. 1984, 4, 529. |
| 33. | Shaabani, A.; Seyyedhamzeh, M.; Maleki, A.; Hajishaabanha, F. Tetrahedron 2010, 66, 4040. doi:10.1016/j.tet.2010.04.028 |
© 2014 Wan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)