
Abstract
New cyclodextrin (CD)-grafted polymers functionalized with water-soluble phosphanes were synthesized in three steps starting from polyNAS. Once characterized by NMR spectroscopy and size-exclusion chromatography, they were used as additives in Rh-catalyzed hydroformylation of 1-hexadecene. The combined supramolecular and coordinating properties of these polymers allowed increasing the catalytic activity of the reaction without affecting the selectivities.
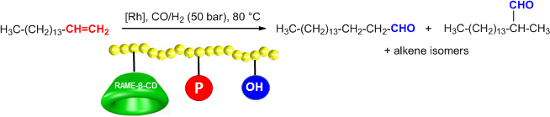
Graphical Abstract
Introduction
Although aqueous organometallic catalysis has been developed long before P. T. Anastas and J. C. Warner set out the foundations of Green Chemistry [1], the very essence of this concept relies on several of the twelve fundamental principles. As such, the use of effective catalysts and water as a solvent greatly contribute to the development of eco-friendly industrial processes. The most impressive example of industrial application that makes use of aqueous organometallic catalysis is undoubtedly the Ruhrchemie/Rhône-Poulenc process which allows conversion of propene mainly into linear butyraldehyde using a rhodium catalyst immobilized in the aqueous phase by coordination of the famous water-soluble ligand TPPTS (trisodium salt of the trisulfonated triphenylphosphane) [2]. However, while propene is partially soluble in water, terminal alkenes containing more than 6 carbon atoms cannot be converted using this process due to obvious insolubility problems. To overcome mass transfer limitations occurring between the catalyst-containing aqueous phase and the substrate-containing organic phase, various solutions have been implemented. For example, other solvents such as supercritical CO2, ionic liquids or fluorous phases have been used to immobilize the organometallic catalyst [3-5]. Co-solvents, surfactants, amphiphilic phosphanes, molecular receptors, polymers or dispersed particles have also been investigated to favour contacts between the aqueous and the organic compartments [6]. We especially developed biphasic catalytic systems in which cyclodextrin (CD) derivatives acted as interfacial additives [7,8]. These torus-like macrorings proved to be appropriate to supramolecularly recognize C8–C10 alkenes within their cavity and convert them into their corresponding aldehydes. Recently, the question arouse about alkenes containing more than 10 carbons. Effective solutions were thus developed using CD-dimers [9], CD-based hydrogels [10,11] and CD-based polymers [12]. In the present study, the concept has been taken a step further. While the CD-based polymer and TPPTS were added separately in the aqueous catalytic solution in our previous study, we synthesized a CD-substituted polymer functionalized with water-soluble phosphanes. The idea was to increase the local concentration of interfacial additive and phosphane-coordinated Rh catalyst at the aqueous/oganic interface to favour the substrate conversion. Herein, we detailed the synthesis and characterisation of this polymer and its catalytic behaviour in Rh-catalyzed hydroformylation of 1-hexadecene.
Results and Discussion
A wide range of CD-based polymers have already been described in the literature [13-18]. To access the expected CD-substituted polymer functionalized with water-soluble phosphanes, a sulfonation step of a commercially available phosphane was first required. 2-(Diphenylphosphino)ethanamine was sulfonated in an oleum/H2SO4 mixture at room temperature over a period of 15 days. Once cold distilled water and trioctylamine (dissolved in chloroform) have been added, phosphane 1 was fractionally collected using a diluted solution of NaOH. The fractionalization allowed for a step-by-step removal of the phosphane oxide. After work-up, the sulfonated phosphane 1 (Scheme 1) was isolated in 55% yield as white crystals. The meta-sulfonation on the aromatic rings was confirmed by NMR. The COSY spectrum (Supporting Information File 1) was especially indicative of the meta-substitution. Indeed, H4 (Scheme 1) did not correlate with any other proton and appeared as an upfield shifted signal due to the electro-withdrawing effect of both the sulfonate group and the phosphorus. Moreover, H4 was detected as a doublet due to the 3J scalar coupling with the phosphorus. H6 also appeared as a doublet (due to the coupling with H7) but its resonance was a little bit more shielded than H4 as H6 did not benefit from the cumulative electron-withdrawing effects of both the sulfonate and the phosphorus. Contrary to H4, H6 showed cross-peaks with H7 and H8 whose doublets of doublets overlapped in the 1H spectrum and strongly correlated in the COSY spectrum.
