Abstract
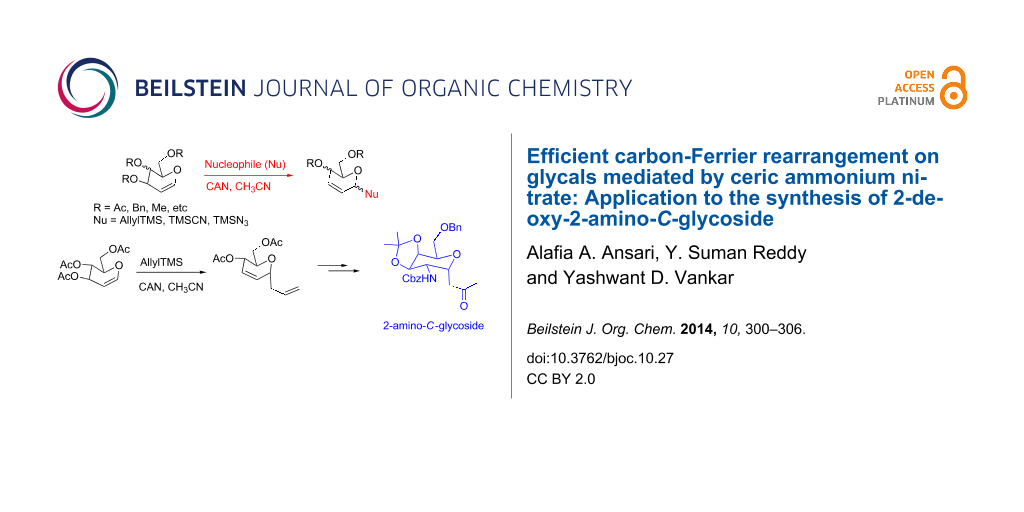
A carbon-Ferrier rearrangement on glycals has been performed by using ceric ammonium nitrate to obtain products in moderate to good yields with high selectivity. The versatility of this method has been demonstrated by applying it to differently protected glycals and by employing several nucleophiles. The obtained C-allyl glycoside has been utilized for the synthesis of a orthogonally protected 2-amino-2-deoxy-C-glycoside.

Graphical Abstract
Introduction
The growing significance of C-glycosides can be attributed to their potential use as inhibitors of carbohydrate-processing enzymes [1-3], their extraordinary stability compared to O-glycosides, and their widespread applicability as intermediates in the synthesis of biologically important molecules [4-8]. C-glycosides are also subunits of several biologically active natural products [9-11]. Consequently, numerous reports are available in literature on the synthesis of C-glycosides [12-14]. Among these, the Ferrier rearrangement [15] of glycals with protic acids [5,16,17] or Lewis acids [18-22] and carbon nucleophiles such as allylsilanes [23], silylacetylenes [24], silyl enol ethers [25], olefins [26], and organozinc reagents [27] has emerged as a popular method. In particular, C-allyl glycosides, glycosyl cyanides, and glycosyl azides have received considerable attention, as the allyl, cyanide and azide moieties can be readily converted into a variety of other functional groups [6,28-30]. Furthermore, the resulting C-pseudoglycals are also important as the double bond can also be easily functionalized in a variety of ways.
Although there is an abundance of methods reporting on the carbon-Ferrier rearrangement of glycals [9], efforts are ongoing for improvements in terms of efficiency, selectivity, time and yields of reaction. In view of our continued interest in the development of novel methods for the synthesis of O- and C-glycosides [31-36], we here report on the carbon-Ferrier rearrangement of glycals by using ceric ammonium nitrate. Ceric ammonium nitrate (CAN) is a versatile and efficient reagent, which has been well-explored for a variety of reactions in literature [37,38]. CAN was utilized successfully in carbohydrate chemistry for important transformations such as the azidonitration of glycals and the formation of 2-C-branched glycosides from glycals [39-41]. In this paper, we report on the addition of carbon nucleophiles onto differently protected glycals by using CAN. The corresponding Ferrier rearrangement products were obtained in moderate to fairly good yields and with a high selectivity.
Results and Discussion
We initially performed the carbon-Ferrier rearrangement on 3,4,6-tri-O-acetyl-D-glucal (1a) by using 3 equivalents of allyltrimethylsilane and 2 equivalents of CAN in anhydrous acetonitrile at room temperature. The reaction proceeded smoothly over 1 hour and exclusively furnished the α-C-allyl glycoside 2a in 88% yield (Table 1) [42]. We attempted the same reaction with a catalytic amount (10–30 mol %) of CAN. However, under these conditions the reaction showed complicated TLC profiles and did not complete even after overnight stirring or heating under reflux for several hours. Changing the solvent to dichloromethane slowed down the reaction considerably due to the poor solubility of CAN, whereas in acetone the reaction gave lower yields. After several optimization experiments, the reaction was observed to be most efficient when 2 equivalents of silane and one equivalent of CAN, with respect to the glycal, was used in acetonitrile.
