Abstract
A synthetic protocol for the construction of new meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins is described starting from 5-(4-amino-3-nitrophenyl)-10,15,20-triphenylporphyrin. The reaction of this porphyrin with 2,5-dimethoxytetrahydrofuran, followed by the reduction of the nitro group in the presence of NiCl2/NaBH4 afforded 5-(3-amino-4-(pyrrol-1-yl)phenyl)-10,15,20-triphenylporphyrin. This triphenylporphyrin underwent a Pictet–Spengler cyclization after the reaction with various aromatic aldehydes followed by in situ KMnO4 oxidation to form target porphyrin analogues in good yields. The structures of all synthesized products were established on the basis of spectral data and elemental analyses.

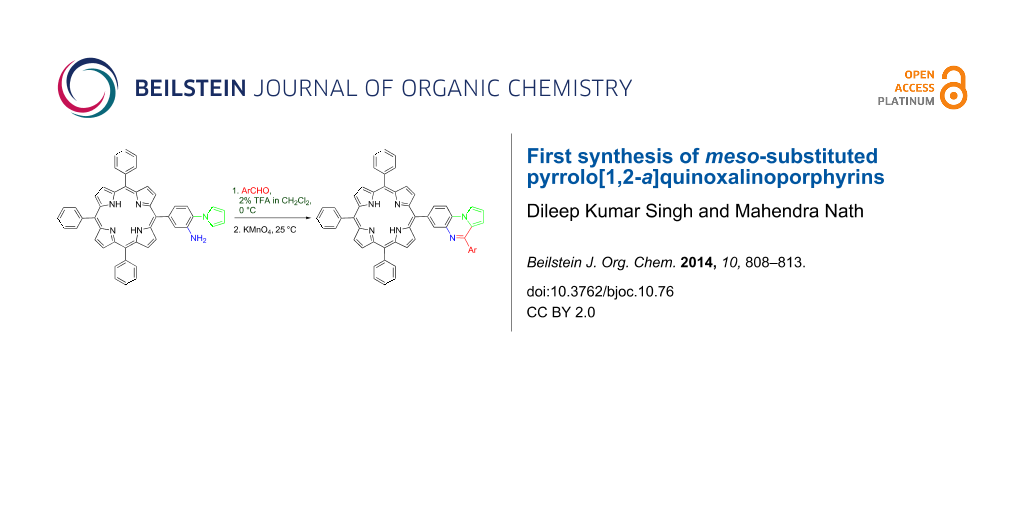
Graphical Abstract
Introduction
Many natural porphyrins are known to play essential roles in a number of biological processes including oxygen transport [1], solar energy conservation [2-4] and photosynthesis [5]. Owing to the expanded π-conjugation system as well as good thermal stabilities, various artificial porphyrins have been prepared as promising materials for organic photonic and electronic applications [6-9]. In addition, porphyrins fused with external aromatic systems exhibit a broad range of applications in diverse areas such as molecular devices [10-13], organic light emitting diodes [14,15], near infrared dyes [16-18], hybrid solar cells [19-22], and biosensors [23-25] due to their intense optical absorptions and photoluminescence characteristics. On the other hand, compounds containing a pyrrolo[1,2-a]quinoxaline subunit display a wide spectrum of biological profiles as antagonists [26,27], PARP-1 inhibitors [28], anticancer agents [29,30], anti-HIV agents [31], and antimalarial agents [32,33]. These molecules are also important intermediates for the construction of 5-HT3 receptor agonists [34,35] and are useful as fluorescent materials for various applications [36,37].
In recent years, numerous covalent or non-covalent supra-porphyrin arrays, based on donor–acceptor architectures have been constructed for mimicking the natural photosynthetic light harvesting systems [38-40]. Additionally, a variety of biologically important functional groups were also introduced on the periphery of meso-substituted porphyrins to develop efficient photosensitizers for photodynamic therapy applications [41-43]. However, the porphyrins with a pyrrolo[1,2-a]quinoxaline moiety at the meso-positions have not been synthesized and their photophysical properties have not been evaluated yet. By considering the biological and fluorescent properties of these two classes of heterocycles, we envisaged to combine both porphyrin and pyrrolo[1,2-a]quinoxaline units in a single molecular framework to generate novel meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrin analogues. Such hybrid molecules may prove useful for various biological studies and in the development of new photodynamic agents. Therefore, in continuation of our efforts to develop simple and efficient methods [44-48] for the synthesis of diverse porphyrin derivatives from meso-tetraarylporphyrins, we wish to report herein the first synthesis and spectroscopic properties of a novel series of meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins.