Once the sulfonated phosphine synthesized, a polyNAS sample (DP n = 45) [10] reacted with the mono-amino randomly methylated β-CD (RAME-β-CD-NH2) [10] in DMF at 60 °C for 48 h under vigorous stirring (1500 rounds per minutes). The CD-substituted polymer then reacted with 1 in DMF at 60 °C for 24 h. Two different CD/1 ratios have been considered for comparison with systems where the water-soluble phosphane and the polymer were added separately. Depending on the amount of 1, CD-substituted phosphane-functionalized polymers 2b and 2c (Scheme 1) were obtained. To ensure the water solubility of the resulting polymers and remove the remaining succinimide groups from the polymer chains, a subsequent reaction of 2b and 2c with aminoethanol for 12 h in the same experimental conditions led to the trisubstituted polymers 3b and 3c respectively (Scheme 1) in 85% yield as pale yellow powders.
![[1860-5397-10-276-i1]](/bjoc/content/inline/1860-5397-10-276-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of CD-substituted polymers 3a, 3b and 3c.
Scheme 1: Synthesis of CD-substituted polymers 3a, 3b and 3c.
Polymers 3a, 3b and 3c were characterized by 1H NMR both in D2O and DMF-d7. Integration of the polymer chain protons and the H-1 CD protons showed that 33% CDs were grafted onto the polymer chains of 3b and 3c. Integration of the H-1 CD protons and the aromatic protons of 1 in D2O allowed confirming the CD/phosphane ratio onto polymers 3b and 3c (Figure 1).
![[1860-5397-10-276-1]](/bjoc/content/figures/1860-5397-10-276-1.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectrum of 3c in D2O at 25 °C.
Figure 1: 1H NMR spectrum of 3c in D2O at 25 °C.
16.5% and 33% phosphanes were also incorporated onto 3b and 3c, respectively. The purity of the phosphane-containing polymers 3b and 3c was subsequently confirmed by 31P NMR. Spectra of 3b (Figure 2a) and 3c recorded in DMF-d7 clearly revealed a broad 31P resonance (2 ppm wide) indicative of different chemical environments of the phosphorus. The broadening of the 31P NMR signal was even more marked in D2O (Figure 2b). Several factors might be involved and be responsible for such disparity in phosphorus resonances. First, phosphanes could be included into CD cavities or not (“free” phosphanes). Moreover, as these polymers were mixtures of compounds, polymers differing from their chain length could have different chemical shifts.
![[1860-5397-10-276-2]](/bjoc/content/figures/1860-5397-10-276-2.png?scale=1.4&max-width=1024&background=FFFFFF)
Figure 2: 31P NMR spectrum of 3b in a) DMF-d7 at 25 °C and b) D2O at 25 °C.
Figure 2: 31P NMR spectrum of 3b in a) DMF-d7 at 25 °C and b) D2O at 25 °C.
In addition, each polymer chain contained three asymmetric carbon atoms, and therefore many diastereomers were present, all of them resulting in a slightly different 31P chemical shift. However, a simplification of the 31P spectra occurred at 60 °C. Only three peaks could be observed at −20.88, −21.85 and −21.98 ppm (Figure 3). We believed that the increase in temperature disfavoured the CD/phosphane inclusion complexes, thus greatly reducing the number of possible chemical environments. The remaining peaks could be attributed to three different diastereoisomers.
Note that, whatever the solvent, no resonance corresponding to 1 could be observed indicative of the total grafting of 1 onto the polymer chains (see Figure S5, (Supporting Information File 1) for details on a physical mixture of 1 and 3b in DMF-d7).
![[1860-5397-10-276-3]](/bjoc/content/figures/1860-5397-10-276-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectrum of 3b (10 mM) in D2O at 25 °C (below) and 60 °C (above).
