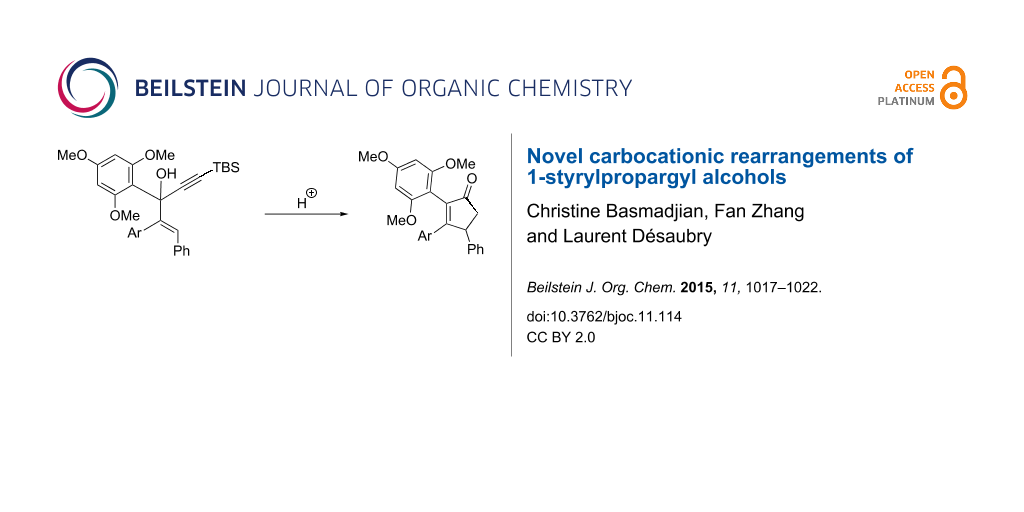
Abstract
The dehydration and subsequent cyclization reactions of 1-styrylpropargyl alcohols was examined. In the course of these studies, numerous scaffolds were synthesized, including a furan, a cyclopentenone, an acyclic enone and even a naphthalenone. The diversity of these structural motifs lies in novel cascades of reactions originating from a common carbocationic manifold.

Graphical Abstract
Introduction
In the course of our medicinal program on a new class of anticancer agents [1-3], we developed a novel synthesis of cyclopentenones substituted by three different aryl groups (Scheme 1) [4]. This approach combines a molybdenum(VI)-catalyzed etherification of allylic alcohol with a gold(I)-catalyzed intramolecular cyclization process [5,6].
![[1860-5397-11-114-i1]](/bjoc/content/inline/1860-5397-11-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Described synthesis of cyclopentenone 4 using a combination of Mo(VI) and Au(I)-catalyzed reactions and serendipitous discovery of a direct conversion of alcohol 1 into 4 [4].
Scheme 1: Described synthesis of cyclopentenone 4 using a combination of Mo(VI) and Au(I)-catalyzed reactions...
During the optimization process of this synthesis, we examined several catalysts to transform allylic alcohol 1 into ether 2, including Re2O7, which is described to efficiently catalyze this type of transformation [7]. Unexpectedly, instead of obtaining ether 2, we observed the formation of cyclopentenone 4 in 32% yield. We noticed that this reaction only occurs when Re2O7 is heated at 45 °C for 15 minutes in MeOH prior to the addition of the substrate. As far as we know, this type of reaction has not been described before. It provides a useful alternative to the Rautenstrauch rearrangement, the main limitation of which lies on the necessity to have the alcohol esterified (Scheme 2) [8]. Indeed, in many cases, this esterification occurs in low yield, or may even be impossible to achieve [4,8,9].
![[1860-5397-11-114-i2]](/bjoc/content/inline/1860-5397-11-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The Rautenstrauch rearrangement.
Scheme 2: The Rautenstrauch rearrangement.
The importance of cyclopentenones as intermediates to the synthesis of bioactive compounds prompted us to explore the synthetic potential of this novel rearrangement of 1-styrylpropargyl alcohols (Table 1). Toward this purpose, a number of other substrates were synthesized from the readily prepared acyl chloride 5 and ketone 6 [4] (Scheme 3).
![[1860-5397-11-114-i3]](/bjoc/content/inline/1860-5397-11-114-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1-styrylpropargyl alcohols.
Scheme 3: Synthesis of 1-styrylpropargyl alcohols.
Results and Discussion
We began our study by applying the reaction conditions developed for alcohol 1 using Re2O7 (1.5%), MeOH (8 equiv) at 45 °C in DCE (entry 1, Table 1). Gratifyingly, replacing the TMS group by a TBS group improved the yield to 44% (which is superior to the overall yield of the previous three steps synthesis). Along with cyclopentenone 4, we observed the formation of furan 13 (27% isolated yield).