Table 1: Optimization of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-27-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Silane (equiv) | CAN (equiv) | Solvent | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 3.0 | 2.0 | CH3CN | rt | 1 h | 88 |
| 2 | 3.0 | 0.2 | CH3CN | reflux | 12 h | 38 |
| 3 | 3.0 | 0.5 | CH3CN | reflux | 12 h | 45 |
| 4 | 3.0 | 1.0 | CH3CN | rt | 1.5 h | 82 |
| 5 | 2.0 | 1.0 | CH3CN | rt | 2 h | 88 |
| 6 | 2.0 | 1.0 | CH3COCH3 | rt | 6 h | 53 |
| 7 | 2.0 | 1.0 | CH2Cl2 | rt | 12 h | 20 |
The reaction was then carried out with several glycals (Table 2). and yielded a single product in all cases studied. In the case of glycals 1a–d, only the α-anomer was obtained as has been reported by Danishefsky et al. in the first report on the C-glycosidation of glycals with allyltrimethylsilane by using TiCl4 [23]. The spectral data of the obtained C-allyl glycosides supported the known data [42-44]. In general, D-glucals were found to react faster than D-galactals and provided the rearranged products in less time and better yields. Moreover, even the benzylated glycals 1c and 1d readily underwent the Ferrier rearrangement under these conditions. The pentose glycals 1e–h exclusively gave the anti-products, confirming previous investigations [45]. Glycals 1i, with a TBDPS protecting group, and 1k, with a methyl protecting group, were also found to undergo the reaction in good yields affording 2i (see Supporting Information File 1) and 2k [42], respectively. These findings indicate that the protecting groups were not affected by this reagent system. D-rhamnal 1j and D-ribose derived glycal 1l [46] yielded hitherto unknown C-allyl glycosides 2j and 2l, respectively (see Supporting Information File 1). However, the same reaction with benzylidene or isopropylidene glucals resulted in a complex mixture of products and the desired product was not isolated.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-27-i4.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-27-i5.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-27-i6.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-27-i7.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-27-i8.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-27-i9.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-27-i10.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-27-i11.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-27-i12.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-27-i13.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-27-i14.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-27-i15.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-27-i16.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-27-i17.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-27-i18.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-27-i19.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-27-i20.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-27-i21.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-27-i22.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-27-i23.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-27-i24.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-27-i25.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-27-i26.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-10-27-i27.svg?max-width=637&scale=1.0)
Furthermore, the scope of this reaction was extended by subjecting glycals to reactions with other nucleophiles. Thus, glycals 1a and 1b subjected to treatment with Me3SiCN afforded a 5:1 and 4:1 mixture of glycosyl cyanides 3 and 4 in 77% and 62% yields, respectively (Table 3). The reaction of glycals 1a and 1b with Me3SiN3 furnished a mixture of C-1 and C-3 substituted glycosyl azides (5/5' and 6/6'). This observation is in conformity with the report of Hayashi and co-workers [47], which described the obtainment of Ferrier products along with C-3 substituted products. On the other hand, glucal 1a on reaction with triethylsilane afforded the product 7 in 43% yield, while galactal 1b gave a complex mixture of products, which could not be analyzed.
![[Graphic 26]](/bjoc/content/inline/1860-5397-10-27-i28.svg?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-10-27-i29.svg?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-10-27-i30.svg?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-10-27-i31.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-10-27-i32.svg?max-width=637&scale=1.0)
Proposed mechanism
The proposed mechanism of the formation of the C-allyl glycosides is shown in Scheme 1. The ring oxygen could donate an electron to the Ce3+ leading to the formation of a radical cation A. Subsequent migration of the double bond and loss of the acetyl radical could result in the formation of the delocalized carbocation B. The acetate radical could accept an electron from Ce2+ thereby forming an acetate anion, which could in turn attack the silyl moiety of allyltrimethylsilane leading to the formation of trimethylsilyl acetate and an allyl anion. The allyl anion could then attack at the anomeric position so that the C-allyl pseudoglycoside is fomed. However, we were not able to isolate Me3SiOAc. As the acetate ion and the acetyl radical are better leaving groups the formation of the acyl radical seems to be more facile than the formation of the alkoxy radicals (e.g., benzyloxy and methoxy). This explains faster reaction times in the case of acetylated glycals compared to benzyl or methyl glycals. It is well-known in the literature that the participation of a neighboring group is possible in the case of glucals thereby leading to a higher reactivity than the corresponding galactals [48,49].
![[1860-5397-10-27-i1]](/bjoc/content/inline/1860-5397-10-27-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Having obtained the α-C-glycosides in an efficient manner, we explored their synthetic utility to synthesize a 2-deoxy-2-amino-α-C-glycoside. 2-Deoxy-2-amino-α-C-glycosides have received considerable attention in recent years due to their use in the synthesis of glycopeptides [50,51], glycolipids [51] and glycosyl amino acids [52] to name but a few. Amongst the various reported methods to prepare these compounds [53-55], the most common method is via C-glycosylation of 2-aminosugars [56-58], which is challenging owing to the incompatibility of protic or Lewis acids with amino and amido functionality. In particular, very few reports on the synthesis of orthogonally protected C-glycosides appeared in literature [59]. Therefore, there is a need for the development of efficient methods to allow easy access to this important class of compounds.