Figure 3: 1H NMR spectrum of 3b (10 mM) in D2O at 25 °C (below) and 60 °C (above).
To get further information on the exact conformations of 3b and 3c in D2O, 2D T-ROESY measurements have been performed to reveal potential interactions between the phosphane aromatic protons and the inner CD protons H-3 and H-5. None of the spectra revealed any correlation between these protons. Indeed, as the peaks were wide (as usually observed for polymers), the NMR signals were drowned in the background noise (under the detection limit). However, an inclusion of the phosphane moiety within the CD cavity could not be excluded at this stage as a 2D T-ROESY spectrum of a 1/5 mixture of RAME-β-CD and 1 clearly indicated cross-peaks between 1 and the inner protons of RAME-β-CD (see Supporting Information File 1). Knowing that sulfonated phosphanes could by partially included into CD cavities, DOSY experiments were then carried out to get insights on the intramolecular interactions existing between the grafted phosphanes and the grafted CDs of 3b. To evaluate the strength of the interaction, RAME-β-CD was added as a competitor in the solution (Supporting Information File 1). In DMSO-d6, RAME-β-CD and 3b showed different diffusion coefficient (D = 1.78.10−10 and 8.05.10−11 m2/s, respectively) when they were analysed in two different NMR tubes. However, when they were mixed together, only one diffusion coefficient could be measured whatever the additional amount of RAME-β-CD, indicative of the interaction of RAME-β-CD with the polymer. Two different diffusion coefficients would have been observed otherwise. Similar behaviours were observed in D2O and CDCl3. Accordingly, the intramolecular interaction between the grafted phosphanes and the grafted CDs was not very strong as the recognition process between them could be easily displaced by a competitor.
The number average molecular weights Mn and dispersity Đ of 3b and 3c were determined by size-exclusion chromatography (SEC). Mn were 13100 and 13700 g·mol−1 and Đ were 1.25 and 1.22 for 3b and 3c, respectively (Figure 4). For comparison (see below the catalytic experiments); a CD-substituted polymer 3a that did not contain any phosphane has also been synthesized (Mn = 11400 g·mol−1, Đ = 1.23).
![[1860-5397-10-276-4]](/bjoc/content/figures/1860-5397-10-276-4.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 4: Size-exclusion chromatography of polyNAS and polymers 3a, 3b and 3c.
Figure 4: Size-exclusion chromatography of polyNAS and polymers 3a, 3b and 3c.
The catalytic performances of polymers 3a, 3b and 3c have been evaluated in a Rh-catalyzed hydroformylation of 1-hexadecene at 80 °C under 50 bar CO/H2 in a 1/1 stoichiometric ratio. The results were gathered in Table 1.
Table 1: Rh-catalyzed hydroformylation of 1-hexadecene.a
| Entry | Polymer/phosphane |
Conversion
(%) |
Chemoselectivity
(% aldehydes) |
Regioselectivity (l/b ratio) |
|---|---|---|---|---|
| 1 | 1 | 3 | 28 | 2.7 |
| 2 | 1 + RAME-β-CD | 34 | 63 | 1.6 |
| 3 | 1 + 3a | 81 | 61 | 1.8 |
| 4 | 3b | 98 | 62 | 1.9 |
| 5 | 3c | 8 | 57 | 1.7 |
aConditions: Rh:substrate ratio: 1/140, Rh:phosphane ratio: 1/0 (3a), 1/5 (3b) or 1/10 (3c), 80 °C, 50 bar CO/H2, reaction time = 1 h. Conversions and selectivities were determined by GC and 1H NMR.