Table 1: Rearrangement of 1-styrylpropargyl alcohols 7–10, 15, and 16.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-114-i7.svg?max-width=637&scale=1.0)
|
|||
| Entry | R | Isolated productsa | Yield (%)b |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-114-i8.svg?max-width=637&scale=1.0)
10c |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-114-i9.svg?max-width=637&scale=1.0)
4 |
44 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-114-i10.svg?max-width=637&scale=1.0)
13 |
27 | ||
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-114-i11.svg?max-width=637&scale=1.0)
15c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-114-i12.svg?max-width=637&scale=1.0)
14 |
20 |
| 3 |
Ph
16c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-114-i13.svg?max-width=637&scale=1.0)
17 |
35 |
| 4 |
H
7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-114-i14.svg?max-width=637&scale=1.0)
18 |
54 |
| 5 |
n-Bu
8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-114-i15.svg?max-width=637&scale=1.0)
19 |
47 |
| 6 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-114-i16.svg?max-width=637&scale=1.0)
9 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-114-i17.svg?max-width=637&scale=1.0)
20 |
31 |
aRe2O7 (1.5%), MeOH (8 equiv), 45 °C, DCE, 16 h. bIsolated yields. cSynthesis described in ref [4]. An = p-anisyl.
The proposed mechanism for this reaction is shown in Scheme 4. Both 4 and 13 are expected to arise through the formation of the stabilized carbocation 11 that may evolve through two pathways. This intermediate may either undergo a ring closure due to the nucleophilic character of the silylated alkyne (pathway A) or react with an oxygenated nucleophile, such as the perrhenate anion, to generate the transient intermediate 12 that undergoes an oxo-cyclization to afford an oxonium en route to furan 13 (pathway B). Alternatively, intermediate 12 may result from the allylic [1,3]-transposition of a perrhenate ester [10].
![[1860-5397-11-114-i4]](/bjoc/content/inline/1860-5397-11-114-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Postulated mechanism for the formation of cyclopentenone 4 and furan 13 (entry 1, Table 1; An= p-anisyl).
Scheme 4: Postulated mechanism for the formation of cyclopentenone 4 and furan 13 (entry 1, Table 1; An= p-anisyl).
Replacement of the highly electron-donating 2,4,6-trimethoxyphenyl group by a 4-chlorophenyl substituent reduced the yield of cyclopentenone formation to 20% (entry 2, Table 1). No furan or other compound could be isolated. As a result, we turned to the examination of substrate 16 [4], whose unsubstituted phenyl ring is even less amenable to stabilize a carbocationic intermediate. Unsurprisingly, we could not detect any cyclopentenone. Instead, we were amazed to isolate the rearranged ketone 17 (entry 3, Table 1). Indeed, 4,4-diarylnaphthalen-1-ones are highly unusual compounds, whose synthesis is rarely described in literature [11]. The structure of 17 was confirmed by NOE and key HMBC correlations (Figure S1, Supporting Information File 1). Although highly speculative and without any experimental support, a putative mechanism for this unprecedented reaction is proposed in Supporting Information File 1 (Scheme S1).
Remarkably, removal of the trimethoxyphenyl group suppressed the formation of cyclopentanone but promoted the formation of furan 18 (54% yield, entry 4, Table 1). Interestingly, substitution of the carbinol part by an n-butyl substituent provided an alternate type of product: the acyclic enone was the sole product isolated from the reaction medium (47% yield, entry 5, Table 1). It could be envisioned that this compound results from the rearrangement of an allene oxide (Scheme 5). Interestingly the dipropargylic alcohol 9 afforded the rearranged allylic ether 20 as the only isolated product (entry 6, Table 1).
![[1860-5397-11-114-i5]](/bjoc/content/inline/1860-5397-11-114-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for the formation of enone 19.
Scheme 5: Proposed mechanism for the formation of enone 19.
In order to improve the yields of these reactions and gain insight in the reaction mechanisms, we explored different reaction conditions starting from alcohol 21 (Table 2). 1,2-Dichloroethane and dichloromethane (entries 1 and 2, Table 2) gave better yields (27–31% for 22 and 10–16% for 23) compared to THF (entry 3, Table 2). In an effort to understand the role of Re2O7, the catalyst was changed to ReO4SiPh3 (entry 5, Table 2) and ReO4H (entry 6, Table 2). In both cases, the results were similar to those obtained with Re2O7. Removal of methanol from the medium suppressed the formation of 22 (entry 7, Table 2). In this case only furan 23 and rearranged alcohol 25 could be isolated as traces (5%).