For this purpose, Ferrier rearranged product 2a was subjected to deacetylation by using K2CO3/MeOH (Scheme 2) followed by the selective protection of the primary hydroxy group as a benzyl ether by using Bu2SnO and benzyl bromide in the presence of triethylamine and tetrabutylammonium iodide to afford alcohol 8 in 69% yield over 2 steps. The terminal olefin in compound 8 was then subjected to a Wacker oxidation [60] by using a catalytic amount of PdCl2 and an excess of CuCl under O2 atmosphere to obtain the methyl ketone 9 in a good yield. The formation of the product was confirmed by the disappearance of the external olefinic protons and the appearance of a sharp singlet at δ 2.11, corresponding to methyl protons in the 1H NMR spectrum, as well as a peak at δ 207 ppm in the 13C NMR spectrum corresponding to the carbonyl group (see Supporting Information File 1). The allylic alcohol 9 was converted to the corresponding trichloroacetimidate, which in crude form was heated under reflux in xylene, to afford the single isomer 10 in 72% yield by an Overman rearrangement [61-63].
![[1860-5397-10-27-i2]](/bjoc/content/inline/1860-5397-10-27-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2-deoxy-2-amino-C-glycoside 12 from Ferrier product 2a.
Scheme 2: Synthesis of 2-deoxy-2-amino-C-glycoside 12 from Ferrier product 2a.
The trichloroacetamide group was hydrolyzed by heating under reflux in 6 N HCl, and the obtained free amine was protected as a benzyloxycarbamate group by using benzyl chloroformate and Na2CO3. The protected amide 11 was obtained in 74% yield over 2 steps. The regioselectivity of the Overman rearrangement step was determined from the 1H NMR and COSY experiments of compound 11. These experiments showed that the anomeric proton (H-1) at δ 4.40 correlated with the H-2 proton at δ 4.00, which in turn was adjacent to the amide proton at δ 4.85, thereby indicating that the amide moiety is present at the C-2 position (see Supporting Information File 1). The internal olefin was subjected to a dihydroxylation under the Upjohn conditions [64], followed by an acetonide protection of the resulting diol by using 2,2-dimethoxypropane and a catalytic amount of para-toluenesulfonic acid (PTSA) to furnish the desired orthogonally protected 2-deoxy-2-amino-C-glycoside 12 as a single isomer in 82% yield (over 2 steps).
The stereochemistry of the newly generated stereocenters in compound 12 was determined with the help of 1H NMR, COSY, nOe and decoupling experiments (Figure 1). Thus, the irradiation of the signal at δ 3.93 corresponding to H-3 led to an enhancement of the H-5 signal at δ 4.25 by 2.5%, while the peak at δ 4.63, corresponding to H-1, was not enhanced. This implies that the H-3 proton is cis to H-5 and trans to H-1 and thus axially oriented. Meanwhile, the irradiation of the H-1 signal at δ 4.63 did not give any enhancement of the H-3 and H-5 peaks at δ 3.93 and δ 4.25, respectively. Moreover, the homonuclear decoupling of the H-3 signal at δ 3.93 gave the coupling constant of H-4 and H-5 as J = 7.3 Hz, which indicates an axial-equatorial relationship. The decoupling of the H-1' signals at δ 2.55 provided the coupling constant of H-1 and H-2 as J = 6.4 Hz, indicating an axial-equatorial relationship (see Supporting Information File 1). This suggests that the rearrangement took place from the axial side, as expected, since the hydroxy group at C-4 is axially oriented in compound 9. Moreover, the dihydroxylation took place from the side opposite to the amino group at C-2, which can be attributed to the steric hindrance from the bulky amide group.
![[1860-5397-10-27-1]](/bjoc/content/figures/1860-5397-10-27-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: nOe and decoupling experiments of compound 12.
Figure 1: nOe and decoupling experiments of compound 12.
A D-galacto-configured C-glycoside 12 was obtained from D-glucal 1a, which may serve as a versatile synthetic intermediate, since the carbonyl moiety and the double bond functionality in this compound can be synthetically manipulated in a variety of ways.
Conclusion
We have developed an efficient method for the Ferrier rearrangement of glycals by using ceric ammonium nitrate and several carbon nucleophiles. We have successfully employed the obtained C-allyl glycoside 2a for the stereoselective synthesis of a orthogonally protected 2-deoxy-2-amino-C-glycoside 12 via an Overman rearrangement as a key step.
Supporting Information
| Supporting Information File 1: Analytical data and copies of the 1H NMR and 13C NMR spectra of all new compounds. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
We thank the Department of Science and Technology, New Delhi, for a J. C. Bose National Fellowship (JCB/SR/S2/JCB-26/2010) and the Council of Scientific and Industrial Research, New Delhi, for financial support [Grant No. 01(2298)/09/EMR-II] to Y.D.V. A.A.A. and Y.S.R. gratefully acknowledge a Senior Research Fellowship from the Council of Scientific Industrial Research, New Delhi.
