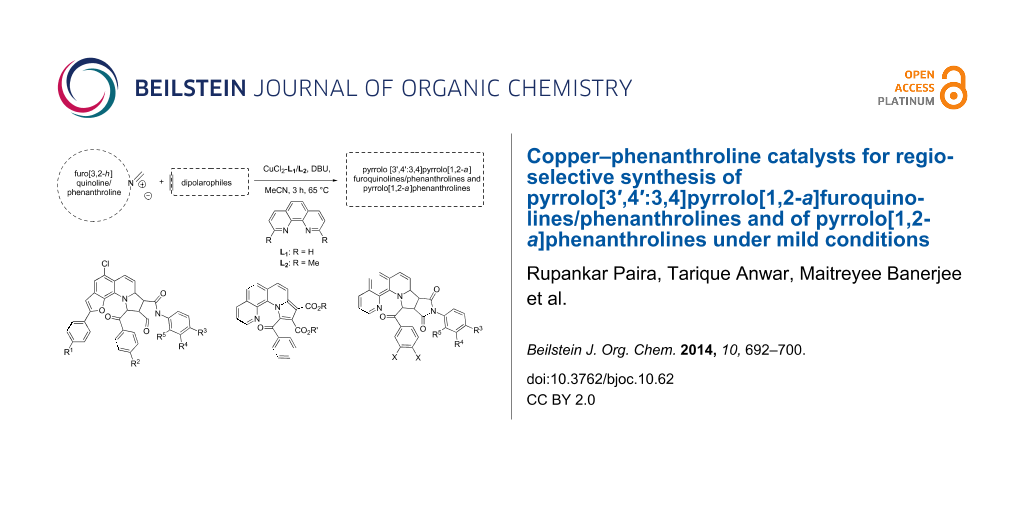
Abstract
A new series of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinolines/phenanthrolines and pyrrolo[1,2-a]phenanthrolines were efficiently built up from an 8-hydroxyquinoline derivative or phenanthroline via 1,3-dipolar cycloaddition reaction involving non-stabilized azomethine ylides, generated in situ from the parent furo[3,2-h]quinoliniums/phenanthroliums, in presence of a copper(II) chloride–phenanthroline catalytic system. The methodology combines general applicability with high yields.

Graphical Abstract
Introduction
The chemistry of the 1,3-dipolar cycloaddition has always been a fascinating undertaking especially when azomethine ylides are involved as the key component [1-9]. These ylides, both stabilized or non-stabilized 1,3-dipoles, can easily enter the reaction independent of their stability and lead to the formation of a pyrrolidine core, a structural motif of immense interest from both chemical and pharmacological points of view [10]. In recent years, there have been many attempts to synthesize diversely modified pyrrolidines, both symmetric and asymmetric in nature. Several of these attempts involved 1,3-dipolar cycloaddition reactions involving azomethine ylides to give cycloadducts, which were further explored as potential antiviral, antifungal, antitumor and anti-HIV candidates [1-15]. We have also been pursuing the cycloaddition methodology for several years and established some synthetic routes towards indolizines, pyrrolo[1,2-a]quinolines/isoquinolines, oxazadicyclopenta[a,h]naphthalenes etc., some of which have been evaluated as potential antibacterial and antifungal agents [16-19]. In order to explore the possibility of using structurally more complex dipoles and dipolarophiles to construct more interesting structural networks, we replaced simple alkenes/alkynes with maleimide derivatives. Our preference for this dienophile was dictated by a recent patent on the lifespan altering properties of cycloadducts involving maleimide dienophiles [20]. Moreover, very recently we have characterized some furo[3,2-h]quinoliniums as potent non-detergent spermicides [19], which encouraged us for further modification and derivatization of furoquinoline analogues in search for more potent agents. Thus we became interested in the construction of a number of structurally complex pentacyclic pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinolines, for possible identification of new antibacterial/antifungal/spermicidal/lifespan altering agents, where furo[3,2-h]quinoliniums were employed as dipole precursors and maleimide derivatives as dipolarophiles.
We initially employed the protocols from our recently developed green methodologies [21-25], which however failed to give any promising outcome and forced us to explore new catalytic systems. While searching for this goal, we were attracted by the possible application of copper-catalysis, which has always been an effective tool especially with Diels–Alder reactions [26,27]. Thus, we studied the effect of a number of catalytic systems and after an extensive screening, we found copper(II) chloride–phenanthroline as the best catalytic pair for this purpose. Herein we wish to present the results of our recent synthetic efforts to synthesize a series of unique pentacyclic pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinolines/phenanthrolines using the above catalytic system. To the best of our knowledge, this is the first report of the application of this catalyst for the regioselective 1,3-dipolar cycloaddition reaction, involving azomethine ylides derived from structurally complex quinoline-based N-heterocycles.