Table 2: Optimization of the acid-catalyzed rearrangements of enynol 2.
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-114-i18.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | R | Catalyst | Solvent | T (°C) | 22 (%)a | 23 (%)a | 24 (%)a | 25 (%)a | 26 (%)a |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CH3 | Re2O7 | DCE | 45 | 31 | 16 | – | – | – |
| 2 | CH3 | Re2O7 | CH2Cl2 | 45 | 27 | 10 | – | – | – |
| 3 | CH3 | Re2O7 | THF | 45 | 11 | 9 | – | 12 | – |
| 4 | CH3 | Re2O7 | THF | rt | 12 | – | – | 4 | – |
| 5 | CH3 | ReO4SiPh3 | DCE | 45 | 28 | 17 | 6 | – | – |
| 6 | CH3 | ReO4H | DCE | 45 | 30 | 17 | – | – | – |
| 7 | – | ReO4H | DCE | 45 | – | 5 | – | 5b | – |
| 8 | CH3 | F3CSO3H | DCE | 45 | 36 | 12 | – | – | – |
| 9 | CH3 | AcOH | DCE | 45 | – | – | – | 51 | 47 |
| 10c | – | F3CSO3H | DCE | 45 | – | – | – | – | – |
| 11c | CF3CH3 | F3CSO3H | DCE | 45 | – | – | – | – | – |
| 11 | CH3 | F3CSO3H | CH3NO2 | 45 | 30 | – | – | – | – |
| 12c | CH3 | F3CSO3H | CH3NO2 | rt | – | – | – | – | – |
aIsolated yields. bAn allylic alcohol is generated (25, R = H). cDegradation of the reaction mixture.
To confirm that the reaction is catalyzed by an acid, we changed its nature and tested two Brønsted acids. With F3CSO3H (entry 8, Table 2), we obtained comparable yields with 36% of cyclopentenone 22 and 12% of furan 23. When using a weaker acid such as acetic acid (entry 9, Table 2) only the two acyclic ethers 25 and 26 were isolated. This observation strongly suggests that perrhenic acid is the active reagent generated from Re2O7 by methanolysis or hydrolysis with traces of water [12].
When removing methanol or replacing it with trifluoroethanol, the reaction mixture degraded in presence of triflic acid (entries 10 and 11, Table 2). Thus, the presence of methanol seems necessary for the cyclization to occur, perhaps by stabilizing an intermediate. Replacement of dichloroethane by nitromethane slightly decreased the yield of 22, and suppressed the formation of furan 23.
To determine whether the protection of the alkyne group was necessary for a rearrangement to take place, the reaction was carried out on compound 27. In the presence of perrhenic acid, 16% of the starting material was recovered along with 36% of diketone 28 (Scheme 6). No cyclization product was isolated. This observation strengthened the hypothesis that the silyl group stabilizes the carbocation intermediate which seems necessary for the cyclization step.
![[1860-5397-11-114-i6]](/bjoc/content/inline/1860-5397-11-114-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Rearrangement of unprotected propargylic carbinol 27.
Scheme 6: Rearrangement of unprotected propargylic carbinol 27.
Conclusion
The discovery of new reaction manifolds often provides a good opportunity to discover novel reactivity in related systems. In this article, we demonstrate the delicate balance among several mechanistic pathways by a minor change of substrate in acid-catalyzed rearrangements of 1-styrylpropargyl alcohols. Indeed, these compounds may generate a furan (18), an enone (19), an allylic ether or even a naphthalenone (17). The formation of the latter is quite intriguing because it suggests an unprecedented cascade of reactions.