Results and Discussion
The synthetic strategy for targeted meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins (4a–h) is depicted in Scheme 1. At first, 5-(4-amino-3-nitrophenyl)-10,15,20-triphenylporphyrin (1) was synthesized from 5,10,15,20-tetraphenylporphyrin (TPP) after a series of reactions [46,49] in five steps. The Clauson–Kaas reaction of porphyrin (1) with 2,5-dimethoxytetrahydrofuran in toluene/acetic acid mixture afforded novel 5-(3-nitro-4-(pyrrol-1-yl)phenyl)-10,15,20-triphenylporphyrin (2) in 89% yield. The reduction of nitroporphyrin 2 was initially carried out by using Sn/HCl, SnCl2·2H2O/HCl, and Pd/C–NaBH4 as reducing agents but the reaction was found to be sluggish and provided an inseparable mixture of products. Instead, nitroporphyrin 2 was successfully reduced to 5-(3-amino-4-(pyrrol-1-yl)phenyl)-10,15,20-triphenylporphyrin (3) in the presence of nickel boride, generated in situ by the reaction of NiCl2 and NaBH4 in a CH2Cl2/MeOH mixture at 25 °C. Finally, the synthesis of novel meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins (4a–h) began via the Pictet–Spengler cyclization reaction [50,51] of 5-(3-amino-4-(pyrrol-1-yl)phenyl)-10,15,20-triphenylporphyrin (3) with various aromatic aldehydes by using 2% TFA in dichloromethane as an acidic catalyst at 0 °C for 5 minutes, followed by aromatization in the presence of KMnO4 at room temperature (Scheme 1).
![[1860-5397-10-76-i1]](/bjoc/content/inline/1860-5397-10-76-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pyrrolo[1,2-a]quinoxalinoporphyrins (4a–h).
Scheme 1: Synthesis of pyrrolo[1,2-a]quinoxalinoporphyrins (4a–h).
The target products were purified by column chromatography over neutral alumina and obtained in 60–76% isolated yields. Furthermore, the π electron-rich free-base porphyrin dyads (4g and 4h) were converted to the corresponding zinc(II) porphyrins (5 and 6) in 84 and 87% yields, respectively, after the treatment with Zn(OAc)2·2H2O in CHCl3/MeOH mixture for 30 minutes at room temperature (Scheme 2).
![[1860-5397-10-76-i2]](/bjoc/content/inline/1860-5397-10-76-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of zinc(II) pyrrolo[1,2-a]quinoxalinoporphyrins 5 and 6.
Scheme 2: Synthesis of zinc(II) pyrrolo[1,2-a]quinoxalinoporphyrins 5 and 6.
All synthesized porphyrins (2, 3, 4a–h, 5 and 6) were characterized on the basis of NMR, IR, UV–vis and mass spectral data in addition to elemental analysis. The proton NMR of newly prepared free-base meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins (4a–h) showed a characteristic singlet around δ −2.7 ppm for two NH protons of the porphyrin core. The β-pyrrolic protons of the porphyrin ring appeared in the downfield region between δ 8.85–9.01 ppm. A characteristic doublet at δ 8.9 and a double doublet at δ 8.3 ppm were assigned to the C-2 and C-6 protons of the meso-phenyl ring fused with the pyrroloquinoxaline moiety. The C-5 proton was found to be merged with nine other meso-phenyl protons and appeared as a multiplet between δ 7.75–7.77 ppm. The remaining six meso-phenyl protons appeared as a multiplet between δ 8.20–8.25 ppm along with a pyrrolic C-1′ proton. In the case of porphyrins (4a–f), the two pyrrolic C-2′ and C-3′ protons of the pyrroloquinoxaline ring appeared as a double doublet at around δ 7.06 ppm and a doublet at around δ 7.19 ppm, respectively. The 1H NMR spectrum of porphyrin 4g displayed these pyrrolic C-2′ and C-3′ protons as a double doublet at δ 7.01 ppm and a doublet at around δ 6.74 ppm, whereas these pyrrolic protons appeared as multiplets between δ 7.09–7.24 ppm in the case of porphyrin 4h. In addition, porphyrin 4h and 6 showed a characteristic singlet for the CH2 protons of the fluorenyl moiety at δ 4.0 and 3.9 ppm, respectively. The IR spectra of all the free-base pyrrolo[1,2-a]quinoxalinoporphyrins showed a peak between 3317–3318 cm−1 due to the NH bond stretching. The structures of porphyrins (2, 3, 4a–h, 5 and 6) were further supported by mass spectral analysis, which revealed the molecular ion peak to be [M + H]+. The electronic absorption and emission data of all the synthesized compounds are presented in Table 1.