Results and Discussion
Our studies started with the preparation (Scheme 1 and Scheme 2) of maleimides 4a–c employing maleic anhydride (1) and different aromatic amines 2a–c, and of furo[3,2-h]quinoliniums 9a–d from 5-chloro-8-hydroxy-7-iodoquinoline 5 [19,24]. However, attempted condensation of 4a with the 1,3-dipole generated in situ from 9a, employing protocols from our recently developed methodologies, did not succeed, affording products only in low yields (10–19%, Table 1, entries 1–7).
![[1860-5397-10-62-i1]](/bjoc/content/inline/1860-5397-10-62-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of maleimide dipolarophiles 4a–c.
Scheme 1: Preparation of maleimide dipolarophiles 4a–c.
![[1860-5397-10-62-i2]](/bjoc/content/inline/1860-5397-10-62-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of 1,3-dipole precursors 9a–d.
Scheme 2: Preparation of 1,3-dipole precursors 9a–d.
Table 1: Optimization of conditions of reaction between 4a and 9a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-62-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalytic system | Solventa | Time (h) | Temp. (°C) | Yieldb |
|---|---|---|---|---|---|
| 1 | Basic alumina | None | 0.3 | 80 | 10c |
| 2 | Amberlite IRA 402 (OH) | H2O | 10 | 90 | 16 |
| 3 | K-10 clay | None | 0.3 | 80 | 12c |
| 4 | Triton X-114 (60 mM) | H2O | 3 | rt | 17 |
| 5 | SDS (60 mM) | H2O | 3 | rt | 15 |
| 6 | TTAB (80 mM) | H2O | 6 | rt | 19 |
| 7 | CTAB (90 mM) | H2O | 5 | rt | 12 |
| 8 | SnCl4 | Toluene | 8 | 80 | NR |
| 9 | Sc(OTf)3 | DCM | 10 | rt | NR |
| 10 | Sc(OTf)3 | Toluene | 10 | 80 | NR |
| 11 | Mg(ClO4)3 | DCM | 10 | rt | NR |
| 12 | Mg(ClO4)3 | Toluene | 10 | 80 | 11 |
| 13 | Cu(OTf)2 | DCM | 8 | rt | 15 |
| 14 | Cu(OTf)2 | Toluene | 8 | 80 | 28 |
| 15 | Cu(OAc)2 | DCM | 8 | rt | 12 |
| 16 | Cu(OAc)2 | Toluene | 8 | 80 | 32 |
| 17 | Cu(OAc)2 | MeCN | 8 | 65 | 47 |
| 18 | CuCl2 | DCM | 6 | rt | 23 |
| 19 | CuCl2 | Toluene | 6 | 80 | 45 |
| 20 | CuCl2 | MeCN | 3 | 65 | 57 |
| 21 | CuCl2/PPh3 | MeCN | 3 | 65 | 58 |
| 22 | CuCl2/PMePh2 | MeCN | 3 | 65 | 58 |
| 23 | CuCl2/PCy3 | MeCN | 3 | 65 | 56 |
| 24 | CuCl2/P(3-ClC6H4)3 | MeCN | 3 | 65 | 58 |
| 25 | CuCl2/P(3-OMeC6H4)3 | MeCN | 3 | 65 | 55 |
| 26 | CuCl2/DPEphos | MeCN | 3 | 65 | 65 |
| 27 | CuCl2/xantphos | MeCN | 3 | 65 | 67 |
| 28 | CuCl2/pyphos | MeCN | 3 | 65 | 65 |
| 29 | CuCl2/bis-oxazocine | MeCN | 3 | 65 | 74 |
| 30 | CuCl2/pyrimidine | MeCN | 3 | 65 | 71 |
| 31 | CuCl2/phenanthroline (L1) | MeCN | 3 | 65 | 94 |
| 32 | CuCl2/phenanthroline (L2) | MeCN | 3 | 65 | 94 |
aAll the reactions were performed in presence of DBU; bIsolated yield; cThe reactions were performed under microwave irradiation at 180 W.
We then studied the feasibility of a metal-catalyzed 1,3-dipolar cycloaddition strategy. A thorough screening of different catalysts, as summarized in Table 1, revealed the supremacy of copper catalysts in this particular reaction over the others; CuCl2 appeared to be the catalyst of choice (Table 1, entries 8–32). In order to explore the effect of ligands, a number of phosphines, bis-oxazocines, pyrazolyl-pyrimidines and phenanthroline analogues were employed (Figure 1). As represented in Table 1, the monodentate ligands are in general less effective (55–58% yield; Table 1, entries 21–25) than bi-/tridentate ligands (65–94% yield; Table 1, entries 26–32), and phosphines in general proved less effective in terms of product yield (Table 1, entries 21–28). Bis-oxazocines and pyrazolyl-pyrimidines on the other hand showed some promising results (Table 1, entries 29 and 30). However, both in terms of yield and cleaner reaction profile, 1,10-phenanthrolines (L1, L2) were identified as the best partners for CuCl2 (Table 1, entries 31 and 32). Thus, the 1,3-dipolar cycloaddition reaction between 9a (1 equiv) and 4a (1.1 equiv) yielded 94% of 10a within 3 h, when 5 mol % CuCl2 and 5 mol % of either L1 or L2 were employed with DBU as the base in acetonitrile solvent at 65 °C. This was taken as the best conditions to perform the reaction.