Supporting Information
| Supporting Information File 1: Experimental procedures for the synthesis of compounds 7–9, 13, 14 and 17–28. | ||
| Format: PDF | Size: 531.6 KB | Download |
References
-
Boussemart, L.; Malka-Mahieu, H.; Girault, I.; Allard, D.; Hemmingsson, O.; Tomasic, G.; Thomas, M.; Basmadjian, C.; Ribeiro, N.; Thuaud, F.; Mateus, C.; Routier, E.; Kamsu-Kom, N.; Agoussi, S.; Eggermont, A. M.; Désaubry, L.; Robert, C.; Vagner, S. Nature 2014, 513, 105–109. doi:10.1038/nature13572
Return to citation in text: [1] -
Basmadjian, C.; Thuaud, F.; Ribeiro, N.; Désaubry, L. Future Med. Chem. 2013, 5, 2185–2197. doi:10.4155/fmc.13.177
Return to citation in text: [1] -
Ribeiro, N.; Thuaud, F.; Bernard, Y.; Gaiddon, C.; Cresteil, T.; Hild, A.; Hirsch, E. C.; Michel, P. P.; Nebigil, C. G.; Désaubry, L. J. Med. Chem. 2012, 55, 10064–10073. doi:10.1021/jm301201z
Return to citation in text: [1] -
Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093
Return to citation in text: [1] [2] [3] [4] [5] [6] -
An, S. E.; Jeong, J.; Baskar, B.; Lee, J.; Seo, J.; Rhee, Y. H. Chem. – Eur. J. 2009, 15, 11837–11841. doi:10.1002/chem.200901824
Return to citation in text: [1] -
Yang, H.; Fang, L.; Zhang, M.; Zhu, C. Eur. J. Org. Chem. 2009, 666–672. doi:10.1002/ejoc.200800976
Return to citation in text: [1] -
Herrmann, A. T.; Saito, T.; Stivala, C. E.; Tom, J.; Zakarian, A. J. Am. Chem. Soc. 2010, 132, 5962–5963. doi:10.1021/ja101673v
Return to citation in text: [1] -
Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d
Return to citation in text: [1] [2] -
Caruana, P. A.; Frontier, A. J. Tetrahedron 2007, 63, 10646–10656. doi:10.1016/j.tet.2007.08.008
Return to citation in text: [1] -
Volchkov, I.; Lee, D. Chem. Soc. Rev. 2014, 43, 4381–4394. doi:10.1039/c4cs00036f
Return to citation in text: [1] -
Terao, Y.; Kametani, Y.; Wakui, H.; Satoh, T.; Miura, M.; Nomura, M. Tetrahedron 2001, 57, 5967–5974. doi:10.1016/S0040-4020(01)00555-5
Return to citation in text: [1] -
Bellemin-Laponnaz, S. ChemCatChem 2009, 1, 357–362. doi:10.1002/cctc.200900206
Return to citation in text: [1]
| 1. | Boussemart, L.; Malka-Mahieu, H.; Girault, I.; Allard, D.; Hemmingsson, O.; Tomasic, G.; Thomas, M.; Basmadjian, C.; Ribeiro, N.; Thuaud, F.; Mateus, C.; Routier, E.; Kamsu-Kom, N.; Agoussi, S.; Eggermont, A. M.; Désaubry, L.; Robert, C.; Vagner, S. Nature 2014, 513, 105–109. doi:10.1038/nature13572 |
| 2. | Basmadjian, C.; Thuaud, F.; Ribeiro, N.; Désaubry, L. Future Med. Chem. 2013, 5, 2185–2197. doi:10.4155/fmc.13.177 |
| 3. | Ribeiro, N.; Thuaud, F.; Bernard, Y.; Gaiddon, C.; Cresteil, T.; Hild, A.; Hirsch, E. C.; Michel, P. P.; Nebigil, C. G.; Désaubry, L. J. Med. Chem. 2012, 55, 10064–10073. doi:10.1021/jm301201z |
| 7. | Herrmann, A. T.; Saito, T.; Stivala, C. E.; Tom, J.; Zakarian, A. J. Am. Chem. Soc. 2010, 132, 5962–5963. doi:10.1021/ja101673v |
| 4. | Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093 |
| 5. | An, S. E.; Jeong, J.; Baskar, B.; Lee, J.; Seo, J.; Rhee, Y. H. Chem. – Eur. J. 2009, 15, 11837–11841. doi:10.1002/chem.200901824 |
| 6. | Yang, H.; Fang, L.; Zhang, M.; Zhu, C. Eur. J. Org. Chem. 2009, 666–672. doi:10.1002/ejoc.200800976 |
| 12. | Bellemin-Laponnaz, S. ChemCatChem 2009, 1, 357–362. doi:10.1002/cctc.200900206 |
| 4. | Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093 |
| 4. | Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093 |
| 4. | Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093 |
| 4. | Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093 |
| 11. | Terao, Y.; Kametani, Y.; Wakui, H.; Satoh, T.; Miura, M.; Nomura, M. Tetrahedron 2001, 57, 5967–5974. doi:10.1016/S0040-4020(01)00555-5 |
| 4. | Basmadjian, C.; Zhao, Q.; Désaubry, L. Tetrahedron Lett. 2015, 56, 727–730. doi:10.1016/j.tetlet.2014.12.093 |
| 8. | Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d |
| 9. | Caruana, P. A.; Frontier, A. J. Tetrahedron 2007, 63, 10646–10656. doi:10.1016/j.tet.2007.08.008 |
| 8. | Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d |
| 10. | Volchkov, I.; Lee, D. Chem. Soc. Rev. 2014, 43, 4381–4394. doi:10.1039/c4cs00036f |
© 2015 Basmadjian et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)