Abstract
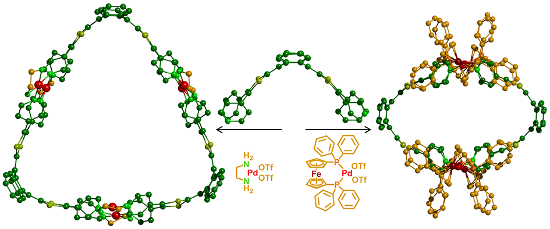
The synthesis of a new Pd coordination-driven self-assembled ring M6L3 constructed from a concave tetrapyridyl π-extended tetrathiafulvalene ligand (exTTF) is described. The same ligand is also able to self-assemble in a M4L2 mode as previously described. Herein, we demonstrate that the bulkiness of the ancillary groups in the Pd complex allows for modulating the size and the shape of the resulting discrete self-assembly, which therefore incorporate two (M4L2) or three (M6L3) electroactive exTTF sidewalls.
Graphical Abstract
Findings
The coordination-driven approach is a well-established method that has been extensively used to reach more and more sophisticated cage-like discrete molecules [1-19], including redox-active ones [20]. In this context and since this strategy results from one single chemical step (metal to ligand assembly), there is a great interest in controlling the parameters which govern the final size and geometry of the resulting discrete self-assembled structures. Some general trends have first to be considered: i) as awaited from a lower kinetic stability, the ligand exchange process in the case of square-planar Pd(II) complexes is faster than with Pt(II) analogues; ii) the most thermodynamically stable species is formed along the assembly process, but if no evident energetic advantage exists for one structure, a dynamic equilibrium between two or more macrocyclic entities may be observed in solution [21-26]. This is in particular the case with flexible (including long) linear ditopic ligands, which favor the formation of triangle species whereas shorter ones shift the equilibrium towards molecular squares for which the enthalpic gain (less steric constraint) compensates for the entropic penalty. Beside the conformational flexibility of the ligand, parameters such as temperature, concentration and solvent type can influence the equilibrium. Isolation of the species from a given equilibrium has not been often carried out [27,28]. We were able in our case to operate the separation of a mixture of a triangle and a square [29]. The triangle–square dynamic equilibrium also depends on the nature of the ancillary ligand on the metal corner [21,22,26,30-33]. In particular, steric repulsions due to the ancillary ligand may displace the equilibrium towards the triangular species since the latter offers more space around the metal center. A change in the ancillary group can also lead to a modification of the cavity volume for a given cage [34]. Beyond those results, additional important issues still need to be addressed and concern in particular the possibility to obtain, from one given ligand, one single and stable assembly whose cavity size can be controlled.
We recently depicted the preparation and properties of redox-active rings [29,35] and cages [36-40] integrating the tetrathiafulvalene (TTF) skeleton. In particular, we described self-assembled containers prepared from an electron-rich ligand precursor based on the extended-TTF framework (exTTF) [39].
On this basis, we report herein that the size and the shape of coordination-driven self-assembled redox-active cages, constructed from a exTTF-based tetratopic ligand, can be tuned by modulating the bulkiness of the ancillary group on the metal complex precursor.
The tetrapyridyl-exTTF ligand L1 (Scheme 1, Figure 1a) was synthesized through a palladium catalysed C–H arylation from the naked exTTF [39]. We already reported that the self-assembly process of this tetratopic ligand with cis-M(dppf)(OTf)2 (M = Pd or Pt; dppf = 1,1’-bis(diphenylphosphino)ferrocene; OTf = trifluoromethanesulfonate) in nitromethane at 40 °C converged into a single symmetrical M4L2 discrete species (Scheme 1, Figure 1b) [39]. It is worth noting that the through space interaction between the phenyl rings of the bulky 1,1’-bis(diphenylphosphino) ferrocene (dppf) coligand and the pyridine moieties force the exTTF unit to increase significantly its curvature in comparison to ligand L1 (56° vs 86° respectively between the 1,3-dithiol-2-ylidene mean planes (Figure 1)). This leads to the formation of the compact M4L2 assembly in which the pyridyl units are wedged between the dppf units, producing therefore a robust assembly affording an oblate spheroidal cavity. On this basis and considering the relative flexibility of the large exTTF moiety, we assumed that the bulkiness of the metal complex coligand could be adjusted to tune the macrocycle size and shape.