References
-
Zou, W. Curr. Top. Med. Chem. 2005, 5, 1363–1391. doi:10.2174/156802605774642999
Return to citation in text: [1] -
Sutherlin, D. P.; Stark, T. M.; Hughes, R.; Armstrong, R. W. J. Org. Chem. 1996, 61, 8350–8354. doi:10.1021/jo960119j
Return to citation in text: [1] -
Weatherman, R. V.; Mortell, K. H.; Chervenak, M.; Kiessling, L. L.; Toone, E. J. Biochemistry 1996, 35, 3619–3624. doi:10.1021/bi951916z
Return to citation in text: [1] -
Danishefsky, S. J.; DeNinno, S.; Lartey, P. J. Am. Chem. Soc. 1987, 109, 2082–2089. doi:10.1021/ja00241a028
Return to citation in text: [1] -
Paterson, I.; Keown, L. E. Tetrahedron Lett. 1997, 38, 5727–5730. doi:10.1016/S0040-4039(97)01257-4
Return to citation in text: [1] [2] -
Kira, K.; Isobe, M. Tetrahedron Lett. 2000, 41, 5951–5955. doi:10.1016/S0040-4039(00)00988-6
Return to citation in text: [1] [2] -
Gallagher, B. M., Jr.; Zhao, H.; Pesant, M.; Fang, F. G. Tetrahedron Lett. 2005, 46, 923–926. doi:10.1016/j.tetlet.2004.12.056
Return to citation in text: [1] -
Sasaki, M.; Tsubone, K.; Shoji, M.; Oikawa, M.; Shimamoto, K.; Sakai, R. Bioorg. Med. Chem. Lett. 2006, 16, 5784–5787. doi:10.1016/j.bmcl.2006.08.082
Return to citation in text: [1] -
Fraser-Reid, B. Acc. Chem. Res. 1985, 18, 347–354. doi:10.1021/ar00119a004
Return to citation in text: [1] [2] -
Kirshning, A.; Chen, G.-w.; Dräger, G.; Schuberth, I.; Tietze, L. F. Bioorg. Med. Chem. 2000, 8, 2347–2354. doi:10.1016/S0968-0896(00)00166-8
Return to citation in text: [1] -
Faul, M. M.; Huff, B. E. Chem. Rev. 2000, 100, 2407–2474. doi:10.1021/cr940210s
Return to citation in text: [1] -
Dondoni, A.; Marra, A. Chem. Rev. 2000, 100, 4395–4422. doi:10.1021/cr9903003
Return to citation in text: [1] -
Du, Y.; Linhardt, R. J.; Vlahov, I. R. Tetrahedron 1998, 54, 9913–9959. doi:10.1016/S0040-4020(98)00405-0
Return to citation in text: [1] -
Ansari, A. A.; Lahiri, R.; Vankar, Y. D. ARKIVOC 2013, No. ii, 316–362. doi:10.3998/ark.5550190.0014.223
Return to citation in text: [1] -
Ferrier, R. J. J. Chem. Soc. 1964, 5443–5449. doi:10.1039/JR9640005443
Return to citation in text: [1] -
Yadav, J. S.; Satyanarayana, M.; Balanarsaiah, E.; Raghavendra, S. Tetrahedron Lett. 2006, 47, 6095–6098. doi:10.1016/j.tetlet.2006.06.084
Return to citation in text: [1] -
Tiwari, P.; Agnihotri, G.; Misra, A. K. Carbohydr. Res. 2005, 340, 749–752. doi:10.1016/j.carres.2004.12.022
Return to citation in text: [1] -
De Las Heras, F. G.; Felix, A. S.; Fernández-Resa, P. Tetrahedron 1983, 39, 1617–1620. doi:10.1016/S0040-4020(01)88571-9
Return to citation in text: [1] -
Ghosh, R.; De, D.; Shown, B.; Maiti, S. B. Carbohydr. Res. 1999, 321, 1–3. doi:10.1016/S0008-6215(99)00219-0
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Chand, P. K. Tetrahedron Lett. 2001, 42, 4057–4059. doi:10.1016/S0040-4039(01)00629-3
Return to citation in text: [1] -
Balamurugan, R.; Koppolu, S. R. Tetrahedron 2009, 65, 8139–8142. doi:10.1016/j.tet.2009.07.087
Return to citation in text: [1] -
Takhi, M.; Rahman, A. A.-H. A.; Schmidt, R. R. Tetrahedron Lett. 2001, 42, 4053–4056. doi:10.1016/S0040-4039(01)00628-1
Return to citation in text: [1] -
Danishefsky, S. J.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 3803–3805. doi:10.1021/jo00140a053
Return to citation in text: [1] [2] -
Ichikawa, Y.; Isobe, M.; Konobe, M.; Goto, T. Carbohydr. Res. 1987, 171, 193–199. doi:10.1016/S0008-6215(00)90886-3
Return to citation in text: [1] -
Dawe, R. D.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1981, 1180–1181. doi:10.1039/C39810001180
Return to citation in text: [1] -
Gemmell, N.; Meo, P.; Osborn, H. M. I. Org. Lett. 2003, 5, 1649–1652. doi:10.1021/ol030023t
Return to citation in text: [1] -
Steinhuebel, D. P.; Fleming, J. J.; Du Bois, J. Org. Lett. 2002, 4, 293–295. doi:10.1021/ol010273e
Return to citation in text: [1] -
Hanessian, S.; Pernet, A. G. Adv. Carbohydr. Chem. Biochem. 1976, 33, 111–188. doi:10.1016/S0065-2318(08)60281-4
Return to citation in text: [1] -