Compared to 1 all the synthesized CD-substituted polymers led to better activities and aldehyde selectivity. A slight decrease in the regioselectivity was also observed and corroborated previous studies dealing with the role of the CD at the aqueous/organic interface [10]. Effects of the phosphanes grafted onto the polymer backbone on the catalytic performances were much more intricate. Indeed, the conversion was greatly dependent upon the CD:phosphane ratio. A significant increase in the conversion (17%) was observed when comparing 3a and 3b (81 vs 98% conversion, respectively). These results highlighted the benefit resulting from the phosphane grafting onto the polymer backbone. Indeed, as the phosphane coordinated the metallic species, the catalyst and the CD were in close vicinity. Hence, when the substrate was supramolecularly recognized by the CD cavity, it could then rapidly react with the catalyst to be hydroformylated. The closeness of the protagonists (CD, catalyst and substrate) was clearly determining in this process. However, while a 2:1 CD/phosphane ratio had a positive effect on the catalytic activity, only 8% 1-hexadecene were converted using a stoichiometric CD/phosphane ratio. The explanation of such as difference between the 2:1 and 1:1 ratios lies in the supramolecular interaction existing between the CD cavity and 1. Indeed, we previously demonstrated that, concurrently to the substrate inclusion, 1 could also be included within the CD cavities. While 3b still had available CD cavities (twice more CDs than phosphanes) to recognize the substrate, the CD cavities of 3c (equal number of CDs and phosphanes) were mainly occupied by the grafted phosphanes and could not efficiently recognize the substrate at the aqueous/organic interface. Excess CDs regarding 1 was thus required for the polymer to be effective in catalytic conditions.
Conclusion
To sum up, we synthesized a new CD-grafted polymer functionalized with water-soluble phosphane moieties which acted as a very effective tool in the aqueous Rh-catalyzed hydroformylation of 1-hexadecene. Both the supramolecular properties of the CD and the coordination ability of the phosphane were combined into the same molecular object. During the course of the reaction, the closeness of the three main protagonists (substrate, CD, phosphane) led to a significant increase in the conversion compared to a catalytic system where the CD and the phosphane were not grafted on the same polymer chain. These interfacial polymer-based additives paved the way to the development of new catalytic systems for the conversion of very hydrophobic substrates.
Experimental
2-(Bis(m-sulfonatophenyl)phosphino)ethanamine, sodium salt (1)
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-276-i2.svg?max-width=637&scale=1.18182)
In a Schlenk tube were stirred 3.4 mL H2SO4 and 0.85 mL oleum at room temperature for 5 min. Once the solution has been degassed under nitrogen, degassed 2-(diphenylphosphino)ethanamine (2.0 g, 8.8 mmol) were then canulated on the acidic solution previously cooled in an ice bath. 7.1 mL oleum (4.4 equiv per phenyl moiety) were then added under nitrogen. The resulting solution was stirred at 800 rpm. Brought back to room temperature, the solution was stirred for 15 days. The Schlenk tube was then placed in an ice bath and 43 mL distilled water were added dropwise. The resulting solution was diluted in 200 mL cold water. 9.2 g (26 mmol) trioctylamine were added along with 45 mL chloroform. After the recovered organic phase has been washed with water, phosphane 1 was fractionally collected using a diluted solution of NaOH (400 µL NaOH 2 N in 4 mL water). Water was evaporated in a rotary evaporizer. 2.1 g of 1 were isolated as white crystals in 55% yield. 31P NMR (121.49 MHz, 21 °C, D2O) δ 36.27 (P=O, 5%), −21.48 (P, 95%); 1H NMR (300.13 MHz, 21 °C, D2O) δ 7.88 (d, J = 7.2 Hz, 2H, Hp), 7.78 (d, J = 7.8 Hz, 2H, Ho’), 7.47–7.59 (m, 4H, Hm and Ho), 3.03 (m, 2H, CH2(1)), 2.52 (m, 2H, CH2(2)); 13C{1H} NMR (75.5 MHz, 21 °C, D2O) δ 142.7 (d, 3J(P,C) = 6.20 Hz, C5), 136.8 (d, 1J(P,C) = 11.93 Hz, C7), 135.7 (d, 2J(P,C) = 19.49 Hz, C8), 129.54 (d, 3J(P,C) = 6.50 Hz, C3), 128.84 (d, 2J(P,C) = 20.21 Hz, C4), 126.48 (s, C6), 36.54 (d, 2J(P,C) = 25.86 Hz, C2), 24.74 (d, 1J(P,C) = 12.93 Hz, C1).
