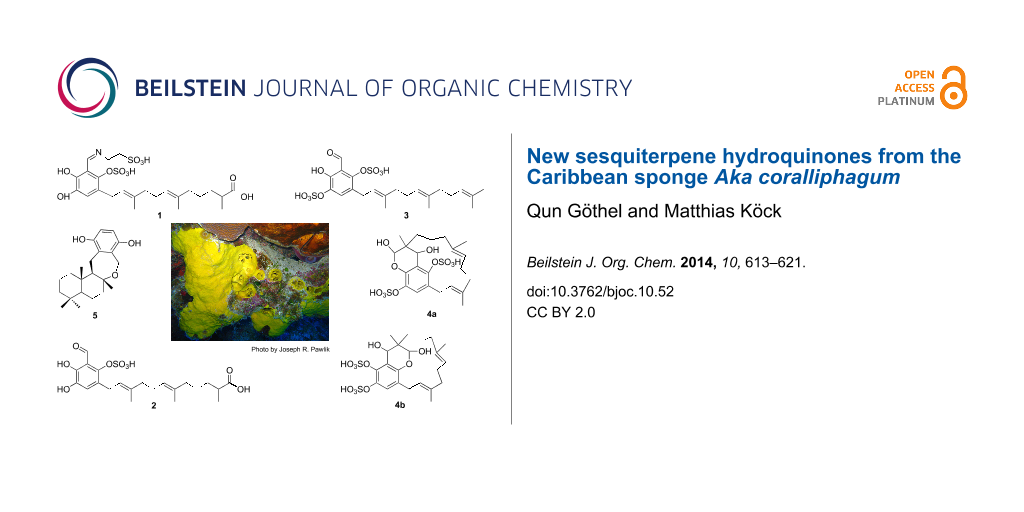
Abstract
Four new sulfated sesquiterpene hydroquinones siphonodictyals E1–E4 (1–4) and cyclosiphonodictyol A (5) were isolated from a sample of the Caribbean sponge Aka coralliphagum collected off the coast of San Salvador in the Bahamas. The structures of the new compounds were elucidated on the basis of mass spectrometric and NMR spectroscopic analysis. Compounds 1–4 are derivatives of siphonodictyal E (9). Siphonodictyal E4 (4) exhibited mild antiproliferation activity against L929 mouse fibroblast, KB-31 epidermoid carcinoma, and MCF-7 breast cancer cell lines, while siphondictyal E3 (3) and cyclosiphonodictyol A (5) showed moderate activity against Gram-positive bacteria.

Graphical Abstract
Introduction
Aka coralliphagum (Siphonodictyon coralliphagum) is known to have four distinct morphological forms: forma typica, f. tubulosa, f. obruta, and f. incrustans [1]. This sponge has the ability to burrow into live coral heads, leaving only the oscular chimney protruding (typica) or the flat crusts (incrustans) exposed. The oscular chimneys or the flat crusts are encircled by a so-called "dead zone" which protects the sponge from overgrowth [2]. Sullivan and Faulkner have proposed that the coral polyps are killed by the viscous mucus containing the toxic secondary metabolites which are produced by the sponge tissue [3]. This ecological observation inspired the chemical investigation of this sponge by different research groups. Thus many secondary metabolites have been reported, such as the sesquiterpene phenolic aldehydes, siphonodictyals B, C, D, E, the monosulfated siphonodictyols G and H, the disulfated siphonodictyal B3, and siphonodictyoic acid [2,4,5]. The sponge under our investigation is of the growth form incrustans, which was collected off the coast of San Salvador in the Bahamas. Fractionation of the aqueous n-BuOH extract yielded the new compounds siphonodictyals E1–E4 (1–4) and cyclosiphonodictyol A (5). In this article, we describe the isolation, structural elucidation, and bioactivity of compounds 1–5 from the Aka growth form incrustans, as well as discuss the biosynthetic pathway of related compounds.
Results and Discussion
The aqueous n-BuOH extract from Aka coralliphagum was subjected to solvent partitioning, followed by gel chromatography and reversed phase (C18) HPLC to yield siphonodictyals E1–E4 (1–4) and cyclosiphonodictyol A (5) (Figure 1). The structure elucidation was based on 1D and 2D NMR as well as HRMS-ESI(−) experiments. The 1H and 13C NMR chemical shifts for the five new compounds are given in Table 1.
![[1860-5397-10-52-1]](/bjoc/content/figures/1860-5397-10-52-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of the new compounds siphonodictyals E1–E4 (1–4) and cyclosiphonodictyol A (5) isolated from the sponge Aka coralliphagum. Since the structure of siphonodictyal E4 (4) could not be unambiguously assigned, the two possible constitutional isomers (4a and 4b) are shown.
Figure 1: Structures of the new compounds siphonodictyals E1–E4 (1–4) and cyclosiphonodictyol A (5) isolated ...
