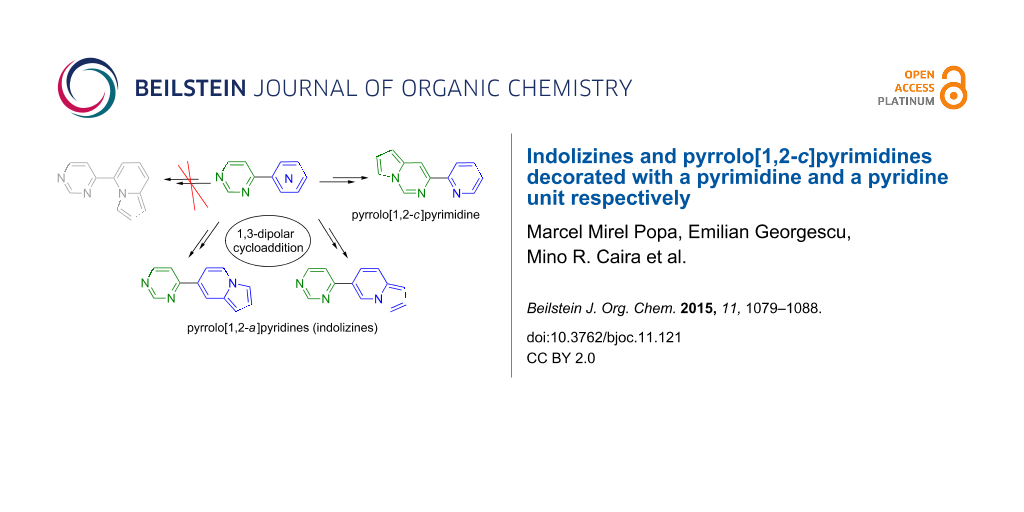
Abstract
The three possible structural isomers of 4-(pyridyl)pyrimidine were employed for the synthesis of new pyrrolo[1,2-c]pyrimidines and new indolizines, by 1,3-dipolar cycloaddition reaction of their corresponding N-ylides generated in situ from their corresponding cycloimmonium bromides. In the case of 4-(3-pyridyl)pyrimidine and 4-(4-pyridyl)pyrimidine the quaternization reactions occur as expected at the pyridine nitrogen atom leading to pyridinium bromides and consequently to new indolizines via the corresponding pyridinium N-ylides. However, in the case of 4-(2-pyridyl)pyrimidine the steric hindrance directs the reaction to the pyrimidinium N-ylides and, subsequently, to the formation of the pyrrolo[1,2-c]pyrimidines. The new pyrrolo[1,2-c]pyrimidines and the new indolizines were structurally characterized through NMR spectroscopy. The X-ray structures of two of the starting materials, 4-(2-pyridyl)pyrimidine and 4-(4-pyridyl)pyrimidine, are also reported.

Graphical Abstract
Introduction
Two heteroarenes linked through a single bond [1] proved to be versatile structural motifs for a broad range of compounds with applications as advanced fluorescent materials [2,3], ligands [4,5], bioactive compounds [6], and for the design of dynamic chemical devices [7-10]. The most accessible route to hybrid heteroarenes is the direct coupling [1,11,12]. However, in some cases alternative routes could be useful to achieve structural variety [13]. An alternative route is to start from accessible simple heteroarenes linked by a single bond and to transform one of them or both into more complex structures [14,15].
Nitrogen-containing compounds from the class of indolizines and azaindolizines are known as fluorescent materials [2-4,15] with applications as chemosensors [16] and as bioactive compounds [17-20]. For example, starting from bispyridyl derivatives such as 1 and 3, fluorescent compounds from the class of indolizines, as for example compounds 2, 4 and 5 (Figure 1) were obtained [14,15].
![[1860-5397-11-121-1]](/bjoc/content/figures/1860-5397-11-121-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of hybrid heteroarenes from the class of indolizines.
Figure 1: Examples of hybrid heteroarenes from the class of indolizines.
Based on our previous experience [21-25] we investigated the synthesis and structural characterization of new pyrrolo[1,2-c]pyrimidines that are 3-substituted with a pyridine unit and new indolizines substituted in positions 6 or 7 with a pyrimidine. Herein, we report the efficient synthesis of small libraries of structurally diverse pyrrolo-fused heterocycles starting from the 4-pyridylpyrimidine isomers via 1,3-dipolar cycloaddition of corresponding N-ylides with several activated alkynes. The biologic and luminescent properties of newly synthesized 3-pyridylpyrrolo[1,2-c]pyrimidine and 7-pyrimidinylindolizine derivatives are under investigation.
Results and Discussion
Our purpose was to obtain new hetarylpyridines and hetarylpyrimidines. Thus, starting from the 4-pyridylpyrimidine isomers 6–8 (Figure 2) we have obtained new pyrrolo[1,2-c]pyrimidines or pyrimidinyl-substituted indolizines. The synthetic strategy was the 1,3-dipolar cycloaddition reaction of the corresponding N-ylides [21-25]. The structural variety of the compounds was determined by the series of dipolarophiles employed. The new final compounds were structurally characterized and our preliminary investigations suggest that some of them possess relevant degrees of fluorescence.
![[1860-5397-11-121-2]](/bjoc/content/figures/1860-5397-11-121-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Starting 4-pyridylpyrimidines 6–8.
Figure 2: Starting 4-pyridylpyrimidines 6–8.
For the synthesis of 4-pyridylpyrimidines 6–8 there are some synthetic methods reported in the literature [26-29]. 4-(2-Pyridyl)pyrimidine (6) was obtained according to Bejan et al. [28] in 44% yield while the reaction for obtaining 4-(3-pyridyl)pyrimidine did not work. However, we devised an appropriate method for the synthesis of 4-pyridylpyrimidines, analogous to that reported for the synthesis of 4-phenylpyrimidines [30]. Thus, by the reaction of trisformylaminomethane and corresponding substituted acetylpyridines in the presence of p-toluenesulfonic acid the 4-pyridylpyrimidines were obtained in 20–25% yields. In the case of 4-(4-pyridyl)pyrimidine (8) the reaction was carried out starting from trisformylaminomethane and the 4-acetylpyridine hydrochloride, the reaction yield increasing to 41%. The compounds were crystallized from methylcyclohexane resulting in waxy crystals of 4-(2-pyridyl)-pyrimidine with mp 75–78 °C (lit: [28], 77–80 °C), 4-(3-pyridyl)pyrimidine with mp 85–88 °C (lit: [26], 89 °C) and 4-(4-pyridyl)pyrimidine with mp 131–133 °C (lit: [29], 132–133 °C).