Daves, G. D., Jr.; Cheng, C. C. Prog. Med. Chem. 1976, 13, 303–349. doi:10.1016/S0079-6468(08)70141-3
Return to citation in text: [1] -
Poonian, M. S.; Nowoswiat, E. F. J. Org. Chem. 1980, 45, 203–208. doi:10.1021/jo01290a002
Return to citation in text: [1] -
Gupta, A.; Vankar, Y. D. Tetrahedron 2000, 56, 8525–8531. doi:10.1016/S0040-4020(00)00775-4
Return to citation in text: [1] -
Pachamuthu, K.; Vankar, Y. D. J. Org. Chem. 2001, 66, 7511–7513. doi:10.1021/jo0103322
Return to citation in text: [1] -
Agarwal, A.; Rani, S.; Vankar, Y. D. J. Org. Chem. 2004, 69, 6137–6140. doi:10.1021/jo049415j
Return to citation in text: [1] -
Babu, J. L.; Khare, A.; Vankar, Y. D. Molecules 2005, 10, 884–892. doi:10.3390/10080884
Return to citation in text: [1] -
Gupta, P.; Kumari, N.; Agarwal, A.; Vankar, Y. D. Org. Biomol. Chem. 2008, 6, 3948–3956. doi:10.1039/b810654a
Return to citation in text: [1] -
Jayakanthan, K.; Vankar, Y. D. Carbohydr. Res. 2005, 340, 2688–2692. doi:10.1016/j.carres.2005.07.024
Return to citation in text: [1] -
Sridharan, V.; Menéndez, J. C. Chem. Rev. 2010, 110, 3805–3849. doi:10.1021/cr100004p
Return to citation in text: [1] -
Nair, V.; Balagopal, L.; Rajan, R.; Mathew, J. Acc. Chem. Res. 2004, 37, 21–30. doi:10.1021/ar030002z
Return to citation in text: [1] -
Agarwal, A.; Vankar, Y. D. Proc. Indian Natl. Sci. Acad., Part A 2005, 71, 309–326.
Return to citation in text: [1] -
Linker, T.; Sommermann, T.; Kahlenberg, F. J. Am. Chem. Soc. 1997, 119, 9377–9384. doi:10.1021/ja971084n
Return to citation in text: [1] -
Yin, J.; Sommermann, T.; Linker, T. Chem.–Eur. J. 2007, 13, 10152–10167. doi:10.1002/chem.200701151
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lin, H.-C.; Du, W.-P.; Chang, C.-C.; Lin, C.-H. Tetrahedron Lett. 2005, 46, 5071–5076. doi:10.1016/j.tetlet.2005.05.061
Return to citation in text: [1] [2] -
Hosokawa, S.; Kirschbaum, B.; Isobe, M. Tetrahedron Lett. 1998, 39, 1917–1920. doi:10.1016/S0040-4039(98)00047-1
Return to citation in text: [1] [2] [3] -
Brawn, R. A.; Panek, J. S. Org. Lett. 2010, 12, 4624–4627. doi:10.1021/ol1019629
Return to citation in text: [1] [2] [3] [4] [5] -
Kawabata, H.; Kubo, S.; Hayashi, M. Carbohydr. Res. 2001, 333, 153–158. doi:10.1016/S0008-6215(01)00128-8
Return to citation in text: [1] [2] [3] -
Ferrier, R. J.; Zubkov, O. A. Org. React. 2004, 569–736.
Return to citation in text: [1] -
Gómez, A. M.; Lobo, F.; Uriel, C.; López, J. C. Eur. J. Org. Chem. 2013, 7221–7262. doi:10.1002/ejoc.201300798
See for a recent review.
Return to citation in text: [1] -
Wang, L.-X.; Fan, J.-Q.; Lee, Y. C. Tetrahedron Lett. 1996, 37, 1975–1978. doi:10.1016/0040-4039(96)00261-4
Return to citation in text: [1] -
McGarvey, G. J.; Benedum, T. E.; Schmidtmann, F. W. Org. Lett. 2002, 4, 3591–3594. doi:10.1021/ol0264861
Return to citation in text: [1] [2] -
Westermann, B.; Walter, A.; Diedrichs, N. Angew. Chem., Int. Ed. 1999, 38, 3384–3386. doi:10.1002/(SICI)1521-3773(19991115)38:22<3384::AID-ANIE3384>3.0.CO;2-7
Return to citation in text: [1] -
Cipolla, L.; La Ferla, B.; Lay, L.; Peri, F.; Nicotra, F. Tetrahedron: Asymmetry 2000, 11, 295–303. doi:10.1016/S0957-4166(99)00480-2
Return to citation in text: [1] -
Cipolla, L.; Lay, L.; Peri, F.; Nicotra, F. J. Org. Chem. 1997, 62, 6678–6681. doi:10.1021/jo970127f
Return to citation in text: [1] -
Petrušová, M.; BeMiller, J. N.; Petruš, L. Tetrahedron Lett. 1996, 37, 2341–2344. doi:10.1016/0040-4039(96)00280-8
Return to citation in text: [1] -
SanMartin, R.; Tavassoli, B.; Walsh, K. E.; Walter, D. S.; Gallagher, T. Org. Lett. 2000, 2, 4051–4054. doi:10.1021/ol006682c
Return to citation in text: [1] -
Urban, D.; Skrydstrup, T.; Beau, J.-M. Chem. Commun. 1998, 955–956. doi:10.1039/a801196f
Return to citation in text: [1] -
Urban, D.; Skrydstrup, T.; Beau, J.-M. J. Org. Chem. 1998, 63, 2507–2516. doi:10.1021/jo971727h
Return to citation in text: [1] -
Bouvet, V. R.; Ben, R. N. J. Org. Chem. 2006, 71, 3619–3622. doi:10.1021/jo051938j
And references therein.