![[1860-5397-11-108-i1]](/bjoc/content/inline/1860-5397-11-108-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of ligand L1, self-assemblies M4L2 and M6L3.
Scheme 1: Synthesis of ligand L1, self-assemblies M4L2 and M6L3.
![[1860-5397-11-108-1]](/bjoc/content/figures/1860-5397-11-108-1.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structures of: (a) ligand L1, (b) self-assembly M4L2. For clarity, H atoms and TfO− counteranions have been omitted.
Figure 1: X-ray crystal structures of: (a) ligand L1, (b) self-assembly M4L2. For clarity, H atoms and TfO− c...
Complexation of ligand L1 with precursor Pd(en)(OTf)2 (en = 1,2-ethylenediamine) was carried out in DMSO at 40 °C and monitored by 1H NMR. In 30 min, the reaction converged into a unique symmetrical discrete species that could be isolated in more than 90% yield after precipitation in ethyl acetate. In contrast with assembly M4L2 for which the presence of through-space interactions (Figure 4a) between the coligand phenyl units (dppf) and pyridyl groups result in an upfield shift of their signals (Figure 2b) compared to L1, the pyridyl protons are in this case shifted downfield (Figure 2c), as expected from coordination to a metal center. The corresponding DOSY NMR shows only one alignment of signals and confirms the formation of one unique species diffusing in solution with a D value of 6.35 × 10−11 m²·s−1 (Figure 2e). An estimated hydrodynamic radius (RH) of 17.2 Å could be calculated from the Stokes–Einstein equation (T = 298 K) for this new discrete system [41]. This result indicates that the latter is larger than the already described M4L2 container (RH = 10.8 Å (Figure 2d)), and that the corresponding size is compatible with the formation of a M6L3 assembly (Scheme 1).
![[1860-5397-11-108-2]](/bjoc/content/figures/1860-5397-11-108-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR, downfield region (α and β signals correspond respectively to α and β pyridyl protons): (a) L1 (DMSO-d6), (b) M4L2 (CD3NO2), (c) M6L3 (DMSO-d6), (d) DOSY NMR of M4L2 (CD3NO2) and (e) DOSY NMR of M6L3 (DMSO-d6).
Figure 2: 1H NMR, downfield region (α and β signals correspond respectively to α and β pyridyl protons): (a) ...
ESI–MS mass spectroscopy experiments were carried out in acetone and agree with a M6L3 stoichiometry in the gas phase for the new assembly, with multicharged isotopic patterns at m/z = 2278.3, 1468.9, 1064.4, 821.8, corresponding respectively to [M6L3-10TfO−]2+, [M6L3-9TfO−]3+, [M6L3-8TfO−]4+, [M6L3-7TfO−]5+ species and matching perfectly with theoretical ones (Supporting Information File 1, Figure S8).
Single crystals of assembly M6L3 were grown by slow diffusion of ethyl acetate in DMSO and allowed for determining unambiguously the solid-state structure by a synchrotron X-ray diffraction study (Figure 3). Whereas the sterically demanding dppf moiety leads to a M4L2 structure characterized by i) exTTF moieties which are highly distorted and ii) short interplanar distances between the phenyl units of the ancillary dppf complex and the pyridyl rings (3.5 Å) (Figure 4a), a much less constrained system is observed in the case of the M6L3 complex, characterizing a prismatic structure which is not driven by steric effects but which is mainly governed by thermodynamic aspects.
![[1860-5397-11-108-3]](/bjoc/content/figures/1860-5397-11-108-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of self-assembly M6L3. For clarity, H atoms and TfO− counteranions have been omitted.
Figure 3: X-ray crystal structure of self-assembly M6L3. For clarity, H atoms and TfO− counteranions have bee...