Table 1: Electronic absorption and emission data of porphyrins (2, 3, 4a–h, 5 and 6).
| Compound | Absorptiona λmax, nm (ε × 10−4, M−1 cm−1) | Fluorescencea,b (λem/nm) |
|---|---|---|
| 2 | 421 (56.24), 517 (2.82), 551 (1.26), 593 (0.41), 647 (0.54) | 651, 717 |
| 3 | 421 (45.66), 517 (2.56), 550 (1.28), 597 (0.31), 647 (0.80) | 653, 717 |
| 4a | 422 (39.00), 517 (2.93), 550 (1.84), 597 (0.33), 647 (0.97) | 653, 717 |
| 4b | 422 (58.51), 517 (3.11), 551 (1.63), 594 (0.37), 647 (0.71) | 652, 717 |
| 4c | 422 (51.51), 517 (2.36), 552 (1.16), 594 (0.41), 648 (0.59) | 652, 716 |
| 4d | 422 (57.74), 517 (3.49), 552 (1.81), 596 (0.30), 648 (0.76) | 652, 715 |
| 4e | 422 (61.39), 517 (3.05), 552 (1.53), 597 (0.19), 648 (0.68) | 652, 716 |
| 4f | 423 (63.00), 517 (3.50), 551 (1.91), 597 (0.34), 647 (0.97) | 652, 717 |
| 4g | 422 (59.97), 517 (3.32), 552 (1.82), 596 (0.32), 648 (0.87) | 652, 717 |
| 4h | 423 (73.28), 517 (3.90), 552 (2.07), 596 (0.38), 648 (0.96) | 652, 717 |
| 5 | 425 (104.90), 554 (3.80), 594 (1.00) | 605, 652 |
| 6 | 425 (117.50), 553 (4.20), 594 (1.20) | 606, 654 |
aAbsorption and emission data were taken for CHCl3 solutions of porphyrins at 298 K. bThe excitation wavelength for emission data is 420 nm.
The UV–vis spectra of newly prepared meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins (4a–h) in chloroform exhibited a typical intense Soret band at ~422 nm and four weaker Q bands at ~517, 552, 596 and 647 nm. In contrast, the zinc(II) pyrrolo[1,2-a]quinoxalinoporphyrin analogues 5 and 6 showed an intense Soret band at ~425 nm and two weaker Q bands at ~553 and 594 nm. In comparison to the TPP and Zn–TPP, the UV–vis spectra of free-base porphyrins 4a–h and zinc porphyrins (5 and 6) were found to be red-sifted by 3 to 4 nm. The electronic absorption spectra of selected free-base porphyrins (4f, 4g, 4h and TPP) and zinc(II) porphyrins (5, 6 and Zn–TPP) are shown in Figure 1a,b. Besides the Soret and Q bands in porphyrins 4g, 4h, 5 and 6, an additional absorption peak originates at 280 and 320 nm due to the presence of pyrene and fluorene units, respectively. Thus, the electronic absorption spectra of these compounds demonstrated the features of both porphyrin and pyrene or fluorene subunits and suggest that there is no significant interaction between the attached chromophore and the porphyrin ring in the ground state.
![[1860-5397-10-76-1]](/bjoc/content/figures/1860-5397-10-76-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Electronic absorption spectra of free-base porphyrins 4f, 4g, 4h and TPP in CHCl3 (1 × 10−6 mol L−1) at 298 K. (b) Electronic absorption spectra of zinc porphyrins (5, 6 and Zn–TPP) in CHCl3 (2 × 10−6 mol L−1) at 298 K. The inset in both (a) and (b) shows the Q bands. (c) Fluorescence spectra of porphyrins 4f, 4g, 4h and TPP in CHCl3 (1 × 10−6 mol L−1) at 298 K, λex = 420 nm. (d) Fluorescence spectra of zinc porphyrins (5, 6, and Zn–TPP) in CHCl3 (2 × 10−6 mol L−1) at 298 K, λex = 420 nm.
Figure 1: (a) Electronic absorption spectra of free-base porphyrins 4f, 4g, 4h and TPP in CHCl3 (1 × 10−6 mol...
The fluorescence spectra of porphyrins 4f, 4g, 4h, 5 and 6 were recorded in CHCl3 at the excitation wavelength of 420 nm and are shown in Figure 1c,d. The free-base pyrrolo[1,2-a]quinoxalinoporphyrins 4f, 4g and 4h displayed an emission band and a weak shoulder at ~652 and ~717 nm. These emission bands are found to be slightly intense in comparison to the TPP (Figure 1c). Similarly, the zinc(II) pyrrolo[1,2-a]quinoxalinoporphyrins 5 and 6 showed two fluorescence bands at ~605 and ~652 nm, which are also found to be slightly intense when compared to the emission bands of Zn–TPP (Figure 1d).
Conclusion
In summary, the synthesis of two new porphyrin building blocks, 5-(3-nitro-4-(pyrrol-1-yl)phenyl)-10,15,20-triphenylporphyrin (2) and 5-(3-amino-4-(pyrrol-1-yl)phenyl)-10,15,20-triphenylporphyrin (3), has been accomplished in good yields. The porphyrin 3 was successfully utilized as starting material for the construction of a novel series of meso-substituted pyrrolo[1,2-a]quinoxalinoporphyrins in 60–76% yields via TFA-catalyzed Pictet–Spengler cyclization with aromatic aldehydes followed by in situ oxidation in the presence of KMnO4. These porphyrin architectures may be useful as potential candidates for various biological evaluations.