Return to citation in text: [1] -
Tsuji, J. Palladium Reagents and Catalysts; Wiley: Chichester, 2004; pp 29–35.
Return to citation in text: [1] -
Overman, L. E. J. Am. Chem. Soc. 1976, 98, 2901–2910. doi:10.1021/ja00426a038
Return to citation in text: [1] -
Mercer, G. J.; Yang, J.; McKay, M. J.; Nguyen, H. M. J. Am. Chem. Soc. 2008, 130, 11210–11218. doi:10.1021/ja803378k
Return to citation in text: [1] -
Gupta, P.; Vankar, Y. D. Eur. J. Org. Chem. 2009, 1925–1933. doi:10.1002/ejoc.200801301
Return to citation in text: [1] -
VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973–1976. doi:10.1016/S0040-4039(00)78093-2
Return to citation in text: [1]
| 43. | Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01 |
| 45. | Hosokawa, S.; Kirschbaum, B.; Isobe, M. Tetrahedron Lett. 1998, 39, 1917–1920. doi:10.1016/S0040-4039(98)00047-1 |
| 43. | Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01 |
| 45. | Hosokawa, S.; Kirschbaum, B.; Isobe, M. Tetrahedron Lett. 1998, 39, 1917–1920. doi:10.1016/S0040-4039(98)00047-1 |
| 46. | Brawn, R. A.; Panek, J. S. Org. Lett. 2010, 12, 4624–4627. doi:10.1021/ol1019629 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 46. | Brawn, R. A.; Panek, J. S. Org. Lett. 2010, 12, 4624–4627. doi:10.1021/ol1019629 |
| 47. | Kawabata, H.; Kubo, S.; Hayashi, M. Carbohydr. Res. 2001, 333, 153–158. doi:10.1016/S0008-6215(01)00128-8 |
| 43. | Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01 |
| 43. | Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01 |
| 46. | Brawn, R. A.; Panek, J. S. Org. Lett. 2010, 12, 4624–4627. doi:10.1021/ol1019629 |
| 46. | Brawn, R. A.; Panek, J. S. Org. Lett. 2010, 12, 4624–4627. doi:10.1021/ol1019629 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 47. | Kawabata, H.; Kubo, S.; Hayashi, M. Carbohydr. Res. 2001, 333, 153–158. doi:10.1016/S0008-6215(01)00128-8 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 47. | Kawabata, H.; Kubo, S.; Hayashi, M. Carbohydr. Res. 2001, 333, 153–158. doi:10.1016/S0008-6215(01)00128-8 |
| 43. | Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01 |
| 59. |
Bouvet, V. R.; Ben, R. N. J. Org. Chem. 2006, 71, 3619–3622. doi:10.1021/jo051938j
And references therein. |
| 60. | Tsuji, J. Palladium Reagents and Catalysts; Wiley: Chichester, 2004; pp 29–35. |
| 53. | Cipolla, L.; La Ferla, B.; Lay, L.; Peri, F.; Nicotra, F. Tetrahedron: Asymmetry 2000, 11, 295–303. doi:10.1016/S0957-4166(99)00480-2 |
| 54. | Cipolla, L.; Lay, L.; Peri, F.; Nicotra, F. J. Org. Chem. 1997, 62, 6678–6681. doi:10.1021/jo970127f |
| 55. | Petrušová, M.; BeMiller, J. N.; Petruš, L. Tetrahedron Lett. 1996, 37, 2341–2344. doi:10.1016/0040-4039(96)00280-8 |
| 56. | SanMartin, R.; Tavassoli, B.; Walsh, K. E.; Walter, D. S.; Gallagher, T. Org. Lett. 2000, 2, 4051–4054. doi:10.1021/ol006682c |
| 57. | Urban, D.; Skrydstrup, T.; Beau, J.-M. Chem. Commun. 1998, 955–956. doi:10.1039/a801196f |
| 58. | Urban, D.; Skrydstrup, T.; Beau, J.-M. J. Org. Chem. 1998, 63, 2507–2516. doi:10.1021/jo971727h |
| 51. | McGarvey, G. J.; Benedum, T. E.; Schmidtmann, F. W. Org. Lett. 2002, 4, 3591–3594. doi:10.1021/ol0264861 |
| 52. | Westermann, B.; Walter, A.; Diedrichs, N. Angew. Chem., Int. Ed. 1999, 38, 3384–3386. doi:10.1002/(SICI)1521-3773(19991115)38:22<3384::AID-ANIE3384>3.0.CO;2-7 |