PolyNAS functionalization
In a Schlenk tube were degassed 1.5 g (1.1 mmol) RAME-β-CD-NH2 and 558 mg (3.3 mmol) polyNAS. 20 mL degassed anhydrous DMF were canulated into the tube. The solution was vigorously stirred at 1500 rpm under nitrogen at 60 °C for 48 h. the solution was then canulated into another Schlenk tube containing 238 mg (0.55 mmol) or 476 mg (1.1 mmol) phosphane 1 to form polymers 2b and 2c, respectively. The resulting solution was stirred at 1500 rpm under nitrogen at 60 °C for another 24 h. Eventually, 403 mg (6.6 mmol) degassed aminoethanol were added dropwise and the solution was stirred under nitrogen at 60 °C for 12 h. Polymers 3a, 3b and 3c were precipitated in a degassed acetone/Et2O (1:1) mixture. Pale yellowish powders were isolated in 82–85% yield.
Polymer 3a: 1H NMR (300.13 MHz, 21 °C, D2O) δ 5.06 (br. s, 3.5H), 4.87 (br. s, 3.5H), 3.75–3.60 (m, 12.3H), 3.60– 3.53 (m, 32.4H), 3.41 (br. s, 10.7H), 3.39 (br. s, 5.1H), 3.30–3.10 (m, 22.1 H), 2.31 (m, 4H, CH2), 2.22 (m, 4H, CH2), 1.53 (br. s, 3H), 0.95 (br. s, 6H); J-MOD NMR (75.5 MHz, 21 °C, D2O) δ 171.5, 168.8, 101.1, 98.7, 82.9–80.0, 77.4, 60.4, 58.8, 46.0, 41.8, 31.0, 25.5, 11.6.
Polymer 3b: 31P NMR (121.49 MHz, 21 °C, D2O) δ 37.40–37.00 (br m, P=O, 6%), −21.84 (br m, P, 94%); 1H NMR (300.13 MHz, 21 °C, D2O) δ 7.91–7.25 (br. m, 4H), 5.08 (br. s, 3.5H), 4.89 (br. s, 3.5H), 3.75–3.56 (m, 12.3H), 3.58–3.52 (m, 32.4H), 3.44 (br. s, 10.7H), 3.42 (br. s, 5.1H), 3.30–3.10 (m, 22.1 H), 2.94 (m, 1H, CH2), 2.49 (m, 1H, CH2), 2.33 (m, 3H, CH2), 2.24 (m, 3H, CH2), 1.57 (br. s, 3H), 0.97 (br. s, 6H); J-MOD NMR (75.5 MHz, 21 °C, D2O) δ 171.7, 168.7, 165.5, 162.6, 101.4, 98.7, 82.8–80.0, 77.7, 60.5, 58.9, 46.0, 42.0, 31.2, 28.9, 25.8, 11.7.
Polymer 3c: 31P NMR (121.49 MHz, 21 °C, D2O) δ 37.40–37.06 (br m, P=O, 6%), −21.81 (br m, P, 94%); 1H NMR (300.13 MHz, 21 °C, D2O) δ 7.96–7.30 (br. m, 8H), 5.07 (br. s, 3.5H), 4.86 (br. s, 3.5H), 3.74–3.61 (m, 12.3H), 3.59–3.55 (m, 32.4H), 3.46 (br. s, 10.7H), 3.38 (br. s, 5.1H), 3.31–3.13 (m, 22.1 H), 2.92 (m, 2H, CH2), 2.46 (m, 2H, CH2), 2.34 (m, 2H, CH2), 2.24 (m, 2H, CH2), 1.51 (br. s, 3H), 0.96 (br. s, 6H); J-MOD NMR (75.5 MHz, 21 °C, D2O) δ 171.6, 168.9, 165.3, 162.8, 101.4, 98.9, 82.6–80.1, 77.6, 60.3, 58.6, 46.1, 41.9, 31.0, 28.8, 25.7, 11.9.