Table 1: NMR data of compounds 1–5 (δC 150 MHz, δH 600 MHz in DMSO-d6).a
| No. | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 177.8 | – | 177.5 | – | 25.5 | 1.62 (s) | 96.2 | 5.42 (s) | 39.4 | 0.76 (m) |
| 1.91 (m) | ||||||||||
| 2 | 38.7 | 2.28 (m) | 38.6 | 2.28 (m) | 130.6 | – | 37.3 | – | 18.2 | 1.38 (m) |
| 1.60 (m) | ||||||||||
| 3 | 32.9 | 1.25 (m) | 32.8 | 1.25 (m) | 124.2 | 5.05 (t, 7.1) | 31.4 | 1.03 (m) | 41.6 | 1.08 (dt, 4.0, 13.5) |
| 1.47 (m) | 1.47 (m) | 1.13 (m) | 1.33 (m) | |||||||
| 4 | 24.9 | 1.32 (m) | 24.9 | 1.31 (m) | 26.3 | 2.00 (m) | 22.1 | 0.56 (m) | 33.1 | – |
| 1.11 (m) | ||||||||||
| 5 | 38.9 | 1.91 (m) | 39.4 | 1.91 (t, 6.9) | 39.3 | 1.92 (m) | 39.3 | 1.28 (m) | 55.4 | 0.83 (m) |
| 1.75 (m) | ||||||||||
| 6 | 134.4 | – | 134.4 | – | 134.4 | – | 134.9 | – | 19.8 | 1.58 (m) |
| 1.60 (m) | ||||||||||
| 7 | 124.0 | 5.09 (t, 6.5) | 124.0 | 5.09 (t, 6.8) | 124.0 | 5.09 (t, 7.1) | 121.7 | 4.52 (t, 5.9) | 40.1 | 1.43 (m) |
| 1.60 (m) | ||||||||||
| 8 | 26.2 | 2.06 (m) | 26.2 | 2.06 (m) | 26.2 | 2.06 (m) | 23.8 | 1.89 (m) | 78.5 | – |
| 2.01 (m) | ||||||||||
| 9 | 40.1 | 1.98 (m) | 39.8 | 1.99 (t, 7.2) | 40.1 | 1.97 (m) | 39.0 | 1.92 (m) | 58.1 | 1.32 (m) |
| 10 | 134.5 | – | 135.4 | – | 135.5 | – | 131.9 | – | 38.2 | – |
| 11 | 123.7 | 5.19 (t, 7.2) | 122.9 | 5.21 (t, 7.2) | 122.6 | 5.21 (t, 7.0) | 125.7 | 5.10 (t, 7.9) | 33.1 | 0.83 (s) |
| 12 | 27.2 | 3.25 (d, 3.4) | 26.9 | 3.33b,c | 27.1 | 3.35b,c | 29.8 | 2.58 (dd, 6.8, 12.6) | 21.2 | 0.78 (s) |
| 3.99 (dd, 9.6, 12.4) | ||||||||||
| 13 | 17.0 | 1.01 (d, 6.6) | 17.0 | 1.01 (d, 6.9) | 17.6 | 1.54 (s) | 19.9 | 1.04 (s) | 21.6 | 1.31 (s) |
| 14 | 15.7 | 1.53 (s) | 15.6 | 1.53 (s) | 16.0 | 1.55 (s) | 17.2 | 1.36 (s) | 15.3 | 0.79 (s) |
| 15 | 15.9 | 1.65 (s) | 15.9 | 1.66 (s) | 15.8 | 1.66 (s) | 15.1 | 1.62 (s) | 20.8 | 2.33 (dd, 9.6, 15.5) |
| 3.06 (d, 15.5) | ||||||||||
| 16 | 120.5 | – | 125.9 | – | 125.5 | – | 126.5 | – | 129.9 | – |
| 17 | 117.7 | 6.50 (s) | 122.8 | 6.84 (s) | 129.5 | 7.48 (s) | 120.8 | 7.19 (s) | 146.4 | – |
| 18 | 144.0 | – | 141.8 | – | 137.7 | – | 137.2 | – | 113.7 | 6.45 (d, 8.6) |
| 19 | 156.4c | – | 147.8 | – | 151.3 | – | 143.2 | – | 112.4 | 6.38 (d, 8.6) |
| 20 | 110.9c | – | 115.5 | – | 115.9 | – | 120.3 | – | 146.5 | – |
| 21 | 141.8c | – | 144.5 | – | 148.2 | – | 142.9 | – | 127.7 | – |
| 22 | 164.6 | 8.67 (s) | 197.2 | 10.10 (s) | 196.6 | 10.11 (s) | 65.3 | 4.44 (s) | 56.7 | 4.50 (d, 15.0) |
| 4.64 (d, 15.0) | ||||||||||
| 23 | 51.2d | 3.83 (t, 6.5) | ||||||||
| 24 | 51.2d | 2.82 (t, 6.2) | ||||||||
| 1-OH | 7.07 (bs) | |||||||||
| 18-OH | 9.09 | |||||||||
| 19-OH | 11.37 | |||||||||
| 22-OH | 5.09 (bs) | |||||||||
aδ are given in ppm (reference: DMSO-d6, 2.50 ppm, 39.52 ppm), coupling constants are given in Hz; assignments were confirmed by 2D NMR experiments (COSY, HSQC, HMBC). bOverlap with water peak. cChemical shifts were obtained from HMBC spectra. dOverlap with each other.