![[1860-5397-11-108-4]](/bjoc/content/figures/1860-5397-11-108-4.png?scale=1.7&max-width=1024&background=FFFFFF)
Figure 4: Geometry (from XRD) around two Pd centers in M4L2 (a) and M6L3 (b). The exTTF moieties have been cut for clarity.
Figure 4: Geometry (from XRD) around two Pd centers in M4L2 (a) and M6L3 (b). The exTTF moieties have been cu...
The M6L3 assembly forms a trigonal prismatic structure presenting a cavity defined by 17.7 Å high, 19.0 Å edge and 9.5 Å depth. The curvature of the exTTF moiety in the complex (70° between the 1,3-dithiol-2-ylidene mean plans, Figure 3) is intermediate between those observed for the free ligand L1 and the ligand in M4L2, which illustrates a lower ring constraint than in M4L2. In addition to the expected variation of the N(pyridyl)-Pd-N(pyridyl) angle within the distorted square planar – i.e. the dppf complex (M4L2, 86°) and the en one (M6L3, 93°) (Figure 4) –, the change in the ancillary ligand also results in a modification of the rotation angles between the pyridyl units and the 1,3-dithiol-2-ylidene heterocycles. Because of the lower steric demand with the 1,2-ethylenediamine co-ligand, the vicinal pyridyl units are free to rotate around the C-pyridine axis in M6L3, resulting in dihedral angles of 40° and 52° in the crystal (Figure 4b). These values are in the same range as those observed in the free ligand L1 (35° and 63°). By comparison, the pyridyl units in complex M4L2 are tilted with angles of 52° and −69° in the solid (Figure 4a). Those higher values result from the increased steric demand generated by the dppf coligand.
A cyclic voltammetry study of prism M6L3 was carried out in acetonitrile containing 0.1 M NBu4PF6 (Figure 5). Compared to ligand L1 which presents the usual electrochemical behavior of exTTF derivatives, i.e. one poorly electrochemically reversible two-electrons oxidation process, the oxidation of exTTF in self-assembly M6L3 is shifted to higher potential (+0.26 V), which is mainly attributed to the coordination to the Pd center. It can be noted that the redox behavior of M6L3 is very similar to the one of M4L2 [42], with an irreversible oxidation occurring exactly at the same potential (Eox = +0.57 V vs Fc/Fc+), illustrating the fact that the nature of the ancillary ligand does not impact the electronic properties of exTTF in the corresponding self-assemblies.
![[1860-5397-11-108-5]](/bjoc/content/figures/1860-5397-11-108-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Cyclic voltammogram of L1 (c = 10−3 M, CH3CN/CH2Cl2 (v/v 50/50), 0.1 M n-Bu4NPF6, 100 mV·s−1, Pt), M4L2 and M6L3 (c = 10−3 M, CH3CN, 0.1 M n-Bu4NPF6, 100 mV·s−1, Cgr), V vs Fc/Fc+.
Figure 5: Cyclic voltammogram of L1 (c = 10−3 M, CH3CN/CH2Cl2 (v/v 50/50), 0.1 M n-Bu4NPF6, 100 mV·s−1, Pt), M...
In summary, two different coordination-driven discrete self-assemblies varying by the size and the shape can be built from the same tetratopic exTTF-based ligand, simply by changing the ancillary group on the Pd metal center. In particular, whereas a 1,1’-bis(diphenylphosphino)ferrocene co-ligand promotes a clipping of the ligand pyridyl units and leads to a strong curvature of the exTTF moiety integrated in a M4L2 coordination cage, the use of a smaller co-ligand leads to the formation of a larger M6L3 cavity in which the curvature of the exTTF is closer to ligand L1. The new M6L3 system has been fully characterized and exhibits electrochemical properties which are essentially similar to those of M4L2, indicating that the strong π-donating ability of the cavity can be maintained while enlarging its size, and illustrating the high potential of the coordination-driven approach in tuning the size and the shape of a target cavity. This approach constitutes a promising strategy to address the design of organic materials (e.g. for organic photovoltaics or molecular electronic devices). Indeed, mastering the geometry of multicomponent redox-active systems offers a unique opportunity to fine-tuning electronic interactions within the material [43], an issue which is of prime importance for optimizing electron transport in organic materials.