The chemistry of 4-(2-pyridyl)pyrimidine (6) is rather scarce compared to its structural 2,2’-bipyridyl analogue but should raise interest for similar synthetic applicative purposes. Also the isomers of 6, compounds 7 and 8, are of interest for coordination chemistry both as ligands and as bridging compounds. For this reason we paid some attention to their structural characterization. To our knowledge the X-ray structures for the parent compounds 6–8 were not reported previously and we thus attempted to obtain suitable single crystals. Only compounds 6 and 8 could be isolated as monocrystals and their X-ray structures were determined. Disordered models were necessarily invoked to account for the requirement of molecular centrosymmetry imposed by their respective crystallographic space groups. The detailed description of the X-ray structures is given below.
Having at our disposal 4-(2-pyridyl)pyrimidine (6), 4-(3-pyridyl)pyrimidine (7), and 4-(4-pyridyl)pyrimidine (8) as starting materials we managed to obtain new pyrrolo-fused derivatives by 1,3-dipolar cycloaddition reactions of their corresponding N-ylides in 1,2-epoxybutane as reaction medium and deprotonating agent.
Pyrrolo[1,2-c]pyrimidines from 4-(2-pyridyl)pyrimidine (6)
Counterintuitively, starting from 6, we obtained new pyrrolo[1,2-c]pyrimidines 12 instead of the expected indolizines. The nitrogen atom of the pyridine moiety is more reactive and it is expected that the quaternization reaction takes place preferentially at that atom, rather than at one of the nitrogen atoms in the pyrimidine. However due to steric hindrance, in the case of 6 the quaternization reaction takes place at the N1 nitrogen of the pyrimidine leading to the corresponding pyrimidinium bromides 11 (Scheme 1). The compounds 11 and 12 are presented in Table 1.
![[1860-5397-11-121-i1]](/bjoc/content/inline/1860-5397-11-121-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthesis of new pyrimidinium bromides 11 and pyrrolo[1,2-c]pyrimidines 12.
Scheme 1: The synthesis of new pyrimidinium bromides 11 and pyrrolo[1,2-c]pyrimidines 12.
Table 1: The bromides 11 and the new pyrrolo[1,2-c]pyrimidines 12.
| entry | Ar | E | R | yield | mp (°C) |
|---|---|---|---|---|---|
| 11a | C6H5 | — | — | 80 | 227–229 |
| 11b | 4-ClC6H4 | — | — | 87 | 222–223 |
| 11c | 4-BrC6H4 | — | — | 90 | 242–243 |
| 11d | 3-NO2C6H4 | — | — | 80 | 219–221 |
| 11e | 4-NO2C6H4 | — | — | 78 | 226–228 |
| 12a | 4-BrC6H4 | COMe | H | 41 | 263–265 |
| 12b | 3-NO2C6H4 | COMe | H | 46 | 255–257 |
| 12c | C6H5 | CO2Me | H | 52 | 218–219 |
| 12d | 4-BrC6H4 | CO2Me | H | 62 | 234–236 |
| 12e | 3-NO2C6H4 | CO2Me | H | 54 | 233–235 |
| 12f | 4-ClC6H4 | CO2Et | H | 40 | 204–205 |
| 12g | 4-NO2C6H4 | CO2Et | H | 48 | 218–220 |
| 12h | 3-NO2C6H4 | CO2Me | CO2Me | 45 | 246–248 |
No quaternization product at the N3 or at the nitrogen atom of the pyridine is observed. The new bromides 11 were obtained in good yields in acetone at room temperature by reacting 6 with different bromoacetophenones, and they were characterized by NMR spectroscopy which confirmed their structure. The 1H NMR spectra of the bromides 11 confirm that the quaternization reaction took place at the N1 of the pyrimidine ring. This was confirmed by the deshielding effect of the quaternization at the N1 atom of the pyrimidine upon the atoms H-2, H-5 and H-6 (Figure 3).
![[1860-5397-11-121-3]](/bjoc/content/figures/1860-5397-11-121-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The 1H NMR spectra (DMSO-d6) of 4-(2-pyridyl)pyrimidine (6) and the corresponding bromides 11a–e (the aromatic region).
Figure 3: The 1H NMR spectra (DMSO-d6) of 4-(2-pyridyl)pyrimidine (6) and the corresponding bromides 11a–e (t...
The three sets of signals in the pyrimidine moiety appear as expected, thus H-5 and H-6 appear as two doublet of doublets (J = 6.6, 0.8, and J = 6.6, 1.7 Hz, respectively; H-5 may appear as a doublet due to the small value of the coupling constant with H-2). The hydrogen H-2 is the most deshielded and appears at 9.86–9.92 ppm (compared to 9.30 ppm in compound 6, Figure 3) as a broad singlet due to the coupling with H-5 and H-6. The atoms in the pyridine moiety appear as four sets of signals with the expected multiplicities (they were assigned by COSY and HETCOR experiments). The methylene protons appear in the range 6.43–6.67 ppm as a sharp singlet for all the compounds 11a–e. The 13C NMR spectra of 11a–e present all the expected signals. The C=O is observed at about 190 ppm. The most deshielded tertiary carbon atoms are C-2, C-6 and C-3’, which are directly bonded to nitrogen atoms. The aliphatic carbon atom appears at 62.8–63.2 ppm. All the signals in the benzoyl moieties appear as expected.
The pyrimidinium bromides 11 were allowed to react with different activated alkynes in 1,2-epoxybutane under reflux with stirring for 24 h, to obtain the new pyrrolo[1,2-c]pyrimidines 12 decorated with a pyridyl moiety. No trace of the corresponding pyrrolo[1,2-a]pyrimidine was observed as previously reported by us [31,32]. The yields were moderate to good.
The NMR data of compounds 12 show strong evidence for the formation of the new compounds. The main 1H NMR features are the signals in the pyrrolo[1,2-c]pyrimidine which were assigned by COSY and HETCOR 2D experiments. Thus, H-6 appears as a singlet at around 7.83–7.91 ppm confirming the regioselectivity of the cycloaddition reaction in the case of compounds 12a–g. The atoms H-1 and H-4 appear as two doublets with J = 1.4 Hz (1.6 Hz in CDCl3 with TFA added). The signals in the pyridine moiety and benzoyl moiety appear as expected. The 13C NMR exhibits all the carbon atom signals as expected. The CO signals appear in accordance with the nature of the substituents. In the case when acetyl is replaced by a carbomethoxy or a carboethoxy group C-5 is shielded from around 118 ppm (12a,b) to around 108 ppm (12c–h). The carbon C-1 is the most deshielded in all the series of compounds due to its direct bond to two nitrogen atoms.