Catalytic experiments
Aqueous Rh-catalyzed hydroformylation: A mixture of Rh(CO)2(acac) (3 mg, 0.012 mmol. 1 equiv), alkene (1.63 mmol, 140 equiv), 1 (25 mg, 0.058 mmol, 5 equiv) and CDs (0.12 mmol, 10 equiv) or polymer 3a, 3b or 3c (calculated for 10 equiv CDs) dissolved in water (6 mL) was degassed by three freeze-pump-thaw cycles and introduced in a previously purged autoclave. Once a temperature of 80 °C has been reached, the autoclave was pressurized under CO/H2 pressure (50 bar) and the solution was vigorously stirred (1500 rpm). When the reaction was over, the apparatus was allowed to cool to room temperature and depressurized. The organic phase was extracted using diethyl ether. After evaporation of diethyl ether under vacuum, the product was analyzed by GC and by 1H and 13C NMR experiments. All runs have been performed at least twice in order to ensure reproducibility.
Supporting Information
| Supporting Information File 1: NMR spectra. | ||
| Format: PDF | Size: 757.5 KB | Download |
References
-
Anastas, P. T.; Warner, J. C., Eds. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998.
Return to citation in text: [1] -
Kohlpaintner, C. W.; Fischer, R. W.; Cornils, B. Appl. Catal., A 2001, 221, 219–225. doi:10.1016/S0926-860X(01)00791-8
Return to citation in text: [1] -
Koch, T. J.; Desset, S. L.; Leitner, W. Green Chem. 2010, 12, 1719–1721. doi:10.1039/c0gc00299b
Return to citation in text: [1] -
Desset, S. L.; Reader, S. W.; Cole-Hamilton, D. J. Green Chem. 2009, 11, 630–637. doi:10.1039/b822139a
Return to citation in text: [1] -
Zhao, X.; He, D.; Mika, L.; Horváth, I. In Fluorous Chemistry; Horváth, I. T., Ed.; Springer: Berlin/Heidelberg, 2012; Vol. 308, pp 275–289.
Return to citation in text: [1] -
Obrecht, L.; Kamer, P. C. J.; Laan, W. Catal. Sci. Technol. 2013, 3, 541–551. doi:10.1039/c2cy20538f
Return to citation in text: [1] -
Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Catal. Sci. Technol. 2014, 4, 1899–1908. doi:10.1039/c4cy00005f
Return to citation in text: [1] -
Hapiot, F.; Leclercq, L.; Azaroual, N.; Fourmentin, S.; Tilloy, S.; Monflier, E. Curr. Org. Synth. 2008, 5, 162–172. doi:10.2174/157017908784221585
Return to citation in text: [1] -
Six, N.; Menuel, S.; Bricout, H.; Hapiot, F.; Monflier, E. Adv. Synth. Catal. 2010, 352, 1467–1475. doi:10.1002/adsc.201000027
Return to citation in text: [1] -
Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. ACS Catal. 2013, 3, 1618–1621. doi:10.1021/cs4002282
Return to citation in text: [1] [2] [3] [4] -
Potier, J.; Menuel, S.; Monflier, E.; Hapiot, F. ACS Catal. 2014, 4, 2342–2346. doi:10.1021/cs5004883
Return to citation in text: [1] -
Potier, J.; Menuel, S.; Fournier, D.; Fourmentin, S.; Woisel, P.; Hapiot, F.; Monflier, E.; Hapiot, F. ACS Catal. 2012, 2, 1417–1420. doi:10.1021/cs300254t
Return to citation in text: [1] -
Ailincai, D.; Ritter, H. Beilstein J. Nanotechnol. 2014, 5, 651–657. doi:10.3762/bjnano.5.76
Return to citation in text: [1] -
Böhm, I.; Strotmann, F.; Koopmans, C.; Wolf, I.; Galla, H.-J.; Ritter, H. Macromol. Biosci. 2012, 12, 432–437. doi:10.1002/mabi.201100357
Return to citation in text: [1] -