Acknowledgements
We thank the University of Delhi, India for financial support. DKS is grateful to the Council of Scientific and Industrial Research, New Delhi, India for a junior Research Fellowship. The JEOL ECX 400P (400 MHz) NMR facility at USIC, University of Delhi and SAIF, CDRI, Lucknow are acknowledged for providing the NMR and mass spectra, respectively.
References
-
Denisov, I. G.; Makris, T. M.; Sligar, S. G.; Schlichting, I. Chem. Rev. 2005, 105, 2253–2278. doi:10.1021/cr0307143
Return to citation in text: [1] -
Gust, D.; Moore, T. A.; Moore, A. L. Acc. Chem. Res. 2001, 34, 40–48. doi:10.1021/ar9801301
Return to citation in text: [1] -
Lindsey, J. S.; Mass, O.; Chen, C.-Y. New J. Chem. 2011, 35, 511–516. doi:10.1039/c0nj00977f
Return to citation in text: [1] -
Imahori, H. J. Phys. Chem. B 2004, 108, 6130–6143. doi:10.1021/jp038036b
Return to citation in text: [1] -
Barber, J. Chem. Soc. Rev. 2009, 38, 185–196. doi:10.1039/B802262N
Return to citation in text: [1] -
Wong, W.-Y.; Harvey, P. D. Macromol. Rapid Commun. 2010, 31, 671–713. doi:10.1002/marc.200900690
Return to citation in text: [1] -
Chen, Y.-C.; Hsu, C.-Y.; Lin, R. Y.-Y.; Ho, K.-C.; Lin, J. T. ChemSusChem 2013, 6, 20–35. doi:10.1002/cssc.201200609
Return to citation in text: [1] -
Ichiki, T.; Matsuo, Y.; Nakamura, E. Chem. Commun. 2013, 49, 279–281. doi:10.1039/c2cc36988e
Return to citation in text: [1] -
Humphrey, J. L.; Kuciauskas, D. J. Am. Chem. Soc. 2006, 128, 3902–3903. doi:10.1021/ja0588353
Return to citation in text: [1] -
Linke-Schaetzel, M.; Anson, C. E.; Powell, A. K.; Buth, G.; Palomares, E.; Durrant, J. D.; Balaban, T. S.; Lehn, J.-M. Chem.–Eur. J. 2006, 12, 1931–1940. doi:10.1002/chem.200500602
Return to citation in text: [1] -
Liu, Z.; Yasseri, A. A.; Lindsey, J. S.; Bocian, D. F. Science 2003, 302, 1543–1545. doi:10.1126/science.1090677
Return to citation in text: [1] -
Martin, R. E.; Diederich, F. Angew. Chem., Int. Ed. 1999, 38, 1350–1377. doi:10.1002/(SICI)1521-3773(19990517)38:10<1350::AID-ANIE1350>3.0.CO;2-6
Return to citation in text: [1] -
Conklin, D.; Nanayakkara, S.; Park, T.-H.; Lagadec, M. F.; Stecher, J. T.; Chen, X.; Therien, M. J.; Bonnell, D. A. ACS Nano 2013, 7, 4479–4486. doi:10.1021/nn401071d
Return to citation in text: [1] -
Borek, C.; Hanson, K.; Djurovich, P. I.; Thompson, M. E.; Aznavour, K.; Bau, R.; Sun, Y.; Forrest, S. R.; Brooks, J.; Michalski, L.; Brown, J. Angew. Chem., Int. Ed. 2007, 46, 1109–1112. doi:10.1002/anie.200604240
Return to citation in text: [1] -
Graham, K. R.; Yang, Y.; Sommer, J. R.; Shelton, A. H.; Schanze, K. S.; Xue, J.; Reynolds, J. R. Chem. Mater. 2011, 23, 5305–5312. doi:10.1021/cm202242x
Return to citation in text: [1] -
Mori, H.; Tanaka, T.; Osuka, A. J. Mater. Chem. C 2013, 1, 2500–2519. doi:10.1039/c3tc00932g
Return to citation in text: [1] -
Jiao, C.; Huang, K.-W.; Chi, C.; Wu, J. J. Org. Chem. 2011, 76, 661–664. doi:10.1021/jo1019046
Return to citation in text: [1] -
Jiao, C.; Zu, N.; Huang, K.-W.; Wang, P.; Wu, J. Org. Lett. 2011, 13, 3652–3655. doi:10.1021/ol201303h
Return to citation in text: [1] -
Li, L.-L.; Diau, E. W.-G. Chem. Soc. Rev. 2013, 42, 291–304. doi:10.1039/c2cs35257e
Return to citation in text: [1] -