New indolizines starting from 4-(3-pyridyl)pyrimidine and 4-(4-pyridyl)pyrimidine
In the case of 7 and 8 the reaction proceeds as expected, the quaternization reactions taking place at the nitrogen atom in the pyridine moiety compared to 4-(2-pyridyl)pyrimidine (6). The reactions were carried out via a multicomponent approach by mixing all the starting materials in 1,2-epoxybutane under reflux. In some cases the isolation and characterization of the intermediate bromide salts was performed and will be discussed in each case.
In the case of pyridinium ylides generated from 4-(3-pyridyl)pyrimidine (7), new indolizines 14a–f decorated with a pyrimidine moiety were obtained (Scheme 2). The reaction appears to be selective towards the indolizines 14. In some cases the other possible indolizine (14A) was observed also in the bulk reaction mass in low to medium quantities but was not recovered, further work being in progress to investigate this aspect. Due to the fact that the salts 13 are not easy to work up we ran the reaction in a multicomponent approach and only in two cases, 13a,b, we separated the salts and assigned their structures by NMR spectroscopy. The 1H NMR spectra of the bromides 13a and 13b show that the quaternization reaction took place at the nitrogen atom of the pyridine. This was concluded by assigning all the protons by COSY and HETCOR experiments. For the pyrimidine the signals are easy to assign as a doublet of doublets for H-5’ (J = 5.2, 1.3 Hz), H-6’ as a doublet (J = 5.2 Hz) and H-2’ (which is the most deshielded hydrogen atom in the pyrimidine moiety) as a doublet with J = 1.3 Hz. The most deshielded proton is H-2 in the pyridine moiety at about 10 ppm for both 13a and 13b. The multiplicities of the hydrogen atoms in the pyridine moiety are more complex but could be assigned by 2D experiments. Another main feature of the spectra of 13a,b is the singlet of the methylene protons at ca. 6.70 ppm. The 13C NMR spectra present all the expected signals. The C=O is observed at around 190 ppm. The aliphatic carbon atom appears at around 66 ppm.
![[1860-5397-11-121-i2]](/bjoc/content/inline/1860-5397-11-121-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of new pyridinium bromides 13a,b and indolizines 14a–f.
Scheme 2: The synthesis of new pyridinium bromides 13a,b and indolizines 14a–f.
The new indolizines 14a–f were obtained in good yields (Table 2) and were characterized through NMR spectroscopy. The 1H NMR spectra of the indolizines 14 confirm their structure. The discrimination between the two possible isomers 14 and 14A was made on the basis of the multiplicity of the proton signals which is unequivocal. The main features are the signals of H-2 which appears as a singlet and H-5, the most deshielded proton appearing as a multiplet in the range of 10.70–10.75 ppm. For the pyrimidine the spectra show three sets of signals for each proton. These signals are easy to assign for H-5’ and H-6’ as a doublet of doublets (J = 5.2, 1.4 Hz) and a doublet (J = 5.2 Hz) respectively. The hydrogen H-2’ is the most deshielded hydrogen atom in the pyrimidine moiety, as a doublet with J = 1.4 Hz. The 13C NMR spectra present all the expected signals. The most deshielded carbon atoms are C-2’ and C-5’ at 159 and 157 ppm, respectively for all the series of compounds 14a–f. As in the case of pyrrolo[1,2-c]pyrimidines 12, replacing the acetyl group at C-1 with a carbomethoxy or carboethoxy group induces a shielding effect on the C-1 atom from 115 ppm in the case of 14a, to 104.8–108.1 ppm for 14b–f.
Table 2: The bromide salts 13 and the new 6-pyrimidinylindolizines 14.
| entry | Ar | E | R | yield | mp (°C) |
|---|---|---|---|---|---|
| 13a | C6H5 | — | — | 90 | 214–215 |
| 13b | 4-FC6H4 | — | — | 78 | 218–222 |
| 14a | 4-FC6H4 | COMe | H | 64 | 242–244 |
| 14b | C6H5 | CO2Me | H | 68 | 199–200 |
| 14c | 4-NO2C6H4 | CO2Et | H | 48 | 247–249 |
| 14d | 4-ClC6H4 | CO2Et | H | 54 | 212–214 |
| 14e | 4-FC6H4 | CO2Et | H | 60 | 176–178 |
| 14f | C6H5 | CO2Me | CO2Me | 45 | 133–136 |
Similarly, in the case of pyridinium N-ylides generated from 8, the new indolizines 16 were synthesized (Scheme 3) and structurally characterized. Again, in the case of some compounds, the salts were not easily purified and we preferred the multicomponent approach. However, we isolated and characterized the bromides 15a,b (Table 3). The new 7-(pyrimidyl)indolizines were obtained in moderate to good yields and were characterized by NMR spectroscopy. The 1H NMR spectra of the bromides 15 show that the quaternization reaction took place at the pyridine nitrogen atom. The first definitive indications are the signals of the protons in the pyridine moiety, which appear as doublet of doublets either in the starting 4-(4-pyridyl)pyrimidine (8) or after quaternization in 15a,b. The positive charge at the nitrogen induces a deshielding effect for H-2 and H-6 (see numbering in Scheme 3) in 15a,b compared to 8. The four protons were assigned as two doublets with J = 6.9 Hz. For the pyrimidine, the signals are easily assigned similarly as for the indolizines 14. Another main feature of the spectra of 15 is the singlet of the methylene protons at 6.6 ppm. The 13C NMR spectra present all the expected signals. The C=O is observed at around 190 ppm.
![[1860-5397-11-121-i3]](/bjoc/content/inline/1860-5397-11-121-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The synthesis of the new pyridinium bromides 15 and 7-pyrimidylindolizines 16a–f.
Scheme 3: The synthesis of the new pyridinium bromides 15 and 7-pyrimidylindolizines 16a–f.
Table 3: The bromides 15a,b and the new 7-pyrimidinylindolizines 16a–f.
| entry | Ar | E | R | yield | mp (°C) |
|---|---|---|---|---|---|
| 15a | 4-BrC6H4 | — | — | 90 | 264–268 |
| 15b | 4-MeOC6H4 | — | — | 88 | 238–240 |
| 16a | C6H5 | COMe | H | 44 | 236–238 |
| 16b | 4-ClC6H4 | COMe | H | 56 | 287–289 |
| 16c | 4-FC6H4 | COMe | H | 62 | 240–242 |
| 16d | 4-BrC6H4 | CO2Me | H | 50 | 281–282 |
| 16e | 4-MeOC6H4 | CO2Me | H | 54 | 249–250 |
| 16f | 3-NO2C6H4 | CO2Et | H | 42 | 208–210 |
The new compounds 16 were obtained by a multicomponent approach by mixing all the reagents in 1,2-epoxybutane and heating under reflux for 48 h (Scheme 3). In some cases we carried out the reaction in two steps by isolating the salts 15, the yields being similar.