![[1860-5397-10-62-1]](/bjoc/content/figures/1860-5397-10-62-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bi-/tridentate ligands used for the optimization of the reaction conditions.
Figure 1: Bi-/tridentate ligands used for the optimization of the reaction conditions.
The structure of 10a was deduced from the appearance of a new doublet of doublets at δ 5.50 and δ 6.20. Another doublet at δ 3.90 and a new multiplet at δ 3.50 also indicated the success of the reaction, as these signals could be attributed to the new pyrrolidine core. Besides, the cluster in the aromatic region (δ 6.60–8.50) indicated the presence of 19 aromatic protons in the cycloadduct 10a. This interpretation was also well supported by the 13C NMR of 10a. Peaks for the four sp3-methine carbons of the newly formed pyrrolidine motif appeared at δ 47.2, 47.4, 61.0 and 66.5 as expected. Two methine carbon signals appearing at δ 101.6 and 110.8 could be assigned to the CH units present in the dihydroquinoline ring. Moreover, the presence of peaks for three carbonyl carbons at δ 174.9, 176.6, 194.5, and for ten other quaternary carbons and 19 methine carbons in the aromatic region (δ 116.0–159.8) also confirmed the structure. The relative orientations of H-1, H-18, H-19 and H-20 could be determined unambiguously by the single-crystal X-ray diffraction analysis of 10a; the ORTEP diagram is presented in Figure 2. No other diastereomer could be detected.
![[1860-5397-10-62-2]](/bjoc/content/figures/1860-5397-10-62-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram showing the molecular structure of 10a at 30% probability level.
Figure 2: ORTEP diagram showing the molecular structure of 10a at 30% probability level.
The plausible mechanism of this cycloaddition is presented in Scheme 3. The base (DBU) abstracts the acidic proton of furo[3,2-h]quinolinium 9a to generate the 1,3-dipole I. The Cu(II)–phenanthroline system activates the maleimide dipolarophile via coordination with the carbonyl group to undergo a [3 + 2] cycloaddition with the 1,3-dipole to form the cycloadduct 10a, releasing the Cu(II) complex to enter another cycle.
![[1860-5397-10-62-i3]](/bjoc/content/inline/1860-5397-10-62-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible mechanistic pathway for the synthesis of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinolines.
Scheme 3: Plausible mechanistic pathway for the synthesis of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinolines.
In order to establish the general applicability of this protocol, we reacted different furo[3,2-h]quinoliniums 9a–d and maleimide dipolarophiles 4a–c under the standardized reaction conditions. As obvious from the results summarized in Scheme 4, all the reactions proceeded smoothly to give cycloadducts with excellent yields, which were fully characterized by mass and NMR analysis.
![[1860-5397-10-62-i4]](/bjoc/content/inline/1860-5397-10-62-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinoline analogues under the optimized protocol.
Scheme 4: Synthesis of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinoline analogues under the optimized protocol.
In order to test its general applicability further, we replaced furo[3,2-h]quinoliniums with phenanthroliniums 12a,b. These phenanthroliniums were synthesized (Scheme 5) from phenanthroline (11) and 2′-bromoacetophenones 8a and 8d, under basic alumina/microwave (180 W) conditions. These were then subjected to the optimized [3 + 2] cycloaddition protocol with different N-phenylmaleimide dipolarophiles 4a–c, which eventually produced similar cycloadducts 14a–c in high yield. It is interesting to note that every reaction proceeded only up to the cycloaddition stage, without any aromatization of the final cycloadduct. However, a similar reaction of phenanthroliniums 12a,b with alkyne dipolarophiles like acetylenedicarboxylates or monocarboxylates 13a–d proceeded with aromatization of the putative dihydroaromatic intermediates to produce the final cycloaddition products 14d–g.
![[1860-5397-10-62-i5]](/bjoc/content/inline/1860-5397-10-62-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Construction of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]phenanthrolines 14a–c and of pyrrolo[1,2-a]phenanthrolines 14d–g.
Scheme 5: Construction of pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]phenanthrolines 14a–c and of pyrrolo[1,2-a]phenanth...