Balaban, T. S. Acc. Chem. Res. 2005, 38, 612–623. doi:10.1021/ar040211z
Return to citation in text: [1] -
Abdul Almohsin, S.; Cui, J. B. J. Phys. Chem. C 2012, 116, 9433–9438. doi:10.1021/jp301881s
Return to citation in text: [1] -
Mozer, A. J.; Griffith, M. J.; Tsekouras, G.; Wagner, P.; Wallace, G. G.; Mori, S.; Sunahara, K.; Miyashita, M.; Earles, J. C.; Gordon, K. C.; Du, L.; Katoh, R.; Furube, A.; Officer, D. L. J. Am. Chem. Soc. 2009, 131, 15621–15623. doi:10.1021/ja9057713
Return to citation in text: [1] -
Tu, W.; Lei, J.; Wang, P.; Ju, H. Chem.–Eur. J. 2011, 17, 9440–9447. doi:10.1002/chem.201100577
Return to citation in text: [1] -
Wang, Q.; Lei, J.; Deng, S.; Zhang, L.; Ju, H. Chem. Commun. 2013, 49, 916–918. doi:10.1039/c2cc37664d
Return to citation in text: [1] -
Lvova, L.; Di Natale, C.; Paolesse, R. Sens. Actuators, B 2013, 179, 21–31. doi:10.1016/j.snb.2012.10.014
Return to citation in text: [1] -
Szabó, G.; Kiss, R.; Páyer-Lengyel, D.; Vukics, K.; Szikra, J.; Baki, A.; Molnár, L.; Fischer, J.; Keserű, G. M. Bioorg. Med. Chem. Lett. 2009, 19, 3471–3475. doi:10.1016/j.bmcl.2009.05.010
Return to citation in text: [1] -
Guillon, J.; Dallemagne, P.; Pfeiffer, B.; Renard, P.; Manechez, D.; Kervran, A.; Rault, S. Eur. J. Med. Chem. 1998, 33, 293–308. doi:10.1016/S0223-5234(98)80063-9
Return to citation in text: [1] -
Miyashiro, J.; Woods, K. W.; Park, C. H.; Liu, X.; Shi, Y.; Johnson, E. F.; Bouska, J. J.; Olson, A. M.; Luo, Y.; Fry, E. H.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2009, 19, 4050–4054. doi:10.1016/j.bmcl.2009.06.016
Return to citation in text: [1] -
Milne, J.; Normington, K. D.; Milburn, M. Tetrahydroquinoxalinone sirtuin modulators. WO Patent WO2006094210 A2, Sept 8, 2006.
Return to citation in text: [1] -
Desplat, V.; Moreau, S.; Gay, A.; Fabre, S. B.; Thiolat, D.; Massip, S.; Macky, G.; Godde, F.; Mossalayi, D.; Jarry, C.; Guillon, J. J. Enzyme Inhib. Med. Chem. 2010, 25, 204–215. doi:10.3109/14756360903169881
Return to citation in text: [1] -
Fan, L.-L.; Huang, N.; Yang, R.-G.; He, S.-Z.; Yang, L.-M.; Xu, H.; Zheng, Y.-T. Lett. Drug Des. Discovery 2012, 9, 44–47. doi:10.2174/157018012798193026
Return to citation in text: [1] -
Guillon, J.; Mouray, E.; Moreau, S.; Mullié, C.; Forfar, I.; Desplat, V.; Belisle-Fabre, S.; Pinaud, N.; Ravanello, F.; Le-Naour, A.; Léger, J.-M.; Gosmann, G.; Jarry, C.; Déléris, G.; Sonnet, P.; Grellier, P. Eur. J. Med. Chem. 2011, 46, 2310–2326. doi:10.1016/j.ejmech.2011.03.014
Return to citation in text: [1] -
van Heerden, L.; Cloete, T. T.; Breytenbach, J. W.; de Kock, C.; Smith, P. J.; Breytenbach, J. C.; N’Da, D. D. Eur. J. Med. Chem. 2012, 55, 335–345. doi:10.1016/j.ejmech.2012.07.037
Return to citation in text: [1] -
Campiani, G.; Morelli, E.; Gemma, S.; Nacci, V.; Butini, S.; Hamon, M.; Novellino, E.; Greco, G.; Cagnotto, A.; Goegan, M.; Cervo, L.; Dalla Valle, F.; Fracasso, C.; Caccia, S.; Mennini, T. J. Med. Chem. 1999, 42, 4362–4379. doi:10.1021/jm990151g
Return to citation in text: [1] -
Katounina, T.; Besret, L.; Dhilly, M.; Petit-Taboué, M.-C.; Barbelivien, A.; Baron, J.-C.; Dauphin, F.; Barré, L. Bioorg. Med. Chem. 1998, 6, 789–795. doi:10.1016/S0968-0896(98)00035-2
Return to citation in text: [1] -