Acknowledgements
The authors gratefully acknowledge the CNRS, the Région des Pays de la Loire and the MENRT for PhD grants (SB and VC), the PIAM (Univ. Angers) and the CRMPO (Univ. Rennes) technical platforms for their assistance in spectroscopic analyses. An access to the synchrotron Soleil – CRISTAL beamline – has been made possible (project 20130173); Drs Pierre Fertey and Sylvain Ravy are warmly acknowledged for their assistance. Finally, the Johnson-Matthey company is acknowledged for the generous providing of palladium salts.
References
-
Amouri, H.; Desmarets, C.; Moussa, J. Chem. Rev. 2012, 112, 2015–2041. doi:10.1021/cr200345v
Return to citation in text: [1] -
MacGillivray, L. R. Angew. Chem., Int. Ed. 2012, 51, 1110–1112. doi:10.1002/anie.201107282
Return to citation in text: [1] -
Chakrabarty, R.; Mukherjee, P. S.; Stang, P. J. Chem. Rev. 2011, 111, 6810–6918. doi:10.1021/cr200077m
Return to citation in text: [1] -
Inokuma, Y.; Kawano, M.; Fujita, M. Nat. Chem. 2011, 3, 349–358. doi:10.1038/nchem.1031
Return to citation in text: [1] -
Jin, P.; Dalgarno, S. J.; Atwood, J. L. Coord. Chem. Rev. 2010, 254, 1760–1768. doi:10.1016/j.ccr.2010.04.009
Return to citation in text: [1] -
De, S.; Mahata, K.; Schmittel, M. Chem. Soc. Rev. 2010, 39, 1555–1575. doi:10.1039/b922293f
Return to citation in text: [1] -
Therrien, B. Eur. J. Inorg. Chem. 2009, 2445–2453. doi:10.1002/ejic.200900180
Return to citation in text: [1] -
Northrop, B. H.; Zheng, Y.-R.; Chi, K.-W.; Stang, P. J. Acc. Chem. Res. 2009, 42, 1554–1563. doi:10.1021/ar900077c
Return to citation in text: [1] -
Stang, P. J. J. Org. Chem. 2009, 74, 2–20. doi:10.1021/jo801682d
Return to citation in text: [1] -
Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340
Return to citation in text: [1] -
Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j
Return to citation in text: [1] -
Northrop, B. H.; Chercka, D.; Stang, P. J. Tetrahedron 2008, 64, 11495–11503. doi:10.1016/j.tet.2008.08.062
Return to citation in text: [1] -
Northrop, B. H.; Yang, H.-B.; Stang, P. J. Chem. Commun. 2008, 5896–5908. doi:10.1039/b811712h
Return to citation in text: [1] -
Cooke, M. W.; Chartrand, D.; Hanan, G. S. Coord. Chem. Rev. 2008, 252, 903–921. doi:10.1016/j.ccr.2008.01.006
Return to citation in text: [1] -
Dalgarno, S. J.; Power, N. P.; Atwood, J. L. Coord. Chem. Rev. 2008, 252, 825–841. doi:10.1016/j.ccr.2007.10.010
Return to citation in text: [1] -
Zangrando, E.; Casanova, M.; Alessio, E. Chem. Rev. 2008, 108, 4979–5013. doi:10.1021/cr8002449
Return to citation in text: [1] -
Mukherjee, S.; Mukherjee, P. S. Chem. Commun. 2014, 50, 2239–2248. doi:10.1039/c3cc49192g
Return to citation in text: [1] -