Characterization of the products was done via mass and NMR spectral studies. Furthermore, the single-crystal X-ray study of cycloadduct 14e undoubtedly confirmed the structure of these cycloadducts, as obvious from the ORTEP diagram presented in Figure 3.
![[1860-5397-10-62-3]](/bjoc/content/figures/1860-5397-10-62-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP diagram showing the molecular structure of 14e at 30% probability level.
Figure 3: ORTEP diagram showing the molecular structure of 14e at 30% probability level.
Conclusion
In conclusion, a simple CuCl2–phenanthroline catalyzed methodology has been developed to synthesize a series of unique heteroaromatic polycycles 10a–h, 14a–g by a 1,3-dipolar cycloaddition reaction, using furo[3,2-h]quinolinium/phenanthrolinium dipole precursors and maleimide/acetylenecarboxylate dipolarophiles. High atom-economy, good to very good isolated yield of the products, short reaction time and ease of separation coupled with general applicability are the key features of this methodology.
Experimental
General procedure to synthesize pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]furoquinolines 10a–h, pyrrolo[3′,4′:3,4]pyrrolo[1,2-a]phenanthrolines 14a–c, and pyrrolo[1,2-a]phenanthrolines 14d–g: A mixture of 3.3 mmol furo[3,2-h]quinolinium derivatives 9a–d/phenanthroliniums 12a,b and 3.3 mmol N-phenylmalimide derivatives 4a–d/dialkyl acetylenedicarboxylates 13a–b/monoalkyl acetylenemonocarboxylates 13c,d was placed in a round bottomed flask (25 mL). To this MeCN (50 mL) and DBU (1 mmol) were added and the mixture was stirred for 30 min. Then 5 mol % CuCl2 and 5 mol % of either L1, L2 were added to the reaction mixture and stirred continuously for 3 h at 65 °C. After completion of the reaction (monitored by TLC), the reaction mixture was partitioned between brine and ethyl acetate. The organic layer was then evaporated and purified by column chromatography (ethyl acetate:hexane).
NMR data and crystal data of some representative compounds: a) spectral data of 10a: Yellow solid. 94% yield; mp 246–248 °C; Rf (20% ethyl acetate–hexane) 0.35; 1H NMR (300 MHz, CDCl3) δ 3.65 (t, J = 8.1 Hz, 1H), 3.84 (s, 3H), 3.89 (m, 1H), 5.52 (m, 1H), 6.18 (m, 1H), 6.64 (s, 1H), 6.76 (s, 1H), 6.98 (m, 8H), 7.11 (m, 1H), 7.20 (m, 2H), 7.65 (t, J = 7.5 Hz, 2H), 7.76 (d, J = 7.2 Hz, 1H), 8.48 (d, J = 7.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 47. 2 (CH), 47.4 (CH), 55.5 (CH3), 61.0 (CH), 66.5 (CH), 101.6 (CH), 110.8 (CH), 114.5 (2CH), 116.0 (C), 119.7 (CH), 124.0 (CH), 124.6 (2CH), 127.6 (2CH), 127.6 (C), 128.4 (3CH), 128.4 (C), 128.8 (C), 129.3 (C), 129.4 (2CH), 129.6 (2CH), 131.2 (C), 133.1 (C), 134.4 (CH), 140.6 (C), 157.0 (C), 159.8 (C), 174.9 (C), 176.6 (C), 194.5 (C); HRMS (ESI) m/z: [M + Na]+ calcd for C36H25ClN2NaO5+ 623.1344; found, 623.1353. b) Spectral data of 14a: Yellow solid. 81% yield; mp 242–243 °C; Rf (20% ethyl acetate–hexane) 0.31; 1H NMR (300 MHz, CDCl3) δ 2.26 (s, 6H), 3.65 (m, 1H), 3.74 (m, 1H), 5.94 (m, 1H), 6.18 (m, 1H), 6.54 (m, 1H), 7.00 (m, 3H), 7.07 (m, 2H), 7.19 (m, 2H), 7.64 (m, 4H), 7.85 (m, 1H), 8.33 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 19.5 (CH3), 19.7 (CH3), 46.9 (CH), 47.5 (CH), 63.4 (CH), 67.0 (CH), 116.9 (CH), 120.4 (CH), 121.1 (C), 121.6 (CH), 123.7 (CH), 126.4 (CH), 126.9 (CH), 127.3 (CH), 128.2 (C), 128.7 (2CH), 128.8 (2CH), 129.1 (C), 129.4 (C), 130.2 (CH), 132.9 (CH), 134.6 (C), 136.0 (CH), 137.7 (C), 137.9 (C), 138.5 (C), 145.3 (CH), 175.7 (C), 176.9 (C), 196.6 (C); HRMS (ESI) m/z: [M + Na]+ calcd for C32H25N3NaO3+ 522.1788; found, 522.1799. c) Spectral data of 14e: Brown solid. 