Siphonodictyal E1 (1) was identified by NMR data and its accurate mass of 598.1338 [M + Na − 2H]− which indicated the molecular formula C24H35NO11S2. Comparison of the NMR data of 1 and siphonodictyal E (9, Figure 2) [2] revealed the presence of the identical sesquiterpene acyclic chain with one of the terminal methyl groups oxidized to a carboxylic acid. Moreover, the diagnostic values for C-14 (δ 15.7) and C-15 (δ 15.9) confirmed a common 6E, 10E double-bond geometry [6,7]. The NMR spectral data also suggested a similar aromatic ring system as in siphonodictyal E (9). Moreover, the 1H,1H-COSY correlation between the two methylene groups CH2-23 and CH2-24 as well as the 1H,13C-HMBC correlations from H-22 (δ 8.67) to C-18 (δ 144.0), C-19 (δ 156.4), C-20 (δ 110.9), C-21 (δ 141.8), and C-23 (δ 51.2) further indicated that an iminoethanesulfonic acid is connected to the aromatic ring system in 1 (Figure 3), as in siphonodictyal B1 (6, Figure 2) [5].
![[1860-5397-10-52-2]](/bjoc/content/figures/1860-5397-10-52-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of the related known compounds siphonodictyal B1 (6), siphonodictyal B2 (7), siphonodictyal B3 (8), and siphonodictyal E (9) from the sponge Aka coralliphagum.
Figure 2: Structures of the related known compounds siphonodictyal B1 (6), siphonodictyal B2 (7), siphonodict...
![[1860-5397-10-52-3]](/bjoc/content/figures/1860-5397-10-52-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected 1H,13C-HMBC correlations (H → C) and 1H,1H-COSY correlation (bold line) observed for siphonodictyal E1 (1).
Figure 3: Selected 1H,13C-HMBC correlations (H → C) and 1H,1H-COSY correlation (bold line) observed for sipho...
Examination of the NMR data for siphonodictyal E2 (2) revealed a high degree of similarity with the data reported for siphonodictyal E (9). However, the molecular ion peak of compound 2 was shown at m/z 469.1534 ([M − H]− C22H29O9S), which has 80 amu more than that of 9, indicating an additional sulfate ester group in compound 2. The 1H,13C-HMBC correlations from 18-OH to C-17 (δ 122.8), C-18 (δ 141.8), and C-19 (δ 147.8) as well as from 19-OH to C-18 (δ 141.8), C-19 (δ 147.8), and C-20 (δ 115.5) indicated that C-18 and C-19 are connected to hydroxy groups rather than sulfate ester groups (Figure 4). Thus, the sulfate ester group should be at position C-21. Furthermore, the 13C NMR chemical shifts of the three constitutional isomers 2-I to 2-III were calculated using an increment system (Table 2) with additional values for sulfate esters published by Ragan [8]. These calculations (Table 3) allowed us to confirm the position of the sulfate moiety (2-I, Figure 5). The comparison of the NMR data of 2 and siphonodictyal B2 (7, Figure 2) [5] further confirmed the identity of the aromatic moiety of both compounds.
![[1860-5397-10-52-4]](/bjoc/content/figures/1860-5397-10-52-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Selected 1H,13C-HMBC correlations (H → C) observed for siphonodictyal E2 (2).
Figure 4: Selected 1H,13C-HMBC correlations (H → C) observed for siphonodictyal E2 (2).
Table 2: 13C substituent effects Δδ in monosubstituted benzenes relative to the benzene carbon at 128.5 ppm [9].
| Functional group | i [ppm] | o [ppm] | m [ppm] | p [ppm] |
|---|---|---|---|---|
| CHO | 8.4 | 1.2 | 0.5 | 5.7 |
| OH | 26.9 | −12.8 | 1.4 | −7.4 |
| Allyl | 15.3 | 0 | 0.2 | −2.4 |
| OSO3H | 22.0 | −7.0 | 1.5 | −2.0 |
Table 3: Results of the increment calculations for the aromatic moiety of the three constitutional proposals of siphonodictyal E2 (2) and siphonodictyal E3 (3).