The 1H NMR spectra of the indolizines 16a–f confirm their structure. All the atoms in the indolizine moiety were assigned by COSY and HETCOR experiments. The main features are the signal for H-2, which appears as a singlet, and those for H-5 which is the most deshielded proton, appearing as a doublet of doublets with J = 7.4, 0.8 Hz. The pyrimidine moiety was assigned as previously described for the compounds 15. The 13C NMR spectra present all the expected signals. As in the case of pyrrolo[1,2-c]pyrimidines 12 and indolizines 14 replacing the acetyl group at C-1 with a carbomethoxy or carboethoxy group induces a shielding effect on the C-1 atom from 115 ppm for 16a–c, to 108 ppm in the case of 16d–f.
The reaction mechanism in all cases implies the attack of the bromide salt of type 17 on the oxirane ring in the 1,2-epoxybutane (Scheme 4). The reactive intermediate obtained by the ring opening of the 1,2-epoxybutane abstracts a methylene proton from the salt, generating the corresponding N-ylide (18) in situ.
![[1860-5397-11-121-i4]](/bjoc/content/inline/1860-5397-11-121-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The N-ylide reacts further with the acetylenic dipolarophile to give a primary cycloadduct 19 which spontaneously rearranges and dehydrogenates under the reaction conditions, to the final aromatic compounds 20.
X-ray crystal structures of the starting compounds 4-(2-pyridyl)pyrimidine (6) and 4-(4-pyridyl)pyrimidine (8)
Full details of the structural solution and refinements are given in Supporting Information File 1. Structural refinements were non-routine owing to subtle disorder occurring in both crystals. Taking into account the values of Z (the number of molecules per unit cell) for the crystals of 6 and 8, their respective space groups, P21/n and P−1, require that each molecule be located on a centre of symmetry and hence be planar. Since neither molecule possesses a centre of symmetry, this implies that they must both be disordered in their respective crystals. Accordingly, centrosymmetric structural models were proposed by considering the possible planar rotamers for both molecules as well as the effect of adding a centre of inversion to each of these. The disordered models assigned were consistent with the observed difference Fourier electron densities, in particular those of the hydrogen atoms, whose relative values indicated that the sites of their parent atoms were either fully occupied by carbon atoms or by nitrogen atoms, or partially occupied by carbon and nitrogen atoms simultaneously.
Figure 4a and 4b show the molecular and crystal structures of 4-(2-pyridyl)pyrimidine (6). Here, all atoms in the molecule have a site-occupancy factor (s.o.f.) equal to 1 except the atom labeled ‘C4/N4’, where C and N atoms have equal occupancy (s.o.f. = 0.5 for each and for H-4), which is required to satisfy the condition of centrosymmetry. The molecules are linked into large concatenated rings with graph-set descriptor [33] R22(22) by one crystallographically unique, weak C–H…N hydrogen bond.
![[1860-5397-11-121-4]](/bjoc/content/figures/1860-5397-11-121-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular and crystal structures of 6 (a,b) and 8 (c,d). The molecules are located on centers of inversion and their asymmetric units are labelled, disordered atoms being indicated as Cn/Nn.
Figure 4: Molecular and crystal structures of 6 (a,b) and 8 (c,d). The molecules are located on centers of in...
In the centrosymmetric structural model for 4-(4-pyridyl)pyrimidine (8) (Figure 4c), atom N4 is necessarily present with s.o.f. = 1, but the evidence from the X-ray analysis indicated the presence of rotamers, with both the 2- and the 6-positions in the two rings occupied by C and N atoms with s.o.f. values of 0.75 and 0.25, respectively, to account for a net total of three carbon atoms and one nitrogen atom. The crystal of 8 is composed of layers, a portion of such a layer being shown in Figure 4d. Two crystallographically distinct cyclic C–H…N hydrogen bonded motifs with graph-set descriptor R22(6) occur [33] resulting in a more extensive H-bonding in the crystal of 8 than in that of 6. This is consistent with their significantly different melting points (131–133 °C for 8 and 75–78 °C for 6). In both crystals, C–H…N hydrogen bonding is complemented by offset π-stacking, with shortest ring centroid-to-centroid distances of 3.743 Å in 6 and 3.766 Å in 8.
It was ascertained from a search of the Cambridge Structural Database [34] (CSD) that the crystal of 6 is isostructural with that of 4,4’-bipyrimidine as the trans-rotamer (CSD refcode SACPAN). This is not surprising when one compares this molecule with the centrosymmetric model of Figure 4a. Isostructurality was established from closely matching unit cell parameters and the common space group, and confirmed by the near superposition of their simulated powder X-ray patterns (see Supporting Information File 1). No isostructural analogues for 8 were evident.
Conclusion
In conclusion, we have obtained 20 new hybrid heteroarenes from the class of indolizines and pyrrolo[1,2-c]pyrimidines by a simple multicomponent or two-step approach. All compounds, including the 9 intermediate bromide salts, were structurally characterized by NMR spectroscopy. The biological and fluorescence properties of the new synthesized compounds will be intensely investigated further. Reported methods were employed in obtaining the 4-pyridylpyrimidine isomers used as starting materials. X-ray analyses of the 4-pyridylpyrimidines 6 and 8 were non-trivial, requiring refinement of disordered models due to their location on centers of symmetry.
Experimental
General
Melting points were measured using a Boetius hot plate microscope and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Varian Gemini 300BB operating at 300 MHz for 1H and 75 MHz for 13C. The spectra were recorded in CDCl3 or DMSO-d6 at 298 K and the chemical shifts are relative to TMS used as the internal standard. The bidimensional correlations spectra (COSY, HETCOR) were performed for complete assignment of chemical shifts. Fourier-transform IR spectra (for representative compounds) were recorded on a Bruker Vertex 70 spectrometer with horizontal device for attenuated reflectance and diamond crystal, in a spectral window ranging from 4000 to 400 cm−1 or on a Nicolet Impact 410 Spectrometer in KBr pellets. Elemental analysis was performed on a Perkin Elmer CHNS/O analyzer Series II 2400 apparatus and the results were in agreement with the calculated values. All starting materials and solvents were purchased from common commercial suppliers and were used without purification unless otherwise noted.