Mishra, A.; Kang, S. C.; Chi, K.-W. Eur. J. Inorg. Chem. 2013, 5222–5232. doi:10.1002/ejic.201300729
Return to citation in text: [1] -
Cook, T. R.; Vajpayee, V.; Lee, M. H.; Stang, P. J.; Chi, K.-W. Acc. Chem. Res. 2013, 46, 2464–2474. doi:10.1021/ar400010v
Return to citation in text: [1] -
Croué, V.; Goeb, S.; Sallé, M. Chem. Commun. 2015, 51, 7275–7289. doi:10.1039/C5CC00597C
Return to citation in text: [1] -
Uehara, K.; Kasai, K.; Mizuno, N. Inorg. Chem. 2010, 49, 2008–2015. doi:10.1021/ic100011a
Return to citation in text: [1] [2] -
Ferrer, M.; Pedrosa, A.; Rodriguez, L.; Rossell, O.; Vilaseca, M. Inorg. Chem. 2010, 49, 9438–9449. doi:10.1021/ic101150p
Return to citation in text: [1] [2] -
Ghosh, S.; Mukherjee, P. S. Inorg. Chem. 2009, 48, 2605–2613. doi:10.1021/ic802254f
Return to citation in text: [1] -
Sun, Q.-F.; Wong, K. M.-C.; Liu, L.-X.; Huang, H.-P.; Yu, S.-Y.; Yam, V. W.-W.; Li, Y.-Z.; Pan, Y.-J.; Yu, K.-C. Inorg. Chem. 2008, 47, 2142–2154. doi:10.1021/ic701344p
Return to citation in text: [1] -
Yamamoto, T.; Arif, A. M.; Stang, P. J. J. Am. Chem. Soc. 2003, 125, 12309–12317. doi:10.1021/ja0302984
Return to citation in text: [1] -
Fujita, M.; Sasaki, O.; Mitsuhashi, T.; Fujita, T.; Yazaki, J.; Yamaguchi, K.; Ogura, K. Chem. Commun. 1996, 1535–1536. doi:10.1039/cc9960001535
Return to citation in text: [1] [2] -
Maeda, H.; Akuta, R.; Bando, Y.; Takaishi, K.; Uchiyama, M.; Muranaka, A.; Tohnai, N.; Seki, S. Chem. – Eur. J. 2013, 19, 11676–11685. doi:10.1002/chem.201302028
Return to citation in text: [1] -
Schweiger, M.; Seidel, S. R.; Arif, A. M.; Stang, P. J. Inorg. Chem. 2002, 41, 2556–2559. doi:10.1021/ic0112692
Return to citation in text: [1] -
Goeb, S.; Bivaud, S.; Dron, P. I.; Balandier, J.-Y.; Chas, M.; Sallé, M. Chem. Commun. 2012, 48, 3106–3108. doi:10.1039/c2cc00065b
Return to citation in text: [1] [2] -
Holló-Sitkei, E.; Tárkányi, G.; Párkányi, L.; Megyes, T.; Besenyei, G. Eur. J. Inorg. Chem. 2008, 1573–1583. doi:10.1002/ejic.200701189
Return to citation in text: [1] -
Weilandt, T.; Troff, R. W.; Saxell, H.; Rissanen, K.; Schalley, C. A. Inorg. Chem. 2008, 47, 7588–7598. doi:10.1021/ic800334k
Return to citation in text: [1] -
Ferrer, M.; Gutiérrez, A.; Mounir, M.; Rossell, O.; Ruiz, E.; Rang, A.; Engeser, M. Inorg. Chem. 2007, 46, 3395–3406. doi:10.1021/ic062373s
Return to citation in text: [1] -
Ferrer, M.; Mounir, M.; Rossell, O.; Ruiz, E.; Maestro, M. A. Inorg. Chem. 2003, 42, 5890–5899. doi:10.1021/ic034489j
Return to citation in text: [1] -
Fang, Y.; Murase, T.; Sato, S.; Fujita, M. J. Am. Chem. Soc. 2013, 135, 613–615. doi:10.1021/ja311373f
Return to citation in text: [1] -