91% yield; mp 233–234 °C; Rf (20% ethyl acetate–hexane) 0.33; 1H NMR (300 MHz, CDCl3) δ 1.06 (t, J = 7.2 Hz, 3H), 1.38 (t, J = 7.2 Hz, 3H), 3.72 (m, 1H), 3.88 (m, 1H), 4.38 (m, 2H), 7.33 (m, 1H), 7.51 (t, J = 7.4 Hz, 2H), 7.60 (m, 1H), 7.69 (d, J = 9.3 Hz, 1H), 7.83 (m, 2H), 8.01 (m, 1H), 8.17 (d, J = 7.8 Hz, 3H), 8.57 (d, J = 9.3 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 13.6 (CH3), 14.3 (CH3), 60.4 (CH2), 61.4 (CH2), 104.0 (C), 120.2 (CH), 122.5 (CH), 125.3 (CH), 125.6 (C), 126.8 (C), 126.0 (CH), 126.7 (CH), 127.7 (C), 128.1 (2CH), 129.0 (C), 130.0 (2CH), 130.7 (C), 132.2 (CH), 135.9 (CH), 137.3 (C), 137.4 (C), 137.9 (C), 145.7 (CH), 163.5 (C), 165.5 (C), 184.4 (C); HRMS (ESI) m/z: [M + Na]+ calcd for C28H22N2NO5+ 489.1421; found, 489.1437. d) Crystal data for 10a: C36H25N2O5Cl, M = 601.03 , monoclinic, P21/c, a = 15.687(2), b = 19.297(2), c = 9.848(1) Å, β = 99.478(8)°, V = 2940.5(7) Å3, Z = 4, Dc = 1.358 g cm−3, µ = 0.178 mm−1, F(000) = 1248, λ(MoKα) = 0.71073 Å, reddish block, crystal size: 0.7 × 0.25 × 0.19 mm, 37123 reflections measured (Rint = 0.0477), 3620 unique reflections, wR(F2) = 0.155 for all data and conventional R = 0.041 for 2932 F-values with I>2σ(I), (Δ/σ)max = 0.000, S = 0.617 for all data and 398 parameters, Δρmax, min (e/Å3) = 0.214, −0.197. e) Crystal data for 13f: C28H22N2O5, M = 466.48 , monoclinic, P21/c, a = 11.9766(8), b = 17.281(1), c = 11.6975(8) Å, β = 109.484(3)°, V = 2282.4(3) Å3, Z = 4, Dc = 1.358 g cm−3, µ = 0.094 mm−1, F(000) = 976, λ (Mo Kα) = 0.71073 Å, yellowish block, crystal size: 0.17 × 0.11 × 0.09 mm, 25248 reflections measured (Rint = 0.0552), 3534 unique reflections, wR(F2) = 0.1976 for all data and conventional R = 0.0501 for 2741 F-values with I>2σ(I), (Δ/σ)max = 0.000, S = 1.504 for all data and 319 parameters, Δρmax, min (e/Å3) = 0.267, −0.351. Unit cell determinations and intensity data collections for both the compounds were performed on a Bruker KAPPA APEXII CCD diffractometer at 296(2) K. Structure solution by direct methods and refinements by full-matrix-least-squares methods on F2. Programs: APEX2 (Bruker AXS Inc., Madison, Wisconsin, USA) for data collection, SAINT (Bruker AXS Inc., Madison, Wisconsin, USA) for cell refinement and data reduction, SHELXTL (Bruker AXS Inc., Madison, Wisconsin, USA) for structure determination, refinements and molecular graphics calculations. CCDC Numbers: 937560 (for compound 10a) and 937561 (for compound 14e) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033)
Supporting Information
| Supporting Information File 1: Experimental and analytical data and copies of 1H NMR and 13C NMR spectra of all new products. | ||
| Format: PDF | Size: 3.4 MB | Download |
Acknowledgements
We would like to thank The Council of Scientific and Industrial Research (CSIR), New Delhi, for financial support in the form of fellowships to R.P., S.M., A.H. and Emeritus Scientist ship to P.R.M. and N.B.M. We are indebted Dr B. Achari, Ex-Emeritus Scientist, CSIR, for critical suggestions and encouragement.