| 48. | Ferrier, R. J.; Zubkov, O. A. Org. React. 2004, 569–736. |
| 49. |
Gómez, A. M.; Lobo, F.; Uriel, C.; López, J. C. Eur. J. Org. Chem. 2013, 7221–7262. doi:10.1002/ejoc.201300798
See for a recent review. |
| 50. | Wang, L.-X.; Fan, J.-Q.; Lee, Y. C. Tetrahedron Lett. 1996, 37, 1975–1978. doi:10.1016/0040-4039(96)00261-4 |
| 51. | McGarvey, G. J.; Benedum, T. E.; Schmidtmann, F. W. Org. Lett. 2002, 4, 3591–3594. doi:10.1021/ol0264861 |
| 64. | VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973–1976. doi:10.1016/S0040-4039(00)78093-2 |
| 61. | Overman, L. E. J. Am. Chem. Soc. 1976, 98, 2901–2910. doi:10.1021/ja00426a038 |
| 62. | Mercer, G. J.; Yang, J.; McKay, M. J.; Nguyen, H. M. J. Am. Chem. Soc. 2008, 130, 11210–11218. doi:10.1021/ja803378k |
| 63. | Gupta, P.; Vankar, Y. D. Eur. J. Org. Chem. 2009, 1925–1933. doi:10.1002/ejoc.200801301 |
| 1. | Zou, W. Curr. Top. Med. Chem. 2005, 5, 1363–1391. doi:10.2174/156802605774642999 |
| 2. | Sutherlin, D. P.; Stark, T. M.; Hughes, R.; Armstrong, R. W. J. Org. Chem. 1996, 61, 8350–8354. doi:10.1021/jo960119j |
| 3. | Weatherman, R. V.; Mortell, K. H.; Chervenak, M.; Kiessling, L. L.; Toone, E. J. Biochemistry 1996, 35, 3619–3624. doi:10.1021/bi951916z |
| 31. | Gupta, A.; Vankar, Y. D. Tetrahedron 2000, 56, 8525–8531. doi:10.1016/S0040-4020(00)00775-4 |
| 32. | Pachamuthu, K.; Vankar, Y. D. J. Org. Chem. 2001, 66, 7511–7513. doi:10.1021/jo0103322 |
| 33. | Agarwal, A.; Rani, S.; Vankar, Y. D. J. Org. Chem. 2004, 69, 6137–6140. doi:10.1021/jo049415j |
| 34. | Babu, J. L.; Khare, A.; Vankar, Y. D. Molecules 2005, 10, 884–892. doi:10.3390/10080884 |
| 35. | Gupta, P.; Kumari, N.; Agarwal, A.; Vankar, Y. D. Org. Biomol. Chem. 2008, 6, 3948–3956. doi:10.1039/b810654a |
| 36. | Jayakanthan, K.; Vankar, Y. D. Carbohydr. Res. 2005, 340, 2688–2692. doi:10.1016/j.carres.2005.07.024 |
| 12. | Dondoni, A.; Marra, A. Chem. Rev. 2000, 100, 4395–4422. doi:10.1021/cr9903003 |
| 13. | Du, Y.; Linhardt, R. J.; Vlahov, I. R. Tetrahedron 1998, 54, 9913–9959. doi:10.1016/S0040-4020(98)00405-0 |
| 14. | Ansari, A. A.; Lahiri, R.; Vankar, Y. D. ARKIVOC 2013, No. ii, 316–362. doi:10.3998/ark.5550190.0014.223 |
| 37. | Sridharan, V.; Menéndez, J. C. Chem. Rev. 2010, 110, 3805–3849. doi:10.1021/cr100004p |
| 38. | Nair, V.; Balagopal, L.; Rajan, R.; Mathew, J. Acc. Chem. Res. 2004, 37, 21–30. doi:10.1021/ar030002z |
| 9. | Fraser-Reid, B. Acc. Chem. Res. 1985, 18, 347–354. doi:10.1021/ar00119a004 |
| 10. | Kirshning, A.; Chen, G.-w.; Dräger, G.; Schuberth, I.; Tietze, L. F. Bioorg. Med. Chem. 2000, 8, 2347–2354. doi:10.1016/S0968-0896(00)00166-8 |
| 11. | Faul, M. M.; Huff, B. E. Chem. Rev. 2000, 100, 2407–2474. doi:10.1021/cr940210s |
| 6. | Kira, K.; Isobe, M. Tetrahedron Lett. 2000, 41, 5951–5955. doi:10.1016/S0040-4039(00)00988-6 |
| 28. | Hanessian, S.; Pernet, A. G. Adv. Carbohydr. Chem. Biochem. 1976, 33, 111–188. doi:10.1016/S0065-2318(08)60281-4 |
| 29. | Daves, G. D., Jr.; Cheng, C. C. Prog. Med. Chem. 1976, 13, 303–349. doi:10.1016/S0079-6468(08)70141-3 |
| 30. | Poonian, M. S.; Nowoswiat, E. F. J. Org. Chem. 1980, 45, 203–208. doi:10.1021/jo01290a002 |
| 4. | Danishefsky, S. J.; DeNinno, S.; Lartey, P. J. Am. Chem. Soc. 1987, 109, 2082–2089. doi:10.1021/ja00241a028 |