Balandier, J.-Y.; Chas, M.; Goeb, S.; Dron, P. I.; Rondeau, D.; Belyasmine, A.; Gallego, N.; Sallé, M. New J. Chem. 2011, 35, 165–168. doi:10.1039/C0NJ00545B
Return to citation in text: [1] -
Goeb, S.; Bivaud, S.; Croué, V.; Vajpayee, V.; Allain, M.; Sallé, M. Materials 2014, 7, 611–622. doi:10.3390/ma7010611
Return to citation in text: [1] -
Vajpayee, V.; Bivaud, S.; Goeb, S.; Croué, V.; Allain, M.; Popp, B. V.; Garci, A.; Therrien, B.; Sallé, M. Organometallics 2014, 33, 1651–1658. doi:10.1021/om401142j
Return to citation in text: [1] -
Bivaud, S.; Goeb, S.; Balandier, J. Y.; Chas, M.; Allain, M.; Sallé, M. Eur. J. Inorg. Chem. 2014, 2440–2448. doi:10.1002/ejic.201400060
Return to citation in text: [1] -
Bivaud, S.; Goeb, S.; Croué, V.; Dron, P. I.; Allain, M.; Sallé, M. J. Am. Chem. Soc. 2013, 135, 10018–10021. doi:10.1021/ja404072w
Return to citation in text: [1] [2] [3] [4] -
Bivaud, S.; Balandier, J.-Y.; Chas, M.; Allain, M.; Goeb, S.; Sallé, M. J. Am. Chem. Soc. 2012, 134, 11968–11970. doi:10.1021/ja305451v
Return to citation in text: [1] -
Cohen, Y.; Avram, L.; Frish, L. Angew. Chem., Int. Ed. 2005, 44, 520–554. doi:10.1002/anie.200300637
Return to citation in text: [1] -
M4L2 presents an additional reversible redox system located at a higher potential (Eox = +0.80 V vs Fc/Fc+), corresponding to the reversible oxidation of the appended ferrocene Pd co-ligand.
Return to citation in text: [1] -
Schneebeli, S. T.; Frasconi, M.; Liu, Z.; Wu, Y.; Gardner, D. M.; Strutt, N. L.; Cheng, C.; Carmieli, R.; Wasielewski, M. R.; Stoddart, J. F. Angew. Chem., Int. Ed. 2013, 52, 13100–13104. doi:10.1002/anie.201307984
Return to citation in text: [1]
| 1. | Amouri, H.; Desmarets, C.; Moussa, J. Chem. Rev. 2012, 112, 2015–2041. doi:10.1021/cr200345v |
| 2. | MacGillivray, L. R. Angew. Chem., Int. Ed. 2012, 51, 1110–1112. doi:10.1002/anie.201107282 |
| 3. | Chakrabarty, R.; Mukherjee, P. S.; Stang, P. J. Chem. Rev. 2011, 111, 6810–6918. doi:10.1021/cr200077m |
| 4. | Inokuma, Y.; Kawano, M.; Fujita, M. Nat. Chem. 2011, 3, 349–358. doi:10.1038/nchem.1031 |
| 5. | Jin, P.; Dalgarno, S. J.; Atwood, J. L. Coord. Chem. Rev. 2010, 254, 1760–1768. doi:10.1016/j.ccr.2010.04.009 |
| 6. | De, S.; Mahata, K.; Schmittel, M. Chem. Soc. Rev. 2010, 39, 1555–1575. doi:10.1039/b922293f |
| 7. | Therrien, B. Eur. J. Inorg. Chem. 2009, 2445–2453. doi:10.1002/ejic.200900180 |
| 8. | Northrop, B. H.; Zheng, Y.-R.; Chi, K.-W.; Stang, P. J. Acc. Chem. Res. 2009, 42, 1554–1563. doi:10.1021/ar900077c |
| 9. | Stang, P. J. J. Org. Chem. 2009, 74, 2–20. doi:10.1021/jo801682d |
| 10. | Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340 |
| 11. | Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j |