References
-
Pandey, G.; Banerjee, P.; Gadre, S. R. Chem. Rev. 2006, 106, 4484–4517. doi:10.1021/cr050011g
Return to citation in text: [1] [2] -
Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765–2810. doi:10.1021/cr040004c
Return to citation in text: [1] [2] -
Gu, Y. G.; Xu, Y.; Krueger, A. C.; Madigan, D.; Sham, H. L. Tetrahedron Lett. 2002, 43, 955–957. doi:10.1016/S0040-4039(01)02303-6
Return to citation in text: [1] [2] -
Nair, V.; Sheela, K. C.; Rath, N. P.; Eigendorf, G. K. Tetrahedron Lett. 2000, 41, 6217–6221. doi:10.1016/S0040-4039(00)01032-7
Return to citation in text: [1] [2] -
Gothelf, A. S.; Gothelf, K. V.; Hazell, R. G.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2002, 41, 4236–4238. doi:10.1002/1521-3773(20021115)41:22<4236::AID-ANIE4236>3.0.CO;2-W
Return to citation in text: [1] [2] -
Henkel, B.; Stenzel, W.; Schotten, T. Bioorg. Med. Chem. Lett. 2000, 10, 975–977. doi:10.1016/S0960-894X(00)00143-8
Return to citation in text: [1] [2] -
Boruah, M.; Konwar, D.; Sharma, S. D. Tetrahedron Lett. 2007, 48, 4535–4537. doi:10.1016/j.tetlet.2007.04.131
Return to citation in text: [1] [2] -
Tsuge, O.; Kanemasa, S.; Takenaka, S. Bull. Chem. Soc. Jpn. 1985, 58, 3137–3157. doi:10.1246/bcsj.58.3137
Return to citation in text: [1] [2] -
Kanemasa, S.; Takenaka, S.; Watanabe, H.; Tsuge, O. J. Org. Chem. 1989, 54, 420–424. doi:10.1021/jo00263a030
Return to citation in text: [1] [2] -
Najera, C.; Sansano, J. M. Angew. Chem., Int. Ed. 2005, 44, 6272–6276. doi:10.1002/anie.200501074
Return to citation in text: [1] [2] -
Zhang, Z.; Chan, G. K.-L.; Li, J.; Fong, W.-F.; Cheung, H.-Y. Chem. Pharm. Bull. 2008, 56, 1229–1233. doi:10.1248/cpb.56.1229
Return to citation in text: [1] -
Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863–910. doi:10.1021/cr970324e
Return to citation in text: [1] -
Girgis, A. S. Eur. J. Med. Chem. 2009, 44, 91–100. doi:10.1016/j.ejmech.2008.03.013
Return to citation in text: [1] -
Girgis, A. S. Eur. J. Med. Chem. 2009, 44, 1257–1264. doi:10.1016/j.ejmech.2008.09.007
Return to citation in text: [1] -
Murugan, R.; Anbazhagan, S.; Narayanan, S. S. Eur. J. Med. Chem. 2009, 44, 3272–3279. doi:10.1016/j.ejmech.2009.03.035
Return to citation in text: [1] -
Hazra, A.; Mondal, S.; Maity, A.; Naskar, S.; Saha, P.; Paira, R.; Sahu, K. B.; Paira, P.; Ghosh, S.; Sinha, C.; Samanta, A.; Banerjee, S.; Mondal, N. B. Eur. J. Med. Chem. 2011, 46, 2132–2140. doi:10.1016/j.ejmech.2011.02.066
Return to citation in text: [1] -
Mondal, S.; Maity, A.; Paira, R.; Banerjee, M.; Bharitkar, Y.; Hazra, A.; Banerjee, S.; Mondal, N. B. Tetrahedron Lett. 2012, 53, 6288–6291. doi:10.1016/j.tetlet.2012.09.033
Return to citation in text: [1] -
Paira, R.; Mondal, S.; Chowdhury, A.; Banerjee, M.; Maity, A.; Hazra, A.; Mondal, N. B. Tetrahedron Lett. 2013, 54, 3046–3050. doi:10.1016/j.tetlet.2013.03.095
Return to citation in text: [1] -
Banerjee, M.; Kumar, S.; Paira, R.; Ghosh, S.; Karmakar, S.; Mondal, N. B. Lett. Drug Des. Discovery 2014, 11, 104–113. doi:10.2174/15701808113109990046
Return to citation in text: [1] [2] [3] -
Goldfarb, D. S. U.S. Patent US 20090163545 A1 20090625, 2009.