General procedure for obtaining 4-(2-pyridyl)pyrimidinium bromides 11a–e
4-(2-Pyridyl)pyrimidine (10 mmol) and bromoacetophenones (9) (10 mmol) were stirred in 50 mL acetone under reflux for 8 h and then were left at room temperature overnight. The precipitated 4-(2-pyridyl)pyrimidinium bromides 11a–e were removed by filtration.
1-[2-Phenyl-2-oxoethyl]-4-(2-pyridyl)pyrimidinium bromide (11a). Light brown crystals with mp 227–229 °C. Yield 80%. Anal. calcd for C17H14BrN3O: N, 11.80; found N, 12.13; 1H NMR (DMSO-d6, 300 MHz) δ 6.43 (s, 2H, CH2), 7.52–7.68 (m, 2H, H-3”, H-5”), 7.74–7.80 (m, 2H, H-4’, H-4”), 8.06–8.08 (m, 2H, H-2”, H-6”), 8.16 (td, J = 7.7, 1.9 Hz, 1H, H-5’), 8.63–8.65 (m, 1H, H-6’), 8.89 (m, 1H, H-3’), 9.04 (dd, J = 6.6 Hz, 0.8 Hz, 1H, H-5), 9.32 (dd, J = 6.6, 1.7 Hz, 1H, H-6), 9.75 (bs, 1H, H-2); 13C NMR (DMSO-d6, 75 MHz) δ 63.4 (CH2), 119.0 (C-5), 125.0 (C-6’), 128.9 (C-2”, C-6”), 129.3 (C-4’), 129.9 (C-3”, C-5”), 133.7, 150.2, 168.5 (C-4, C-1’, C-1”), 135.7 (C-4”), 139.2 (C-5’), 151.5 (C-3’), 154.7 (C-6), 155.3 (C-2), 190.7 (COAr); IR (KBr, cm−1): 1696, 1627, 1550, 1450, 1341, 1213.
General procedure for obtaining pyrrolo[1,2-c]pyrimidines 12a–h
4-(2-Pyridyl)pyrimidinium bromide (3 mmol) 11a–e previously obtained and acetylenic dipolarophiles 10 (3.5 mmol) were stirred under reflux in 20 mL 1,2-epoxybutane for 24 h. The products were precipitated with ethanol and removed by filtration. Further purification was made by crystallization from ethanol or by column chromatography on Al2O3 using methylene chloride as eluent.
3-(2-Pyridyl)-5-acetyl-7-(4-bromobenzoyl)-pyrrolo[1,2-c]pyrimidine (12a). Pale yellow powder with mp 263–265 °C. Yield 41%. Anal. calcd for C21H14BrN3O2: C, 60.02; H, 3.36; Br, 19.01; N, 10.00; found: C, 60.30; H, 3.65; Br, 19.24; N, 10.22; 1H NMR (CDCl3, 300 MHz) δ 2.60 (CH3), 7.64 (s, 4H, H-2”, H-3”, H-5”, H-6”), 7.80 (s, 1H, H-6), 7.96–8.00 (m, 1H, H-4’), 8.60 (td, J = 7.7, 1.9 Hz, 1H, H-5’), 8.70–8.73 (m, 1H, H-6’), 8.86–8.89 (m, 1H, H-3’), 9.30 (d, J = 1.6 Hz, 1H, H-4), 10.49 (d, J = 1.6 Hz, 1H, H-1); 13C NMR (CDCl3+TFA, 75 MHz) δ 28.0 (CH3), 114.8 (C-4), 117.8 (C-5), 124.6, 128.8, 136.3, 138.5, 142.0, 146.5 (C-3, C-7, C-4a, C-1’, C-1”, C-4”), 124.3 (C-6’), 127.7 (C-4’), 130.3 (C-6), 130.7 (C-2”, C-6”), 132.9 (C-3”, C-5”), 138.6 (C-5’), 142.4 (C-3’), 147.7 (C-1), 185.5 (COAr), 197.5 (CO); IR (ATR, cm−1): 3054, 1650, 1627, 1585, 1469, 1328, 1217, 887.
Procedure for obtaining 4-(3-pyridyl)pyrimidinium bromides 13a,b
4-(3-Pyridyl)pyrimidine (7, 10 mmol) and bromoacetophenone (9) (10 mmol) were stirred in 50 mL acetone under reflux for 8 h and then were left at room temperature overnight. The precipitated bromides 13a,b were removed by filtration.
1-[2-Phenyl-2-oxoethyl]-3-(4-pyrimidinyl)pyridinium bromide (13a). Red powder with mp 214–215 °C. Yield 90%. Anal. calcd for C17H14BrN3O: N, 11.80; found N, 12.07; 1H NMR (DMSO-d6, 300 MHz) δ 6.71 (s, 2H, CH2), 7.65–7.70 (m, 2H, H-3”, H-5”), 7.77–7.83 (m, 1H, H-4”), 8.08–8.12 (m, 2H, H-2”, H-6”), 8.40 (dd, J = 5.2, 1.3 Hz, 1H, H-6’), 8.50 (dd, J = 8.2, 6.0 Hz, 1H, H-5), 9.14 (d, J = 5.2 Hz, 1H, H-5’), 9.24 (dt, J = 6.0, 1.4 Hz, 1H, H-6), 9.44 (d, J = 1.3 Hz, 1H, H-2’), 9.48 (dt, J = 8.2, 1.4 Hz, 1H, H-4), 10.00 (m, 1H, H-2); 13C NMR (DMSO-d6, 75 MHz) δ 66.6 (CH2), 118.5 (C-5’), 128.1 (C-5); 128.3 (C-2”, C-6”), 129.1 (C-3”, C-5”), 133.5, 135.5, 156.9 (C-3, C-4’, C-1”), 134.7 (C-4”), 143.9 (C-4), 145.4 (C-2), 147.6 (C-6), 159.0 (C-2’), 159.4 (C-6’), 190.6 (COAr).
General procedure for obtaining indolizines 14a–f
4-(3-Pyridyl)pyrimidine (7, 3 mmol), bromoacetophenones 9 (3 mmol) and different acetylenic dipolarophiles 10 (3.5 mmol) were stirred at reflux in 20 mL 1,2-epoxybutane for 48 h. The products were precipitated with ethanol and removed by filtration. Further purification was made by crystallization from ethanol or column chromatography on Silicagel-60 (Merck, 70–230 mesh) using methylene chloride as eluent.