| 12. | Northrop, B. H.; Chercka, D.; Stang, P. J. Tetrahedron 2008, 64, 11495–11503. doi:10.1016/j.tet.2008.08.062 |
| 13. | Northrop, B. H.; Yang, H.-B.; Stang, P. J. Chem. Commun. 2008, 5896–5908. doi:10.1039/b811712h |
| 14. | Cooke, M. W.; Chartrand, D.; Hanan, G. S. Coord. Chem. Rev. 2008, 252, 903–921. doi:10.1016/j.ccr.2008.01.006 |
| 15. | Dalgarno, S. J.; Power, N. P.; Atwood, J. L. Coord. Chem. Rev. 2008, 252, 825–841. doi:10.1016/j.ccr.2007.10.010 |
| 16. | Zangrando, E.; Casanova, M.; Alessio, E. Chem. Rev. 2008, 108, 4979–5013. doi:10.1021/cr8002449 |
| 17. | Mukherjee, S.; Mukherjee, P. S. Chem. Commun. 2014, 50, 2239–2248. doi:10.1039/c3cc49192g |
| 18. | Mishra, A.; Kang, S. C.; Chi, K.-W. Eur. J. Inorg. Chem. 2013, 5222–5232. doi:10.1002/ejic.201300729 |
| 19. | Cook, T. R.; Vajpayee, V.; Lee, M. H.; Stang, P. J.; Chi, K.-W. Acc. Chem. Res. 2013, 46, 2464–2474. doi:10.1021/ar400010v |
| 29. | Goeb, S.; Bivaud, S.; Dron, P. I.; Balandier, J.-Y.; Chas, M.; Sallé, M. Chem. Commun. 2012, 48, 3106–3108. doi:10.1039/c2cc00065b |
| 43. | Schneebeli, S. T.; Frasconi, M.; Liu, Z.; Wu, Y.; Gardner, D. M.; Strutt, N. L.; Cheng, C.; Carmieli, R.; Wasielewski, M. R.; Stoddart, J. F. Angew. Chem., Int. Ed. 2013, 52, 13100–13104. doi:10.1002/anie.201307984 |
| 27. | Maeda, H.; Akuta, R.; Bando, Y.; Takaishi, K.; Uchiyama, M.; Muranaka, A.; Tohnai, N.; Seki, S. Chem. – Eur. J. 2013, 19, 11676–11685. doi:10.1002/chem.201302028 |
| 28. | Schweiger, M.; Seidel, S. R.; Arif, A. M.; Stang, P. J. Inorg. Chem. 2002, 41, 2556–2559. doi:10.1021/ic0112692 |
| 21. | Uehara, K.; Kasai, K.; Mizuno, N. Inorg. Chem. 2010, 49, 2008–2015. doi:10.1021/ic100011a |
| 22. | Ferrer, M.; Pedrosa, A.; Rodriguez, L.; Rossell, O.; Vilaseca, M. Inorg. Chem. 2010, 49, 9438–9449. doi:10.1021/ic101150p |
| 23. | Ghosh, S.; Mukherjee, P. S. Inorg. Chem. 2009, 48, 2605–2613. doi:10.1021/ic802254f |
| 24. | Sun, Q.-F.; Wong, K. M.-C.; Liu, L.-X.; Huang, H.-P.; Yu, S.-Y.; Yam, V. W.-W.; Li, Y.-Z.; Pan, Y.-J.; Yu, K.-C. Inorg. Chem. 2008, 47, 2142–2154. doi:10.1021/ic701344p |
| 25. | Yamamoto, T.; Arif, A. M.; Stang, P. J. J. Am. Chem. Soc. 2003, 125, 12309–12317. doi:10.1021/ja0302984 |
| 26. | Fujita, M.; Sasaki, O.; Mitsuhashi, T.; Fujita, T.; Yazaki, J.; Yamaguchi, K.; Ogura, K. Chem. Commun. 1996, 1535–1536. doi:10.1039/cc9960001535 |
| 41. | Cohen, Y.; Avram, L.; Frish, L. Angew. Chem., Int. Ed. 2005, 44, 520–554. doi:10.1002/anie.200300637 |
| 20. | Croué, V.; Goeb, S.; Sallé, M. Chem. Commun. 2015, 51, 7275–7289. doi:10.1039/C5CC00597C |
| 42. | M4L2 presents an additional reversible redox system located at a higher potential (Eox = +0.80 V vs Fc/Fc+), corresponding to the reversible oxidation of the appended ferrocene Pd co-ligand. |