| Carbon No. | Siphonodictyal E2 (2) | 2-I | 2-II | 2-III |
|---|---|---|---|---|
| 16 | 125.9 | 131.3 (5.4) | 130.9 (5.0) | 125.6 (0.3) |
| 17 | 122.8 | 124.3 (1.5) | 124.3 (1.5) | 130.0 (7.2) |
| 18 | 141.8 | 141.3 (0.5) | 141.7 (0.1) | 131.0 (10.8) |
| 19 | 147.8 | 142.9 (4.9) | 137.9 (9.9) | 148.6 (0.8) |
| 20 | 115.5 | 118.7 (3.2) | 118.7 (3.2) | 113.0 (2.5) |
| 21 | 144.5 | 145.7 (1.2) | 150.7 (6.2) | 156.0 (11.5) |
| Σ [ppm] | 16.7 | 25.9 | 33.1 | |
| Carbon No. | Siphonodictyal E3 (3) | 3-I | 3-II | 3-III |
| 16 | 125.5 | 131.4 (5.9) | 136.7 (11.2) | 131.0 (5.5) |
| 17 | 129.5 | 130.1 (0.6) | 124.4 (5.1) | 130.1 (0.6) |
| 18 | 137.7 | 136.4 (1.3) | 147.1 (9.4) | 136.8 (0.9) |
| 19 | 151.3 | 148.7 (2.6) | 138.0 (13.3) | 143.7 (7.6) |
| 20 | 115.9 | 118.8 (2.9) | 124.5 (8.6) | 118.8 (2.9) |
| 21 | 148.2 | 151.1 (2.9) | 145.8 (2.4) | 156.1 (7.9) |
| Σ [ppm] | 16.2 | 50.0 | 25.4 | |
![[1860-5397-10-52-5]](/bjoc/content/figures/1860-5397-10-52-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Possible constitutions for the aromatic moieties of siphonodictyals E2 (2) and E3 (3) (sum over all Δδ(13C): 2-I 16.7 ppm, 2-II 25.9 ppm, 2-III 33.1 ppm, 3-I 16.2 ppm, 3-II 50.0 ppm, 3-III 25.4 ppm); plain arrows in 2-I showed the HMBC correlations from 18-OH and 19-OH to the aromatic carbons; dashed arrows in 2-II and 2-III indicate HMBC correlations which would be expected.
Figure 5: Possible constitutions for the aromatic moieties of siphonodictyals E2 (2) and E3 (3) (sum over all...
The HRMS-ESI(−) spectrum for siphonodictyal E3 (3) displayed the pseudomolecular ion peak [M + Na − 2H]− at m/z 539.1046, indicating the molecular formula C22H30O10S2. The 13C NMR spectrum contained six olefinic signals in addition to the aromatic signals, and the 1H NMR spectrum contained four olefinic methyl signals at δ 1.62 (s), δ 1.54 (s), δ 1.55 (s), and δ 1.66 (s), which suggested a sesquiterpene side chain containing three trisubstituted olefinic bonds. The two sulfate ester groups were assigned to attach to C-18 and C-21 after comparison of the NMR data of the aromatic moieties of 3 with those of siphonodictyal B3 (8, Figure 2). Nevertheless, the results of the increment calculations using the values for sulfate esters from Ragan [8] clearly favours the isomer 3-I (Figure 5), which further supports our proposed structure.
The HRMS-ESI(−) spectrum for siphonodictyal E4 (4) showed the pseudomolecular ion peak [M + Na − 2H]− at m/z 555.0954, confirmed its molecular formula as C22H30O11S2, corresponding to eight double bond equivalents. The sesquiterpenoid moiety of the molecule contained two trisubstituted olefinic bonds (δ 121.7, δ 125.7, δ 131.9, δ 134.9) and a hemiacetal carbon (δ 96.2) at one terminus which differed from the acyclic side chain in 1 and 2. The remaining six carbon signals in the downfield region of the 13C NMR spectrum belonged to the tetrasubstituted aromatic ring system. In contrast to 1 and 2, compound 4 contained an oxymethine group (CH-22, δC 65.3, δH 4.44) which connected the sesquiterpene chain with the aromatic ring. This is supported by the six/seven HMBC correlations from H-22 to C-1 (δ 96.2), C-2 (δ 37.3), C-3 (δ 31.4), C-13 (δ 19.9), C-19 (δ 143.2) / C-21 (δ 142.9), and C-20 (δ 120.3). The HMBC correlations from H-12 (δ 2.58, 3.99) to C-17 (δ 120.8) and C-21 and from H-17 (δ 7.19) to C-18 (δ 137.2) confirmed the position of the unsubstituted CH-17 as shown in the proposed structure (Figure 6). Since the very weak HMBC correlation from H-1 to C-19 or C-21 could not be unambiguously assigned, the constitutional isomers with respect to the position of the oxygen bridge 4a and 4b (Figure 1) are in accordance with the NMR data. This is the first compound isolated from this sponge which contains a macrocycle. Moreover, this macrocycle could probably be formed by an aldol addition from a precursor with an acyclic sesquiterpene side chain (Figure 7). The aromatic ring system of proposed precursor 3-ox in Figure 7 also appeared in the known compound siphonodictyal B3 (8) [5] isolated from the same sponge source as well as the newly identified compound 3. There are no similar compounds known which contained the aromatic ring system with two adjacent sulfate ester groups. Therefore, we assumed that the constitutional isomer 4a is more likely. The δ values of methyl carbons C-14 and C-15 below 20 ppm indicated the E configuration of the double bonds C-6/C-7 and C-10/C-11 of the isoprenoid chain [6,7]. Additionally, in the NOESY spectrum we could observe a weak correlation between H-22 (δ 4.44) and H-3 (δ 1.03) as well as between H-22 (δ 4.44) and H-13 (δ 1.04). Because of the overlapping of the proton signals of H-3 and H-13, this assignment is not unambiguous. Due to the flexibility of the macrocycle of 4 it was not possible to analyse the conformation of the pyrane. Therefore, the configurations at the centers C-1, C-2, and C-22 could not be assigned. Further investigations are hindered by the fact that compound 4 easily degrades.