1-Acetyl-3-(4-fluorobenzoyl)-6-(4-pyrimidinyl)indolizine (14a). Yellow crystals with mp 242–244 °C. Yield 64%. Anal. calcd for C21H14FN3O2: C, 70.19; H, 3.93; N, 11.69; found C, 70.40; H, 4.21; N, 11.88; 1H NMR (CDCl3, 300 MHz) δ 2.56 (s, 3H, CH3), 7.25 (t, J = 8.8 Hz, 1H, H-3”, H-5”), 7.74 (s, 1H, H-2), 7.81 (dd, J = 5.2, 1.4 Hz, 1H, H-6’), 7.85–7.93 (m, 2H, H-2”, H-6”), 8.23 (dd, J = 9.3, 1.6 Hz, 1H, H-7), 8.74 (dd, J = 9.3, 1.1 Hz, 1H, H-8), 8.85 (d, J = 5.2, 1H, H-5’), 9.32 (d, J = 1.4 Hz, 1H, H-2’), 10.71–10.72 (m, 1H, H-5); 13C NMR (CDCl3, 75 MHz) δ 28.0 (CH3), 115.3 (C-1), 115.8 (J = 21.7 Hz, C-3”, C-5”), 116.7 (C-5’), 120.6 (C-8), 126.8 (C-7), 128.8 (C-5), 129.3 (C-2), 131.4 (J = 8.9 Hz, C-2”, C-6”), 122.8, 126.1, 135.7, 139.5, 160.9 (C-3, C-8a, C-6, C-4’, C-1”), 157.9 (C-6’), 159.4 (C-2’), 165.1 (J = 252.3 Hz, C-4”), 184.3 (COAr), 193.1 (CO); IR (KBr, cm−1): 1659, 1630, 1581, 1510, 1485, 1431, 1359, 1213, 1152.
General procedure for obtaining the 4-(4-pyrimidinyl)pyridinium bromides 15
4-(4-Pyridyl)pyrimidine (8) (10 mmol) and different bromoacetophenones 9 (10 mmol) were stirred in 50 mL acetone under reflux for 8 h and then were left at room temperature overnight. The precipitated bromides 15a,b were removed by filtration.
1-[2-(4-Bromophenyl)-2-oxoethyl]-4-(4-pyrimidinyl)pyridinium bromide (15a). Brown powder with mp 264–268 °C. Yield 90%. Anal. calcd for C17H13Br2N3O: N, 9.66; found N, 9.73; 1H NMR (DMSO-d6, 300 MHz) δ 6.61 (s, 2H, CH2), 7.90 (d, J = 8.7 Hz, 2H, H-3”, H-5”), 8.02 (d, J = 8.7 Hz, 2H, H-2”, H-6”), 8.54 (dd, J = 5.3, 1.3 Hz, H-5’), 9.01 (d, J = 6.9 Hz, 2H, H-3, H-5), 9.21–9.24 (m, 3H, H-2, H-6, H-6’), 9.53 (d, J = 1.3 Hz, 1H, H-2’); 13C NMR (DMSO-d6, 75 MHz) δ 66.1 (CH2), 119.8 (C-5’), 125.0 (C-3, C-5), 128.9, 132.6, 151.5, 159.3 (C-4, C-1’, C-1”, C-4”), 130.3 (C-2”, C-6”), 132.3 (C-3”, C-5”), 147.2 (C-2, C-6), 159.6 (C-6’), 159.9 (C-2’), 190.0 (COAr).
General procedure for obtaining the indolizines 16a–f
4-(4-Pyridyl)pyrimidine (8) (3 mmol), bromoacetophenones 9 (3 mmol) and acetylenic dipolarophile 10 (3.5 mmol) in 20 mL 1,2-epoxybutane were stirred under reflux for 48 h. The products were precipitated with ethanol and removed by filtration. Further purification was performed by crystallization from ethanol or column chromatography on Silicagel-60 (Merck) or neutral Al2O3 by using methylene chloride as an eluent.
1-Acetyl-3-benzoyl-7-(4-pyrimidinyl)indolizine (16a). Yellow crystals with mp 236–238 °C. Yield 44%. Anal. calcd for C21H15N3O2: C, 73.89; H, 4.43; N, 12.31; found C, 74.11; H, 4.69; N, 12.59; 1H NMR (CDCl3, 300 MHz) δ 2.51 (s, 3H, Me), 7.48–7.58 (m, 3H, H-3”, H-4”, H-5”), 7.69 (s, 1H, H-2), 7.78–7.81 (m, 2H, H-2”, H-6”), 7.91 (dd, J = 5.3, 1.4 Hz, 1H, H-5’), 7.95 (dd, J = 7.4, 1.9 Hz, 1H, H-6), 8.83 (d, J = 5.4 Hz, 1H, H-6’), 9.28 (dd, J = 1.9, 0.8 Hz, 1H, H-8), 9.30 (d, J = 1.4 Hz, 1H, H-2’), 9.97 (dd, J = 7.4, 0.8 Hz, 1H, H-5); 13C NMR (CDCl3, 75 MHz) δ 28.0 (Me), 114.0 (C-6), 116.5 (C-1), 117.4 (C-5’), 119.0 (C-8), 123.2, 136.5, 138.9, 139.7, 161.0 (C-3, C-8a, C-7, C-1’, C-1”), 129.1 (C-5), 129.3 (C-2), 128.7, 129.1, 132.0 (C-2”, C-6”, C-3”, C-4”, C-5”), 158.1 (C-6’), 159.4 (C-2’), 185.9 (COAr), 193.4 (COMe); IR (KBr, cm−1): 1655, 1616, 1575, 1507, 1394, 1348, 1229, 1193.
Supporting Information
Experimental procedures and structural characterization (1H, 13C NMR data) for all newly synthesized compounds.
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 2.7 MB | Download |
Acknowledgements
MRC is grateful to the NRF (Pretoria) and the University of Cape Town for research support. MP gratefully acknowledges financial support of the Sectorial Operational Programme Human Resources Development 2007-2013 of the Ministry of European Funds through the Financial Agreement POSDRU/159/1.5/S/134398.