Return to citation in text: [1] -
Saha, P.; Naskar, S.; Paira, P.; Hazra, A.; Sahu, K. B.; Paira, R.; Banerjee, S.; Mondal, N. B. Green Chem. 2009, 11, 931–934. doi:10.1039/b902916h
Return to citation in text: [1] -
Paira, R.; Paira, P.; Maity, A.; Mondal, S.; Hazra, A.; Sahu, K. B.; Naskar, S.; Saha, P.; Banerjee, M.; Mondal, N. B. Tetrahedron Lett. 2010, 51, 3200–3204. doi:10.1016/j.tetlet.2010.04.034
Return to citation in text: [1] -
Naskar, S.; Saha, P.; Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Hazra, A.; Paira, P.; Banerjee, S.; Mondal, N. B. Tetrahedron Lett. 2010, 51, 1437–1440. doi:10.1016/j.tetlet.2010.01.030
Return to citation in text: [1] -
Saha, P.; Naskar, S.; Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Paira, P.; Hazra, A.; Bhattacharya, D.; Banerjee, S.; Mondal, N. B. Synthesis 2010, 486–492. doi:10.1055/s-0029-1217143
Return to citation in text: [1] [2] -
Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Naskar, S.; Saha, P.; Hazra, A.; Kundu, S.; Banerjee, S.; Mondal, N. B. Tetrahedron Lett. 2011, 52, 5516–5520. doi:10.1016/j.tetlet.2011.08.080
Return to citation in text: [1] -
Reymond, S.; Cossy, J. Chem. Rev. 2008, 108, 5359–5406. doi:10.1021/cr078346g
Return to citation in text: [1] -
Dias, L. C. J. Braz. Chem. Soc. 1997, 8, 289–332. doi:10.1590/S0103-50531997000400002
Return to citation in text: [1]
| 1. | Pandey, G.; Banerjee, P.; Gadre, S. R. Chem. Rev. 2006, 106, 4484–4517. doi:10.1021/cr050011g |
| 2. | Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765–2810. doi:10.1021/cr040004c |
| 3. | Gu, Y. G.; Xu, Y.; Krueger, A. C.; Madigan, D.; Sham, H. L. Tetrahedron Lett. 2002, 43, 955–957. doi:10.1016/S0040-4039(01)02303-6 |
| 4. | Nair, V.; Sheela, K. C.; Rath, N. P.; Eigendorf, G. K. Tetrahedron Lett. 2000, 41, 6217–6221. doi:10.1016/S0040-4039(00)01032-7 |
| 5. | Gothelf, A. S.; Gothelf, K. V.; Hazell, R. G.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2002, 41, 4236–4238. doi:10.1002/1521-3773(20021115)41:22<4236::AID-ANIE4236>3.0.CO;2-W |
| 6. | Henkel, B.; Stenzel, W.; Schotten, T. Bioorg. Med. Chem. Lett. 2000, 10, 975–977. doi:10.1016/S0960-894X(00)00143-8 |
| 7. | Boruah, M.; Konwar, D.; Sharma, S. D. Tetrahedron Lett. 2007, 48, 4535–4537. doi:10.1016/j.tetlet.2007.04.131 |
| 8. | Tsuge, O.; Kanemasa, S.; Takenaka, S. Bull. Chem. Soc. Jpn. 1985, 58, 3137–3157. doi:10.1246/bcsj.58.3137 |
| 9. | Kanemasa, S.; Takenaka, S.; Watanabe, H.; Tsuge, O. J. Org. Chem. 1989, 54, 420–424. doi:10.1021/jo00263a030 |
| 16. | Hazra, A.; Mondal, S.; Maity, A.; Naskar, S.; Saha, P.; Paira, R.; Sahu, K. B.; Paira, P.; Ghosh, S.; Sinha, C.; Samanta, A.; Banerjee, S.; Mondal, N. B. Eur. J. Med. Chem. 2011, 46, 2132–2140. doi:10.1016/j.ejmech.2011.02.066 |
| 17. | Mondal, S.; Maity, A.; Paira, R.; Banerjee, M.; Bharitkar, Y.; Hazra, A.; Banerjee, S.; Mondal, N. B. Tetrahedron Lett. 2012, 53, 6288–6291. doi:10.1016/j.tetlet.2012.09.033 |
| 18. | Paira, R.; Mondal, S.; Chowdhury, A.; Banerjee, M.; Maity, A.; Hazra, A.; Mondal, N. B. Tetrahedron Lett. 2013, 54, 3046–3050. doi:10.1016/j.tetlet.2013.03.095 |
| 19. | Banerjee, M.; Kumar, S.; Paira, R.; Ghosh, S.; Karmakar, S.; Mondal, N. B. Lett. Drug Des. Discovery 2014, 11, 104–113. doi:10.2174/15701808113109990046 |
| 1. | Pandey, G.; Banerjee, P.; Gadre, S. R. Chem. Rev. 2006, 106, 4484–4517. doi:10.1021/cr050011g |
| 2. | Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765–2810. doi:10.1021/cr040004c |