Çarbas, B. B.; Kivrak, A.; Zora, M.; Önal, A. M. React. Funct. Polym. 2011, 71, 579–587. doi:10.1016/j.reactfunctpolym.2011.02.008
Return to citation in text: [1] -
Achelle, S.; Baudequin, C.; Plé, N. Dyes Pigm. 2013, 98, 575–600. doi:10.1016/j.dyepig.2013.03.030
Return to citation in text: [1] -
Panda, M. K.; Ladomenou, K.; Coutsolelos, A. G. Coord. Chem. Rev. 2012, 256, 2601–2627. doi:10.1016/j.ccr.2012.04.041
Return to citation in text: [1] -
Son, H.-J.; Jin, S.; Patwardhan, S.; Wezenberg, S. J.; Jeong, N. C.; So, M.; Wilmer, C. E.; Sarjeant, A. A.; Schatz, G. C.; Snurr, R. Q.; Farha, O. K.; Wiederrecht, G. P.; Hupp, J. T. J. Am. Chem. Soc. 2013, 135, 862–869. doi:10.1021/ja310596a
Return to citation in text: [1] -
Yang, J.; Yoon, M.-C.; Yoo, H.; Kim, P.; Kim, D. Chem. Soc. Rev. 2012, 41, 4808–4826. doi:10.1039/c2cs35022j
Return to citation in text: [1] -
Sternberg, E. D.; Dolphin, D.; Brückner, C. Tetrahedron 1998, 54, 4151–4202. doi:10.1016/S0040-4020(98)00015-5
Return to citation in text: [1] -
Ballut, S.; Makky, A.; Chauvin, B.; Michel, J.-P.; Kasselouri, A.; Maillard, P.; Rosilio, V. Org. Biomol. Chem. 2012, 10, 4485–4495. doi:10.1039/c2ob25181g
Return to citation in text: [1] -
Stamati, I.; Kuimova, M. K.; Lion, M.; Yahioglu, G.; Phillips, D.; Deonarain, M. P. Photochem. Photobiol. Sci. 2010, 9, 1033–1041. doi:10.1039/c0pp00038h
Return to citation in text: [1] -
Sharma, S.; Nath, M. New J. Chem. 2011, 35, 1630–1639. doi:10.1039/c1nj20248k
Return to citation in text: [1] -
Sharma, S.; Nath, M. J. Heterocycl. Chem. 2012, 49, 88–92. doi:10.1002/jhet.664
Return to citation in text: [1] -
Sharma, S.; Nath, M. Dyes Pigm. 2012, 92, 1241–1249. doi:10.1016/j.dyepig.2011.07.022
Return to citation in text: [1] [2] -
Bhatt, R. K.; Sharma, S.; Nath, M. Monatsh. Chem. 2012, 143, 309–316. doi:10.1007/s00706-011-0625-0
Return to citation in text: [1] -
Sharma, S.; Nath, M. Beilstein J. Org. Chem. 2013, 9, 496–502. doi:10.3762/bjoc.9.53
Return to citation in text: [1] -
Zhang, H.-L.; Shi, W.-M.; Wu, J. Heterocycles 2005, 65, 3001–3006. doi:10.3987/COM-05-10555
Return to citation in text: [1] -
Cox, E. D.; Cook, J. M. Chem. Rev. 1995, 95, 1797–1842. doi:10.1021/cr00038a004
Return to citation in text: [1] -
Agarwal, P. K.; Sawant, D.; Sharma, S.; Kundu, B. Eur. J. Org. Chem. 2009, 292–303. doi:10.1002/ejoc.200800929
Return to citation in text: [1]
| 44. | Sharma, S.; Nath, M. New J. Chem. 2011, 35, 1630–1639. doi:10.1039/c1nj20248k |
| 45. | Sharma, S.; Nath, M. J. Heterocycl. Chem. 2012, 49, 88–92. doi:10.1002/jhet.664 |
| 46. | Sharma, S.; Nath, M. Dyes Pigm. 2012, 92, 1241–1249. doi:10.1016/j.dyepig.2011.07.022 |
| 47. | Bhatt, R. K.; Sharma, S.; Nath, M. Monatsh. Chem. 2012, 143, 309–316. doi:10.1007/s00706-011-0625-0 |
| 48. | Sharma, S.; Nath, M. Beilstein J. Org. Chem. 2013, 9, 496–502. doi:10.3762/bjoc.9.53 |
| 38. | Panda, M. K.; Ladomenou, K.; Coutsolelos, A. G. Coord. Chem. Rev. 2012, 256, 2601–2627. doi:10.1016/j.ccr.2012.04.041 |
| 39. | Son, H.-J.; Jin, S.; Patwardhan, S.; Wezenberg, S. J.; Jeong, N. C.; So, M.; Wilmer, C. E.; Sarjeant, A. A.; Schatz, G. C.; Snurr, R. Q.; Farha, O. K.; Wiederrecht, G. P.; Hupp, J. T. J. Am. Chem. Soc. 2013, 135, 862–869. doi:10.1021/ja310596a |