![[1860-5397-10-52-6]](/bjoc/content/figures/1860-5397-10-52-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Selected 1H,13C-HMBC correlations (H → C) observed for siphonodictyal E4 (4a).
Figure 6: Selected 1H,13C-HMBC correlations (H → C) observed for siphonodictyal E4 (4a).
![[1860-5397-10-52-7]](/bjoc/content/figures/1860-5397-10-52-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Proposed biogenesis of 4a starting from the hypothetical precursor 3-ox with an acyclic sesquiterpenoid part.
Figure 7: Proposed biogenesis of 4a starting from the hypothetical precursor 3-ox with an acyclic sesquiterpe...
The HRMS-ESI(−) spectrum for cyclosiphonodictyol A (5) showed the pseudomolecular ion peak [M + Na − 2H]− at m/z 343.2292, confirmed its molecular formula as C22H32O3. The 13C NMR spectrum displayed six signals in the area of aromatic carbon atoms and 16 signals in the aliphatic region. The 1H,13C-HSQC spectrum indicated four methyl and seven methylene groups. Two of these methylene groups connected directly to the aromatic ring system indicated by the 1H,13C-HMBC correlations H-15 (δ 2.33, 3.06) → C-16 (δ 129.9), C-17(δ 146.4), and C-21 (δ 127.7) as well as H-22 (δ 4.50, 4.64) → C-16, C-20 (δ 146.5), and C-21. The sesquiterpene unit of 5 was identified by 1H,1H-COSY and 1H,13C-HMBC experiments. Furthermore, the HMBC correlations H-15 → C-8 (δ 78.5), C-9 (δ 58.1), and C-10 (δ 38.2) confirmed the connection between C-15 and C-9 and the HMBC correlation H-22 → C-8 indicated the existence of the oxygen bridge between C-22 and C-8. The p-hydroquinone moiety was established on the basis of the 1H and 13C NMR chemical shifts as well as the HMBC correlations mentioned above, namely, H-15 → C-16, C-17, C-21; H-22 → C-16, C-20, C-21. The relative configuration was confirmed by a ROESY experiment. The observed ROESY correlations between H-12 and H-13, H-13 and H-15, H-14 and H-15 suggested that the three methyl groups as well as CH2-15 are axial. Examination of the NMR data for 5 revealed a high degree of similarity with data reported for bis(sulfato)cyclosiphonodictyol A [10]. The difference concerned the absence of the two sulfate ester groups. Compound 5 could be the precursor of bis(sulfato)cyclosiphonodictyol A. Another possibility is that bis(sulfato)cyclosiphonodictyol A lost its sulfate ester group during the isolation procedure.
The new compounds were tested for antimicrobial activity against different Gram-positive and Gram-negative bacteria, fungi, and for antiproliferative activity. The results given in Table 4 show that siphonodictyal E3 (3) and cyclosiphonodictyol A (5) exhibited mild activity against the Gram-positive bacteria Staphylococcus aureus and Micrococcus luteus, while siphonodictyal E4 (4) show cytotoxic activity against the L929 mouse fibroblasts, KB-31 epidermoid carcinoma, and the breast cancer cell line MCF7, although none of them showed activity in the antimicrobial assays against Gram-negative bacteria and the fungus Candida albicans.
Table 4: Results of the antimicrobial and antiproliferation assays of compounds 1–5. Minimum inhibiting concentrations (MIC) for the antimicrobial assays and the IC50 values for the antiproliferation assays are given in µM.
| 1 | 2 | 3 | 4 | 5 | ||
|---|---|---|---|---|---|---|
| Gram-negative bacteria | Pseudomonas aeruginosa | – | – | – | – | – |
| Klebsiella pneumonia | – | – | – | – | – | |
| Gram-positive bacteria | Staphylococcus aureus (MRSA) | – | – | 37 | – | 117 |
| Staphylococcus aureus (MSSA) | – | – | 74 | – | 117 | |
| Micrococcus luteus | – | – | 74 | – | 58 | |
| Fungi | Candida albicans | – | – | – | – | – |
| Cytotoxic activity | L929 mouse fibroblasts | – | – | – | 27 | – |
| KB31 epidermoid carcinoma | – | – | – | 108 | – | |
| MCF7 breast cancer | – | – | – | 54 | 175 | |
| FS4-LTM conditional immortalization human fibroblasts | – | – | – | – | – | |
Many of the isolated compounds from Aka coralliphagum are bicyclic sesquiterpene hydroquinones. However, in the course of our investigation, three linear sesquiterpenes were isolated. From a biosynthetical point of view [11], we propose that the linear sesquiterpenes are the precursors of the bicyclic sesquiterpenes (Figure 8). Therefore, siphonodictyals B1–B3 (6–8) could be derived from siphonodictyals E1–E3 (1–3), respectively.