| 3. | Gu, Y. G.; Xu, Y.; Krueger, A. C.; Madigan, D.; Sham, H. L. Tetrahedron Lett. 2002, 43, 955–957. doi:10.1016/S0040-4039(01)02303-6 |
| 4. | Nair, V.; Sheela, K. C.; Rath, N. P.; Eigendorf, G. K. Tetrahedron Lett. 2000, 41, 6217–6221. doi:10.1016/S0040-4039(00)01032-7 |
| 5. | Gothelf, A. S.; Gothelf, K. V.; Hazell, R. G.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2002, 41, 4236–4238. doi:10.1002/1521-3773(20021115)41:22<4236::AID-ANIE4236>3.0.CO;2-W |
| 6. | Henkel, B.; Stenzel, W.; Schotten, T. Bioorg. Med. Chem. Lett. 2000, 10, 975–977. doi:10.1016/S0960-894X(00)00143-8 |
| 7. | Boruah, M.; Konwar, D.; Sharma, S. D. Tetrahedron Lett. 2007, 48, 4535–4537. doi:10.1016/j.tetlet.2007.04.131 |
| 8. | Tsuge, O.; Kanemasa, S.; Takenaka, S. Bull. Chem. Soc. Jpn. 1985, 58, 3137–3157. doi:10.1246/bcsj.58.3137 |
| 9. | Kanemasa, S.; Takenaka, S.; Watanabe, H.; Tsuge, O. J. Org. Chem. 1989, 54, 420–424. doi:10.1021/jo00263a030 |
| 10. | Najera, C.; Sansano, J. M. Angew. Chem., Int. Ed. 2005, 44, 6272–6276. doi:10.1002/anie.200501074 |
| 11. | Zhang, Z.; Chan, G. K.-L.; Li, J.; Fong, W.-F.; Cheung, H.-Y. Chem. Pharm. Bull. 2008, 56, 1229–1233. doi:10.1248/cpb.56.1229 |
| 12. | Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863–910. doi:10.1021/cr970324e |
| 13. | Girgis, A. S. Eur. J. Med. Chem. 2009, 44, 91–100. doi:10.1016/j.ejmech.2008.03.013 |
| 14. | Girgis, A. S. Eur. J. Med. Chem. 2009, 44, 1257–1264. doi:10.1016/j.ejmech.2008.09.007 |
| 15. | Murugan, R.; Anbazhagan, S.; Narayanan, S. S. Eur. J. Med. Chem. 2009, 44, 3272–3279. doi:10.1016/j.ejmech.2009.03.035 |
| 10. | Najera, C.; Sansano, J. M. Angew. Chem., Int. Ed. 2005, 44, 6272–6276. doi:10.1002/anie.200501074 |
| 19. | Banerjee, M.; Kumar, S.; Paira, R.; Ghosh, S.; Karmakar, S.; Mondal, N. B. Lett. Drug Des. Discovery 2014, 11, 104–113. doi:10.2174/15701808113109990046 |
| 24. | Saha, P.; Naskar, S.; Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Paira, P.; Hazra, A.; Bhattacharya, D.; Banerjee, S.; Mondal, N. B. Synthesis 2010, 486–492. doi:10.1055/s-0029-1217143 |
| 26. | Reymond, S.; Cossy, J. Chem. Rev. 2008, 108, 5359–5406. doi:10.1021/cr078346g |
| 27. | Dias, L. C. J. Braz. Chem. Soc. 1997, 8, 289–332. doi:10.1590/S0103-50531997000400002 |
| 21. | Saha, P.; Naskar, S.; Paira, P.; Hazra, A.; Sahu, K. B.; Paira, R.; Banerjee, S.; Mondal, N. B. Green Chem. 2009, 11, 931–934. doi:10.1039/b902916h |
| 22. | Paira, R.; Paira, P.; Maity, A.; Mondal, S.; Hazra, A.; Sahu, K. B.; Naskar, S.; Saha, P.; Banerjee, M.; Mondal, N. B. Tetrahedron Lett. 2010, 51, 3200–3204. doi:10.1016/j.tetlet.2010.04.034 |
| 23. | Naskar, S.; Saha, P.; Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Hazra, A.; Paira, P.; Banerjee, S.; Mondal, N. B. Tetrahedron Lett. 2010, 51, 1437–1440. doi:10.1016/j.tetlet.2010.01.030 |
| 24. | Saha, P.; Naskar, S.; Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Paira, P.; Hazra, A.; Bhattacharya, D.; Banerjee, S.; Mondal, N. B. Synthesis 2010, 486–492. doi:10.1055/s-0029-1217143 |
| 25. | Paira, R.; Mondal, S.; Maity, A.; Sahu, K. B.; Naskar, S.; Saha, P.; Hazra, A.; Kundu, S.; Banerjee, S.; Mondal, N. B. Tetrahedron Lett. 2011, 52, 5516–5520. doi:10.1016/j.tetlet.2011.08.080 |
| 19. | Banerjee, M.; Kumar, S.; Paira, R.; Ghosh, S.; Karmakar, S.; Mondal, N. B. Lett. Drug Des. Discovery 2014, 11, 104–113. doi:10.2174/15701808113109990046 |
© 2014 Paira et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)