| 40. | Yang, J.; Yoon, M.-C.; Yoo, H.; Kim, P.; Kim, D. Chem. Soc. Rev. 2012, 41, 4808–4826. doi:10.1039/c2cs35022j |
| 41. | Sternberg, E. D.; Dolphin, D.; Brückner, C. Tetrahedron 1998, 54, 4151–4202. doi:10.1016/S0040-4020(98)00015-5 |
| 42. | Ballut, S.; Makky, A.; Chauvin, B.; Michel, J.-P.; Kasselouri, A.; Maillard, P.; Rosilio, V. Org. Biomol. Chem. 2012, 10, 4485–4495. doi:10.1039/c2ob25181g |
| 43. | Stamati, I.; Kuimova, M. K.; Lion, M.; Yahioglu, G.; Phillips, D.; Deonarain, M. P. Photochem. Photobiol. Sci. 2010, 9, 1033–1041. doi:10.1039/c0pp00038h |
| 1. | Denisov, I. G.; Makris, T. M.; Sligar, S. G.; Schlichting, I. Chem. Rev. 2005, 105, 2253–2278. doi:10.1021/cr0307143 |
| 10. | Linke-Schaetzel, M.; Anson, C. E.; Powell, A. K.; Buth, G.; Palomares, E.; Durrant, J. D.; Balaban, T. S.; Lehn, J.-M. Chem.–Eur. J. 2006, 12, 1931–1940. doi:10.1002/chem.200500602 |
| 11. | Liu, Z.; Yasseri, A. A.; Lindsey, J. S.; Bocian, D. F. Science 2003, 302, 1543–1545. doi:10.1126/science.1090677 |
| 12. | Martin, R. E.; Diederich, F. Angew. Chem., Int. Ed. 1999, 38, 1350–1377. doi:10.1002/(SICI)1521-3773(19990517)38:10<1350::AID-ANIE1350>3.0.CO;2-6 |
| 13. | Conklin, D.; Nanayakkara, S.; Park, T.-H.; Lagadec, M. F.; Stecher, J. T.; Chen, X.; Therien, M. J.; Bonnell, D. A. ACS Nano 2013, 7, 4479–4486. doi:10.1021/nn401071d |
| 34. | Campiani, G.; Morelli, E.; Gemma, S.; Nacci, V.; Butini, S.; Hamon, M.; Novellino, E.; Greco, G.; Cagnotto, A.; Goegan, M.; Cervo, L.; Dalla Valle, F.; Fracasso, C.; Caccia, S.; Mennini, T. J. Med. Chem. 1999, 42, 4362–4379. doi:10.1021/jm990151g |
| 35. | Katounina, T.; Besret, L.; Dhilly, M.; Petit-Taboué, M.-C.; Barbelivien, A.; Baron, J.-C.; Dauphin, F.; Barré, L. Bioorg. Med. Chem. 1998, 6, 789–795. doi:10.1016/S0968-0896(98)00035-2 |
| 6. | Wong, W.-Y.; Harvey, P. D. Macromol. Rapid Commun. 2010, 31, 671–713. doi:10.1002/marc.200900690 |
| 7. | Chen, Y.-C.; Hsu, C.-Y.; Lin, R. Y.-Y.; Ho, K.-C.; Lin, J. T. ChemSusChem 2013, 6, 20–35. doi:10.1002/cssc.201200609 |
| 8. | Ichiki, T.; Matsuo, Y.; Nakamura, E. Chem. Commun. 2013, 49, 279–281. doi:10.1039/c2cc36988e |
| 9. | Humphrey, J. L.; Kuciauskas, D. J. Am. Chem. Soc. 2006, 128, 3902–3903. doi:10.1021/ja0588353 |
| 36. | Çarbas, B. B.; Kivrak, A.; Zora, M.; Önal, A. M. React. Funct. Polym. 2011, 71, 579–587. doi:10.1016/j.reactfunctpolym.2011.02.008 |
| 37. | Achelle, S.; Baudequin, C.; Plé, N. Dyes Pigm. 2013, 98, 575–600. doi:10.1016/j.dyepig.2013.03.030 |
| 31. | Fan, L.-L.; Huang, N.; Yang, R.-G.; He, S.-Z.; Yang, L.-M.; Xu, H.; Zheng, Y.-T. Lett. Drug Des. Discovery 2012, 9, 44–47. doi:10.2174/157018012798193026 |
| 2. | Gust, D.; Moore, T. A.; Moore, A. L. Acc. Chem. Res. 2001, 34, 40–48. doi:10.1021/ar9801301 |
| 3. | Lindsey, J. S.; Mass, O.; Chen, C.-Y. New J. Chem. 2011, 35, 511–516. doi:10.1039/c0nj00977f |
| 4. | Imahori, H. J. Phys. Chem. B 2004, 108, 6130–6143. doi:10.1021/jp038036b |
| 32. | Guillon, J.; Mouray, E.; Moreau, S.; Mullié, C.; Forfar, I.; Desplat, V.; Belisle-Fabre, S.; Pinaud, N.; Ravanello, F.; Le-Naour, A.; Léger, J.-M.; Gosmann, G.; Jarry, C.; Déléris, G.; Sonnet, P.; Grellier, P. Eur. J. Med. Chem. 2011, 46, 2310–2326. doi:10.1016/j.ejmech.2011.03.014 |