![[1860-5397-10-52-8]](/bjoc/content/figures/1860-5397-10-52-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Hypothetical biogenesis of the bicyclic sesquiterpenoid moiety from the acyclic precursor of the siphonodictyals in the sponge Aka coralliphagum [11]. The newly formed bonds from the acyclic to the bicyclic sesquiterpenoid moiety are drawn in red. The isomerizing double bond is drawn in blue.
Figure 8: Hypothetical biogenesis of the bicyclic sesquiterpenoid moiety from the acyclic precursor of the si...
Aromatic sulfates occur in a wide range of higher plants and animals, particularly those growing in marine, estuarine, or freshwater environments. This is probably due to the high content of inorganic sulfate in aquatic environments (both limnic and marine environments – seawater contains an average of 28.7 mM inorganic sulfate [12]). Jensen and Ragan have demonstrated the presence of the natural product 1,2,3,5-tetrahydroxybenzene-2,5-disulfate in the marine brown alga Ascophyllum nodosum [13]. Kobayashi et al. isolated halenaquinol sulfate from the marine sponge Xestospongia sapra [14].
In the course of our investigation on the sponge Aka coralliphagum, we isolated a number of compounds containing sulfated phenols. These sulfated metabolites are labile [5], and easily loose the sulfate ester groups by hydrolysis in water [15]. Moreover, the influence of the sulfate esters on the bioactivity has been proven by activity tests: the desulfated compounds tend to be more active [5]. Therefore, we propose that the sulfate esters act as hydroxy protecting groups. The sponges excrete the sesquiterpene hydroquinones primarily in the sulfated form and their activities get increased by hydrolysis of these labile groups. This would lead to a prolonged bioactivity of these metabolites and therefore to a more efficient defense against predators or pathogens.
Experimental
1H and 13C NMR spectra were recorded on a Bruker Avance 600 MHz NMR spectrometer equipped with a cryo platform and a Bruker Avance 400 MHz NMR spectrometer. All experiments were measured at 303 K. HPLC separation was achieved by a Kromasil RP-18 column (16 mm × 250 mm, 5 µm) applying a H2O (5 mM NH4OAc)/MeCN/MeOH gradient. UV spectra were recorded during HPLC separation with a DAD detector (Jasco MD-2010 Plus). Accurate mass spectra were acquired using a Bruker micrOTOFLC mass spectrometer equipped with an ESI source. Mass calibration was performed using a sodium formate cluster prior each experiment.
The sponges were collected by SCUBA diving at a depth of 29 m off the coast of San Salvador in the Bahamas in June 2008. The samples were immediately frozen after collection and kept at −20 °C until extraction. A voucher specimen of this species is deposited in our lab (voucher number: AKA 08/45). The freeze-dried sponge sample (107.6 g) was extracted three times at room temperature with DCM/MeOH (1:1). After filtration, the filtrate was evaporated to yield 35.5 g crude extract which was further partitioned between n-hexane and methanol. The methanol extract was dried and followed with the partitioning between n-BuOH and H2O. The resulting n-BuOH phase was concentrated (9.2 g) and purified by liquid chromatography using RP-18 silica gel with a stepwise gradient from H2O/MeOH (100:0), (85:15), (50:50) to (0:100) yielding fractions A, B, C, and D, respectively. The fraction B was further purified by HPLC using an RP-18 column yielding compounds 1 (2.2 mg, HPLC gradient flow: H2O (5 mM NH4OAc)/MeCN: 0 min 81:19, 40 min 75:25), 2 (3.1 mg, HPLC isocratic flow: H2O (5 mM NH4OAc)/MeCN: 87:13 40 min), 4 (1.3 mg, HPLC gradient flow: H2O (5 mM NH4OAc)/MeCN: 0 min 85:15, 40 min 83:17, 60 min 80:20), and 5 (3.0 mg, HPLC gradient flow: H2O (5 mM NH4OAc)/MeCN: 0 min 85:15, 40 min 83:17, 60 min 80:20). The fraction C was also further purified by HPLC using the same RP-18 column yielding 3 (2.5 mg, HPLC gradient flow: H2O (5 mM NH4OAc)/MeCN: 0 min 76:24, 40 min 74:26, 45 min 70:30)
Antimicrobial assay
Determination of antimicrobial activities was carried out by the Helmholtz Center for Infection Research (HZI), and was based on the microdilution test, using 96-well micro titration plates. The MIC was defined as 50% growth inhibition after 24 hour incubation compared to that in the growth control well.