Jazkewitsch, O.; Ritter, H. Macromolecules 2011, 44, 375–382. doi:10.1021/ma102456n
Return to citation in text: [1] -
Zou, C.; Zhao, P.; Ge, J.; Lei, Y.; Luo, P. Carbohydr. Polym. 2012, 87, 607–613. doi:10.1016/j.carbpol.2011.08.031
Return to citation in text: [1] -
Ohashi, H.; Abe, T.; Tamaki, T.; Yamaguchi, T. Macromolecules 2012, 45, 9742–9750. doi:10.1021/ma3018603
Return to citation in text: [1] -
Bertrand, A.; Stenzel, M.; Fleury, E.; Bernard, J. Polym. Chem. 2012, 3, 377–383. doi:10.1039/c1py00478f
Return to citation in text: [1]
| 1. | Anastas, P. T.; Warner, J. C., Eds. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. |
| 7. | Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Catal. Sci. Technol. 2014, 4, 1899–1908. doi:10.1039/c4cy00005f |
| 8. | Hapiot, F.; Leclercq, L.; Azaroual, N.; Fourmentin, S.; Tilloy, S.; Monflier, E. Curr. Org. Synth. 2008, 5, 162–172. doi:10.2174/157017908784221585 |
| 6. | Obrecht, L.; Kamer, P. C. J.; Laan, W. Catal. Sci. Technol. 2013, 3, 541–551. doi:10.1039/c2cy20538f |
| 3. | Koch, T. J.; Desset, S. L.; Leitner, W. Green Chem. 2010, 12, 1719–1721. doi:10.1039/c0gc00299b |
| 4. | Desset, S. L.; Reader, S. W.; Cole-Hamilton, D. J. Green Chem. 2009, 11, 630–637. doi:10.1039/b822139a |
| 5. | Zhao, X.; He, D.; Mika, L.; Horváth, I. In Fluorous Chemistry; Horváth, I. T., Ed.; Springer: Berlin/Heidelberg, 2012; Vol. 308, pp 275–289. |
| 2. | Kohlpaintner, C. W.; Fischer, R. W.; Cornils, B. Appl. Catal., A 2001, 221, 219–225. doi:10.1016/S0926-860X(01)00791-8 |
| 13. | Ailincai, D.; Ritter, H. Beilstein J. Nanotechnol. 2014, 5, 651–657. doi:10.3762/bjnano.5.76 |
| 14. | Böhm, I.; Strotmann, F.; Koopmans, C.; Wolf, I.; Galla, H.-J.; Ritter, H. Macromol. Biosci. 2012, 12, 432–437. doi:10.1002/mabi.201100357 |
| 15. | Jazkewitsch, O.; Ritter, H. Macromolecules 2011, 44, 375–382. doi:10.1021/ma102456n |
| 16. | Zou, C.; Zhao, P.; Ge, J.; Lei, Y.; Luo, P. Carbohydr. Polym. 2012, 87, 607–613. doi:10.1016/j.carbpol.2011.08.031 |
| 17. | Ohashi, H.; Abe, T.; Tamaki, T.; Yamaguchi, T. Macromolecules 2012, 45, 9742–9750. doi:10.1021/ma3018603 |
| 18. | Bertrand, A.; Stenzel, M.; Fleury, E.; Bernard, J. Polym. Chem. 2012, 3, 377–383. doi:10.1039/c1py00478f |
| 10. | Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. ACS Catal. 2013, 3, 1618–1621. doi:10.1021/cs4002282 |
| 12. | Potier, J.; Menuel, S.; Fournier, D.; Fourmentin, S.; Woisel, P.; Hapiot, F.; Monflier, E.; Hapiot, F. ACS Catal. 2012, 2, 1417–1420. doi:10.1021/cs300254t |
| 10. | Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. ACS Catal. 2013, 3, 1618–1621. doi:10.1021/cs4002282 |
| 10. | Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. ACS Catal. 2013, 3, 1618–1621. doi:10.1021/cs4002282 |
| 11. | Potier, J.; Menuel, S.; Monflier, E.; Hapiot, F. ACS Catal. 2014, 4, 2342–2346. doi:10.1021/cs5004883 |
| 9. | Six, N.; Menuel, S.; Bricout, H.; Hapiot, F.; Monflier, E. Adv. Synth. Catal. 2010, 352, 1467–1475. doi:10.1002/adsc.201000027 |
| 10. | Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. ACS Catal. 2013, 3, 1618–1621. doi:10.1021/cs4002282 |
© 2014 Potier et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