References
-
Zhao, D.; You, J.; Hu, C. Chem. – Eur. J. 2011, 17, 5466–5492. doi:10.1002/chem.201003039
Return to citation in text: [1] [2] -
Koszarna, B.; Matczak, R.; Krzeszewwski, M.; Vakuliuk, O.; Klajn, J.; Tasior, M.; Nowicki, J. T.; Gryko, D. T. Tetrahedron 2014, 70, 225–231. doi:10.1016/j.tet.2013.11.088
Return to citation in text: [1] [2] -
Matczak, R.; Koszarna, B.; Gryko, D. T. Tetrahedron 2014, 70, 7006–7009. doi:10.1016/j.tet.2014.07.060
Return to citation in text: [1] [2] -
Akula, M.; El-Khoury, P. Z.; Nag, A.; Bhattacharya, A. RSC Adv. 2014, 4, 25605–25608. doi:10.1039/c4ra00922c
Return to citation in text: [1] [2] -
Gelman, D.; Dechert, S.; Schumann, H.; Blum, J. Inorg. Chim. Acta 2002, 334, 149–158. doi:10.1016/S0020-1693(02)00743-0
Return to citation in text: [1] -
Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/C2OB27002A
Return to citation in text: [1] -
Barboiu, M.; Vaughan, G.; Kyritsakas, N.; Lehn, J.-M. Chem. – Eur. J. 2003, 9, 763–769. doi:10.1002/chem.200390085
Return to citation in text: [1] -
Barboiu, M.; Lehn, J.-M. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5201–5206. doi:10.1073/pnas.082099199
Return to citation in text: [1] -
Ohkita, M.; Lehn, J.-M.; Baum, G.; Fenske, D. Chem. – Eur. J. 1999, 5, 3471–3481. doi:10.1002/(SICI)1521-3765(19991203)5:12<3471::AID-CHEM3471>3.0.CO;2-5
Return to citation in text: [1] -
Bassani, D. M.; Lehn, J.-M.; Baum, G.; Fenske, D. Angew. Chem., Int. Ed. Engl. 1997, 36, 1845–1847. doi:10.1002/anie.199718451
Return to citation in text: [1] -
Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015
Return to citation in text: [1] -
Qin, X.; Liu, H.; Qion, D.; Wu, Q.; You, J.; Zhao, D.; Guo, Q.; Huang, X.; Lan, J. Chem. Sci. 2013, 4, 1964–1969. doi:10.1039/c3sc22241a
Return to citation in text: [1] -
Yu, D.-G.; de Azambuja, F.; Gensch, T.; Daniliuc, C. G.; Glorius, F. Angew. Chem., Int. Ed. 2014, 53, 9650–9654. doi:10.1002/anie.201403782
Return to citation in text: [1] -
Georgescu, E.; Dumitrascu, F.; Georgescu, F.; Draghici, C.; Barbu, L. J. Heterocycl. Chem. 2013, 50, 78–82. doi:10.1002/jhet.997
Return to citation in text: [1] [2] -
Dinica, R. M.; Druta, I. I.; Pettinari, C. Synlett 2000, 1013–1015. doi:10.1055/s-2000-6657
Return to citation in text: [1] [2] [3] -
Chaichi, M. J.; Ehsani, M.; Asghari, S.; Behboodi, V. Luminescence 2014, 29, 1169–1176. doi:10.1002/bio.2678
Return to citation in text: [1] -
Singh, G. S.; Mmatli, E. E. Eur. J. Med. Chem. 2011, 46, 5237–5257. doi:10.1016/j.ejmech.2011.08.042
Return to citation in text: [1] -
Rise, F.; Wikstroem, H.; Ugland, S.; Dijkstra, D.; Gundersen, L. L.; De Boer, P.; Bast, A.; Haenen, G.; Antonsen, O. G. The use of heterocyclic compounds as antioxidants, radical scavangers, Fe2+ complexing agents, tissue and/or neuroprotectants. WO 9621662 A1 July 18, 1996.
Return to citation in text: [1] -
Ono, M.; Sun, L.; Xia, Z. Q.; Kostik, E.; Koya, K.; Wu, Y.; Nagai, M. Fused pyrrole compounds. WO 2004082606 A2 Sept 30, 2004.
Return to citation in text: [1] -
Mangalagiu, G.; Ungureanu, M.; Grosu, G.; Mangalagiu, I. I.; Petrovanu, M. Ann. Pharm. Fr. 2001, 59, 139–140.
Return to citation in text: [1] -
Nicolescu, A.; Deleanu, C.; Georgescu, E.; Georgescu, F.; Iurascu, A.-M.; Shova, S.; Filip, P. Tetrahedron Lett. 2013, 54, 1486–1488. doi:10.1016/j.tetlet.2013.01.036
Return to citation in text: [1] [2] -
Caira, M. R.; Georgescu, E.; Georgescu, F.; Popa, M. M.; Dumitraşcu, F. ARKIVOC 2009, No. xii, 242–253. doi:10.3998/ark.5550190.0010.c21
Return to citation in text: [1] [2] -
Caira, M. R.; Popa, M. M.; Draghici, C.; Barbu, L.; Dumitrescu, D.; Dumitrascu, F. Tetrahedron Lett. 2014, 55, 5635–5638. doi:10.1016/j.tetlet.2014.08.054
Return to citation in text: [1] [2] -
Georgescu, E.; Nicolescu, A.; Georgescu, F.; Teodorescu, F.; Marinescu, D.; Macsim, A.-M.; Deleanu, C. Beilstein J. Org. Chem. 2014, 10, 2377–2387. doi:10.3762/bjoc.10.248
Return to citation in text: [1] [2] -
Dumitraşcu, F.; Vasilescu, M.; Drăghici, C.; Căproiu, M. T.; Barbu, L.; Dumitrescu, D. G. ARKIVOC 2011, No. x, 338–350. doi:10.3998/ark.5550190.0012.a28
Return to citation in text: [1] [2] -
Bredereck, H.; Gompper, R.; Herlinger, H. Chem. Ber. 1958, 91, 2832–2849. doi:10.1002/cber.19580911240
Return to citation in text: [1] [2] -
Nishiwaki, N.; Yamashita, K.; Azuma, M.; Adachi, T.; Tamura, M.; Ariga, M. Synthesis 2004, 1996–2000. doi:10.1055/s-2004-829195
Return to citation in text: [1] -
Bejan, E.; Haddou, H. A.; Daran, J. C.; Balavoine, G. G. A. Synthesis 1996, 1012–1018. doi:10.1055/s-1996-4330
Return to citation in text: [1] [2] [3] -
Bennett, G. B.; Mason, R. B.; Alden, L. J.; Roach, J. B., Jr. J. Med. Chem. 1978, 21, 623–628. doi:10.1021/jm00205a006
Return to citation in text: [1] [2] -
Bredereck, H.; Gompper, R.; Geiger, B. Chem. Ber. 1960, 93, 1402–1406. doi:10.1002/cber.19600930626
Return to citation in text: [1] -
Georgescu, E.; Georgescu, F.; Draghici, C.; Cristian, L.; Popa, M. M.; Dumitrascu, F. Comb. Chem. High Throughput Screening 2013, 16, 851–857. doi:10.2174/13862073113169990050
Return to citation in text: [1] -
Georgescu, E.; Georgescu, F.; Popa, M. M.; Draghici, C.; Tarko, L.; Dumitrascu, F. ACS Comb. Sci. 2012, 14, 101–107. doi:10.1021/co2002125
Return to citation in text: [1] -
Etter, M. C.; Macdonald, J. C.; Bernstein, J. Acta Crystallogr., Sect. B 1990, 46, 256–262. doi:10.1107/S0108768189012929
Return to citation in text: [1] [2] -
Cambridge Structural Database and Cambridge Structural Database system, Version 5.36; Cambridge Crystallographic Data Centre, University Chemical Laboratory: Cambridge, United Kingdom, 2015.