Cytotoxic assay
The cytotoxicity was determined using WST-1 assays, targeting cell lines L929 mouse fibroblasts, KB-31 epidermoid carcinoma, MCF-7 breast cancer, and FS4-LTM conditional immortalization human fibroblasts, respectively. The FS4-LTM cell line was incubated for 24 hours with the tested substances. The other cell lines were incubated for 5 days with the tested substances.
Experimental data
Siphonodictyal E1 (1): yellow powder; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M + Na − 2H]− calcd for C24H33NO11S2Na, 598.1398; found, 598.1338.
Siphonodictyal E2 (2): yellow powder; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M − H]− calcd for C22H29O9S, 469.1538; found, 469.1561.
Siphonodictyal E3 (3): yellow powder; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M + Na − 2H]− calcd for C22H28O10S2Na, 539.1027; found, 539.1046.
Siphonodictyal E4 (4): yellow powder; CD data {(MeOH) ![[Graphic 1]](/bjoc/content/inline/1860-5397-10-52-i1.svg?max-width=637&scale=1.18182) = +37}; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M + Na − 2H]− calcd for C22H28O11S2Na, 555.0976; found, 555.0954.
= +37}; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M + Na − 2H]− calcd for C22H28O11S2Na, 555.0976; found, 555.0954.
Cyclosiphonodictyol A (5): colorless powder; CD data {(MeOH) ![[Graphic 2]](/bjoc/content/inline/1860-5397-10-52-i2.svg?max-width=637&scale=1.18182) = +163}; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M − H]− calcd for C22H31O3, 343.2279; found, 343.2292.
= +163}; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz) see Table 1; HRMS-ESI(−) m/z: [M − H]− calcd for C22H31O3, 343.2279; found, 343.2292.
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra of the isolated compounds. | ||
| Format: PDF | Size: 538.7 KB | Download |
Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (DFG) under grants Ko 1314/5-1 and 5-2 (DFG research unit FOR 934) is gratefully acknowledged. Sponge collection was carried out by Dr. Gesine Schmidt and Dr. Achim Grube during a scientific expedition to the Bahamas in 2008. We would like to acknowledge the support of Prof. Dr. Joseph R. Pawlik (University of North Carolina, Wilmington, USA) who gave members of the Köck research group the opportunity to participate in the research trips to the Bahamas for the time period from 1998 to 2008. We further thank Dr. Sven Zea (Universidad Nacional de Colombia) for identification of the sponge samples as well as Dr. Florenz Sasse and Dr. Galina Sergeev (HZI Braunschweig) for carrying out the bioassays.
References
-
Rützler, K. Smithsonian Contrib. Zool. 1971, 77, 1–37. doi:10.5479/si.00810282.77
Return to citation in text: [1] -
Sullivan, B. W.; Faulkner, D. J.; Matsumoto, G. K.; He, C. H.; Clardy, J. J. Org. Chem. 1986, 51, 4568–4573. doi:10.1021/jo00374a015
Return to citation in text: [1] [2] [3] -
Sullivan, B. W.; Faulkner, D. J. New Perspectives in Sponge Biology. Third International Sponge Conference, Woods Hole, Massachusetts, Nov 17–23, 1985; Smithsonian Institution Press: Washington, DC, 1985; pp 45–50.
Return to citation in text: [1] -
Sullivan, B.; Djura, P.; Mcintyre, D. E.; Faulkner, D. J. Tetrahedron 1981, 37, 979–982. doi:10.1016/S0040-4020(01)97672-0
Return to citation in text: [1] -
Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Couperus, P. A.; Clague, A. D. H.; van Dongen, J. P. C. M. Org. Magn. Reson. 1976, 8, 426–431. doi:10.1002/mrc.1270080807
Return to citation in text: [1] [2] -
Coates, R. M.; Ley, D. A.; Cavender, P. L. J. Org. Chem. 1978, 43, 4915–4922. doi:10.1021/jo00420a003
Return to citation in text: [1] [2] -
Ragan, M. A. Can. J. Chem. 1978, 56, 2681–2685. doi:10.1139/v78-441
Return to citation in text: [1] [2] -
Kalinowski, H.-O.; Berger, S.; Braun, S. 13C NMR-Spektroskopie; Georg Thieme Verlag: Stuttgart, 1984; pp 685 ff.
Return to citation in text: [1] -
Killday, K. B.; Wright, A. E.; Jackson, R. H.; Sills, M. A. J. Nat. Prod. 1995, 58, 958–960. doi:10.1021/np50120a024
Return to citation in text: [1] -
Fontana, A.; Tramice, A.; Cutignano, A.; d'Ippolito, G.; Gavagnin, M.; Cimino, G. J. Org. Chem. 2003, 68, 2405–2409. doi:10.1021/jo026131v
Return to citation in text: [1] [2] -
Zehnder, A. J. B.; Zinder, S. H. The Sulfur Cycle. In The Handbook of Environmental Chemistry; Hutzinger, O., Ed.; Springer: Berlin, 1980; Vol. 1A, pp 105–145.