| 36. | Goeb, S.; Bivaud, S.; Croué, V.; Vajpayee, V.; Allain, M.; Sallé, M. Materials 2014, 7, 611–622. doi:10.3390/ma7010611 |
| 37. | Vajpayee, V.; Bivaud, S.; Goeb, S.; Croué, V.; Allain, M.; Popp, B. V.; Garci, A.; Therrien, B.; Sallé, M. Organometallics 2014, 33, 1651–1658. doi:10.1021/om401142j |
| 38. | Bivaud, S.; Goeb, S.; Balandier, J. Y.; Chas, M.; Allain, M.; Sallé, M. Eur. J. Inorg. Chem. 2014, 2440–2448. doi:10.1002/ejic.201400060 |
| 39. | Bivaud, S.; Goeb, S.; Croué, V.; Dron, P. I.; Allain, M.; Sallé, M. J. Am. Chem. Soc. 2013, 135, 10018–10021. doi:10.1021/ja404072w |
| 40. | Bivaud, S.; Balandier, J.-Y.; Chas, M.; Allain, M.; Goeb, S.; Sallé, M. J. Am. Chem. Soc. 2012, 134, 11968–11970. doi:10.1021/ja305451v |
| 39. | Bivaud, S.; Goeb, S.; Croué, V.; Dron, P. I.; Allain, M.; Sallé, M. J. Am. Chem. Soc. 2013, 135, 10018–10021. doi:10.1021/ja404072w |
| 29. | Goeb, S.; Bivaud, S.; Dron, P. I.; Balandier, J.-Y.; Chas, M.; Sallé, M. Chem. Commun. 2012, 48, 3106–3108. doi:10.1039/c2cc00065b |
| 35. | Balandier, J.-Y.; Chas, M.; Goeb, S.; Dron, P. I.; Rondeau, D.; Belyasmine, A.; Gallego, N.; Sallé, M. New J. Chem. 2011, 35, 165–168. doi:10.1039/C0NJ00545B |
| 39. | Bivaud, S.; Goeb, S.; Croué, V.; Dron, P. I.; Allain, M.; Sallé, M. J. Am. Chem. Soc. 2013, 135, 10018–10021. doi:10.1021/ja404072w |
| 34. | Fang, Y.; Murase, T.; Sato, S.; Fujita, M. J. Am. Chem. Soc. 2013, 135, 613–615. doi:10.1021/ja311373f |
| 21. | Uehara, K.; Kasai, K.; Mizuno, N. Inorg. Chem. 2010, 49, 2008–2015. doi:10.1021/ic100011a |
| 22. | Ferrer, M.; Pedrosa, A.; Rodriguez, L.; Rossell, O.; Vilaseca, M. Inorg. Chem. 2010, 49, 9438–9449. doi:10.1021/ic101150p |
| 26. | Fujita, M.; Sasaki, O.; Mitsuhashi, T.; Fujita, T.; Yazaki, J.; Yamaguchi, K.; Ogura, K. Chem. Commun. 1996, 1535–1536. doi:10.1039/cc9960001535 |
| 30. | Holló-Sitkei, E.; Tárkányi, G.; Párkányi, L.; Megyes, T.; Besenyei, G. Eur. J. Inorg. Chem. 2008, 1573–1583. doi:10.1002/ejic.200701189 |
| 31. | Weilandt, T.; Troff, R. W.; Saxell, H.; Rissanen, K.; Schalley, C. A. Inorg. Chem. 2008, 47, 7588–7598. doi:10.1021/ic800334k |
| 32. | Ferrer, M.; Gutiérrez, A.; Mounir, M.; Rossell, O.; Ruiz, E.; Rang, A.; Engeser, M. Inorg. Chem. 2007, 46, 3395–3406. doi:10.1021/ic062373s |
| 33. | Ferrer, M.; Mounir, M.; Rossell, O.; Ruiz, E.; Maestro, M. A. Inorg. Chem. 2003, 42, 5890–5899. doi:10.1021/ic034489j |
| 39. | Bivaud, S.; Goeb, S.; Croué, V.; Dron, P. I.; Allain, M.; Sallé, M. J. Am. Chem. Soc. 2013, 135, 10018–10021. doi:10.1021/ja404072w |
© 2015 Bivaud et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)