| 5. | Paterson, I.; Keown, L. E. Tetrahedron Lett. 1997, 38, 5727–5730. doi:10.1016/S0040-4039(97)01257-4 |
| 6. | Kira, K.; Isobe, M. Tetrahedron Lett. 2000, 41, 5951–5955. doi:10.1016/S0040-4039(00)00988-6 |
| 7. | Gallagher, B. M., Jr.; Zhao, H.; Pesant, M.; Fang, F. G. Tetrahedron Lett. 2005, 46, 923–926. doi:10.1016/j.tetlet.2004.12.056 |
| 8. | Sasaki, M.; Tsubone, K.; Shoji, M.; Oikawa, M.; Shimamoto, K.; Sakai, R. Bioorg. Med. Chem. Lett. 2006, 16, 5784–5787. doi:10.1016/j.bmcl.2006.08.082 |
| 24. | Ichikawa, Y.; Isobe, M.; Konobe, M.; Goto, T. Carbohydr. Res. 1987, 171, 193–199. doi:10.1016/S0008-6215(00)90886-3 |
| 26. | Gemmell, N.; Meo, P.; Osborn, H. M. I. Org. Lett. 2003, 5, 1649–1652. doi:10.1021/ol030023t |
| 23. | Danishefsky, S. J.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 3803–3805. doi:10.1021/jo00140a053 |
| 27. | Steinhuebel, D. P.; Fleming, J. J.; Du Bois, J. Org. Lett. 2002, 4, 293–295. doi:10.1021/ol010273e |
| 18. | De Las Heras, F. G.; Felix, A. S.; Fernández-Resa, P. Tetrahedron 1983, 39, 1617–1620. doi:10.1016/S0040-4020(01)88571-9 |
| 19. | Ghosh, R.; De, D.; Shown, B.; Maiti, S. B. Carbohydr. Res. 1999, 321, 1–3. doi:10.1016/S0008-6215(99)00219-0 |
| 20. | Yadav, J. S.; Reddy, B. V. S.; Chand, P. K. Tetrahedron Lett. 2001, 42, 4057–4059. doi:10.1016/S0040-4039(01)00629-3 |
| 21. | Balamurugan, R.; Koppolu, S. R. Tetrahedron 2009, 65, 8139–8142. doi:10.1016/j.tet.2009.07.087 |
| 22. | Takhi, M.; Rahman, A. A.-H. A.; Schmidt, R. R. Tetrahedron Lett. 2001, 42, 4053–4056. doi:10.1016/S0040-4039(01)00628-1 |
| 5. | Paterson, I.; Keown, L. E. Tetrahedron Lett. 1997, 38, 5727–5730. doi:10.1016/S0040-4039(97)01257-4 |
| 16. | Yadav, J. S.; Satyanarayana, M.; Balanarsaiah, E.; Raghavendra, S. Tetrahedron Lett. 2006, 47, 6095–6098. doi:10.1016/j.tetlet.2006.06.084 |
| 17. | Tiwari, P.; Agnihotri, G.; Misra, A. K. Carbohydr. Res. 2005, 340, 749–752. doi:10.1016/j.carres.2004.12.022 |
| 25. | Dawe, R. D.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1981, 1180–1181. doi:10.1039/C39810001180 |
| 23. | Danishefsky, S. J.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 3803–3805. doi:10.1021/jo00140a053 |
| 39. | Agarwal, A.; Vankar, Y. D. Proc. Indian Natl. Sci. Acad., Part A 2005, 71, 309–326. |
| 40. | Linker, T.; Sommermann, T.; Kahlenberg, F. J. Am. Chem. Soc. 1997, 119, 9377–9384. doi:10.1021/ja971084n |
| 41. | Yin, J.; Sommermann, T.; Linker, T. Chem.–Eur. J. 2007, 13, 10152–10167. doi:10.1002/chem.200701151 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 44. | Lin, H.-C.; Du, W.-P.; Chang, C.-C.; Lin, C.-H. Tetrahedron Lett. 2005, 46, 5071–5076. doi:10.1016/j.tetlet.2005.05.061 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 46. | Brawn, R. A.; Panek, J. S. Org. Lett. 2010, 12, 4624–4627. doi:10.1021/ol1019629 |
| 42. | Yadav, J. S.; Reddy, B. V. S. Synthesis 2002, 511–514. doi:10.1055/s-2002-20968 |
| 43. | Ghosh, R.; Chakraborty, A.; Maiti, S. B. ARKIVOC 2004, xiv, 1–9. doi:10.3998/ark.5550190.0005.e01 |
| 44. | Lin, H.-C.; Du, W.-P.; Chang, C.-C.; Lin, C.-H. Tetrahedron Lett. 2005, 46, 5071–5076. doi:10.1016/j.tetlet.2005.05.061 |
| 45. | Hosokawa, S.; Kirschbaum, B.; Isobe, M. Tetrahedron Lett. 1998, 39, 1917–1920. doi:10.1016/S0040-4039(98)00047-1 |
© 2014 Ansari et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)