Return to citation in text: [1] -
Jensen, A.; Ragan, M. A. Tetrahedron Lett. 1978, 19, 847–850. doi:10.1016/S0040-4039(01)85415-0
Return to citation in text: [1] -
Kobayashi, M.; Shimizu, N.; Kyogoku, Y.; Kitagawa, I. Chem. Pharm. Bull. 1985, 33, 1305–1308. doi:10.1248/cpb.33.1305
Return to citation in text: [1] -
Benkovic, S. J. J. Am. Chem. Soc. 1966, 88, 5511–5515. doi:10.1021/ja00975a027
Return to citation in text: [1]
| 14. | Kobayashi, M.; Shimizu, N.; Kyogoku, Y.; Kitagawa, I. Chem. Pharm. Bull. 1985, 33, 1305–1308. doi:10.1248/cpb.33.1305 |
| 12. | Zehnder, A. J. B.; Zinder, S. H. The Sulfur Cycle. In The Handbook of Environmental Chemistry; Hutzinger, O., Ed.; Springer: Berlin, 1980; Vol. 1A, pp 105–145. |
| 13. | Jensen, A.; Ragan, M. A. Tetrahedron Lett. 1978, 19, 847–850. doi:10.1016/S0040-4039(01)85415-0 |
| 1. | Rützler, K. Smithsonian Contrib. Zool. 1971, 77, 1–37. doi:10.5479/si.00810282.77 |
| 2. | Sullivan, B. W.; Faulkner, D. J.; Matsumoto, G. K.; He, C. H.; Clardy, J. J. Org. Chem. 1986, 51, 4568–4573. doi:10.1021/jo00374a015 |
| 11. | Fontana, A.; Tramice, A.; Cutignano, A.; d'Ippolito, G.; Gavagnin, M.; Cimino, G. J. Org. Chem. 2003, 68, 2405–2409. doi:10.1021/jo026131v |
| 2. | Sullivan, B. W.; Faulkner, D. J.; Matsumoto, G. K.; He, C. H.; Clardy, J. J. Org. Chem. 1986, 51, 4568–4573. doi:10.1021/jo00374a015 |
| 4. | Sullivan, B.; Djura, P.; Mcintyre, D. E.; Faulkner, D. J. Tetrahedron 1981, 37, 979–982. doi:10.1016/S0040-4020(01)97672-0 |
| 5. | Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018 |
| 11. | Fontana, A.; Tramice, A.; Cutignano, A.; d'Ippolito, G.; Gavagnin, M.; Cimino, G. J. Org. Chem. 2003, 68, 2405–2409. doi:10.1021/jo026131v |
| 3. | Sullivan, B. W.; Faulkner, D. J. New Perspectives in Sponge Biology. Third International Sponge Conference, Woods Hole, Massachusetts, Nov 17–23, 1985; Smithsonian Institution Press: Washington, DC, 1985; pp 45–50. |
| 6. | Couperus, P. A.; Clague, A. D. H.; van Dongen, J. P. C. M. Org. Magn. Reson. 1976, 8, 426–431. doi:10.1002/mrc.1270080807 |
| 7. | Coates, R. M.; Ley, D. A.; Cavender, P. L. J. Org. Chem. 1978, 43, 4915–4922. doi:10.1021/jo00420a003 |
| 2. | Sullivan, B. W.; Faulkner, D. J.; Matsumoto, G. K.; He, C. H.; Clardy, J. J. Org. Chem. 1986, 51, 4568–4573. doi:10.1021/jo00374a015 |
| 10. | Killday, K. B.; Wright, A. E.; Jackson, R. H.; Sills, M. A. J. Nat. Prod. 1995, 58, 958–960. doi:10.1021/np50120a024 |
| 5. | Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018 |
| 5. | Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018 |
| 5. | Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018 |
| 5. | Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018 |
| 5. | Grube, A.; Assmann, M.; Lichte, E.; Sasse, F.; Pawlik, J. R.; Köck, M. J. Nat. Prod. 2007, 70, 504–509. doi:10.1021/np0603018 |
| 6. | Couperus, P. A.; Clague, A. D. H.; van Dongen, J. P. C. M. Org. Magn. Reson. 1976, 8, 426–431. doi:10.1002/mrc.1270080807 |
| 7. | Coates, R. M.; Ley, D. A.; Cavender, P. L. J. Org. Chem. 1978, 43, 4915–4922. doi:10.1021/jo00420a003 |
| 9. | Kalinowski, H.-O.; Berger, S.; Braun, S. 13C NMR-Spektroskopie; Georg Thieme Verlag: Stuttgart, 1984; pp 685 ff. |
| 15. | Benkovic, S. J. J. Am. Chem. Soc. 1966, 88, 5511–5515. doi:10.1021/ja00975a027 |
© 2014 Göthel and Köck; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)