| 33. | van Heerden, L.; Cloete, T. T.; Breytenbach, J. W.; de Kock, C.; Smith, P. J.; Breytenbach, J. C.; N’Da, D. D. Eur. J. Med. Chem. 2012, 55, 335–345. doi:10.1016/j.ejmech.2012.07.037 |
| 23. | Tu, W.; Lei, J.; Wang, P.; Ju, H. Chem.–Eur. J. 2011, 17, 9440–9447. doi:10.1002/chem.201100577 |
| 24. | Wang, Q.; Lei, J.; Deng, S.; Zhang, L.; Ju, H. Chem. Commun. 2013, 49, 916–918. doi:10.1039/c2cc37664d |
| 25. | Lvova, L.; Di Natale, C.; Paolesse, R. Sens. Actuators, B 2013, 179, 21–31. doi:10.1016/j.snb.2012.10.014 |
| 28. | Miyashiro, J.; Woods, K. W.; Park, C. H.; Liu, X.; Shi, Y.; Johnson, E. F.; Bouska, J. J.; Olson, A. M.; Luo, Y.; Fry, E. H.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2009, 19, 4050–4054. doi:10.1016/j.bmcl.2009.06.016 |
| 19. | Li, L.-L.; Diau, E. W.-G. Chem. Soc. Rev. 2013, 42, 291–304. doi:10.1039/c2cs35257e |
| 20. | Balaban, T. S. Acc. Chem. Res. 2005, 38, 612–623. doi:10.1021/ar040211z |
| 21. | Abdul Almohsin, S.; Cui, J. B. J. Phys. Chem. C 2012, 116, 9433–9438. doi:10.1021/jp301881s |
| 22. | Mozer, A. J.; Griffith, M. J.; Tsekouras, G.; Wagner, P.; Wallace, G. G.; Mori, S.; Sunahara, K.; Miyashita, M.; Earles, J. C.; Gordon, K. C.; Du, L.; Katoh, R.; Furube, A.; Officer, D. L. J. Am. Chem. Soc. 2009, 131, 15621–15623. doi:10.1021/ja9057713 |
| 29. | Milne, J.; Normington, K. D.; Milburn, M. Tetrahydroquinoxalinone sirtuin modulators. WO Patent WO2006094210 A2, Sept 8, 2006. |
| 30. | Desplat, V.; Moreau, S.; Gay, A.; Fabre, S. B.; Thiolat, D.; Massip, S.; Macky, G.; Godde, F.; Mossalayi, D.; Jarry, C.; Guillon, J. J. Enzyme Inhib. Med. Chem. 2010, 25, 204–215. doi:10.3109/14756360903169881 |
| 16. | Mori, H.; Tanaka, T.; Osuka, A. J. Mater. Chem. C 2013, 1, 2500–2519. doi:10.1039/c3tc00932g |
| 17. | Jiao, C.; Huang, K.-W.; Chi, C.; Wu, J. J. Org. Chem. 2011, 76, 661–664. doi:10.1021/jo1019046 |
| 18. | Jiao, C.; Zu, N.; Huang, K.-W.; Wang, P.; Wu, J. Org. Lett. 2011, 13, 3652–3655. doi:10.1021/ol201303h |
| 46. | Sharma, S.; Nath, M. Dyes Pigm. 2012, 92, 1241–1249. doi:10.1016/j.dyepig.2011.07.022 |
| 49. | Zhang, H.-L.; Shi, W.-M.; Wu, J. Heterocycles 2005, 65, 3001–3006. doi:10.3987/COM-05-10555 |
| 14. | Borek, C.; Hanson, K.; Djurovich, P. I.; Thompson, M. E.; Aznavour, K.; Bau, R.; Sun, Y.; Forrest, S. R.; Brooks, J.; Michalski, L.; Brown, J. Angew. Chem., Int. Ed. 2007, 46, 1109–1112. doi:10.1002/anie.200604240 |
| 15. | Graham, K. R.; Yang, Y.; Sommer, J. R.; Shelton, A. H.; Schanze, K. S.; Xue, J.; Reynolds, J. R. Chem. Mater. 2011, 23, 5305–5312. doi:10.1021/cm202242x |
| 26. | Szabó, G.; Kiss, R.; Páyer-Lengyel, D.; Vukics, K.; Szikra, J.; Baki, A.; Molnár, L.; Fischer, J.; Keserű, G. M. Bioorg. Med. Chem. Lett. 2009, 19, 3471–3475. doi:10.1016/j.bmcl.2009.05.010 |
| 27. | Guillon, J.; Dallemagne, P.; Pfeiffer, B.; Renard, P.; Manechez, D.; Kervran, A.; Rault, S. Eur. J. Med. Chem. 1998, 33, 293–308. doi:10.1016/S0223-5234(98)80063-9 |
| 50. | Cox, E. D.; Cook, J. M. Chem. Rev. 1995, 95, 1797–1842. doi:10.1021/cr00038a004 |
| 51. | Agarwal, P. K.; Sawant, D.; Sharma, S.; Kundu, B. Eur. J. Org. Chem. 2009, 292–303. doi:10.1002/ejoc.200800929 |
© 2014 Singh and Nath; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)