Return to citation in text: [1]
| 26. | Bredereck, H.; Gompper, R.; Herlinger, H. Chem. Ber. 1958, 91, 2832–2849. doi:10.1002/cber.19580911240 |
| 30. | Bredereck, H.; Gompper, R.; Geiger, B. Chem. Ber. 1960, 93, 1402–1406. doi:10.1002/cber.19600930626 |
| 28. | Bejan, E.; Haddou, H. A.; Daran, J. C.; Balavoine, G. G. A. Synthesis 1996, 1012–1018. doi:10.1055/s-1996-4330 |
| 1. | Zhao, D.; You, J.; Hu, C. Chem. – Eur. J. 2011, 17, 5466–5492. doi:10.1002/chem.201003039 |
| 7. | Barboiu, M.; Vaughan, G.; Kyritsakas, N.; Lehn, J.-M. Chem. – Eur. J. 2003, 9, 763–769. doi:10.1002/chem.200390085 |
| 8. | Barboiu, M.; Lehn, J.-M. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5201–5206. doi:10.1073/pnas.082099199 |
| 9. | Ohkita, M.; Lehn, J.-M.; Baum, G.; Fenske, D. Chem. – Eur. J. 1999, 5, 3471–3481. doi:10.1002/(SICI)1521-3765(19991203)5:12<3471::AID-CHEM3471>3.0.CO;2-5 |
| 10. | Bassani, D. M.; Lehn, J.-M.; Baum, G.; Fenske, D. Angew. Chem., Int. Ed. Engl. 1997, 36, 1845–1847. doi:10.1002/anie.199718451 |
| 26. | Bredereck, H.; Gompper, R.; Herlinger, H. Chem. Ber. 1958, 91, 2832–2849. doi:10.1002/cber.19580911240 |
| 27. | Nishiwaki, N.; Yamashita, K.; Azuma, M.; Adachi, T.; Tamura, M.; Ariga, M. Synthesis 2004, 1996–2000. doi:10.1055/s-2004-829195 |
| 28. | Bejan, E.; Haddou, H. A.; Daran, J. C.; Balavoine, G. G. A. Synthesis 1996, 1012–1018. doi:10.1055/s-1996-4330 |
| 29. | Bennett, G. B.; Mason, R. B.; Alden, L. J.; Roach, J. B., Jr. J. Med. Chem. 1978, 21, 623–628. doi:10.1021/jm00205a006 |
| 6. | Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/C2OB27002A |
| 28. | Bejan, E.; Haddou, H. A.; Daran, J. C.; Balavoine, G. G. A. Synthesis 1996, 1012–1018. doi:10.1055/s-1996-4330 |
| 4. | Akula, M.; El-Khoury, P. Z.; Nag, A.; Bhattacharya, A. RSC Adv. 2014, 4, 25605–25608. doi:10.1039/c4ra00922c |
| 5. | Gelman, D.; Dechert, S.; Schumann, H.; Blum, J. Inorg. Chim. Acta 2002, 334, 149–158. doi:10.1016/S0020-1693(02)00743-0 |
| 21. | Nicolescu, A.; Deleanu, C.; Georgescu, E.; Georgescu, F.; Iurascu, A.-M.; Shova, S.; Filip, P. Tetrahedron Lett. 2013, 54, 1486–1488. doi:10.1016/j.tetlet.2013.01.036 |
| 22. | Caira, M. R.; Georgescu, E.; Georgescu, F.; Popa, M. M.; Dumitraşcu, F. ARKIVOC 2009, No. xii, 242–253. doi:10.3998/ark.5550190.0010.c21 |
| 23. | Caira, M. R.; Popa, M. M.; Draghici, C.; Barbu, L.; Dumitrescu, D.; Dumitrascu, F. Tetrahedron Lett. 2014, 55, 5635–5638. doi:10.1016/j.tetlet.2014.08.054 |
| 24. | Georgescu, E.; Nicolescu, A.; Georgescu, F.; Teodorescu, F.; Marinescu, D.; Macsim, A.-M.; Deleanu, C. Beilstein J. Org. Chem. 2014, 10, 2377–2387. doi:10.3762/bjoc.10.248 |
| 25. | Dumitraşcu, F.; Vasilescu, M.; Drăghici, C.; Căproiu, M. T.; Barbu, L.; Dumitrescu, D. G. ARKIVOC 2011, No. x, 338–350. doi:10.3998/ark.5550190.0012.a28 |
| 34. | Cambridge Structural Database and Cambridge Structural Database system, Version 5.36; Cambridge Crystallographic Data Centre, University Chemical Laboratory: Cambridge, United Kingdom, 2015. |
| 2. | Koszarna, B.; Matczak, R.; Krzeszewwski, M.; Vakuliuk, O.; Klajn, J.; Tasior, M.; Nowicki, J. T.; Gryko, D. T. Tetrahedron 2014, 70, 225–231. doi:10.1016/j.tet.2013.11.088 |
| 3. | Matczak, R.; Koszarna, B.; Gryko, D. T. Tetrahedron 2014, 70, 7006–7009. doi:10.1016/j.tet.2014.07.060 |
| 21. | Nicolescu, A.; Deleanu, C.; Georgescu, E.; Georgescu, F.; Iurascu, A.-M.; Shova, S.; Filip, P. Tetrahedron Lett. 2013, 54, 1486–1488. doi:10.1016/j.tetlet.2013.01.036 |
| 22. | Caira, M. R.; Georgescu, E.; Georgescu, F.; Popa, M. M.; Dumitraşcu, F. ARKIVOC 2009, No. xii, 242–253. doi:10.3998/ark.5550190.0010.c21 |
| 23. | Caira, M. R.; Popa, M. M.; Draghici, C.; Barbu, L.; Dumitrescu, D.; Dumitrascu, F. Tetrahedron Lett. 2014, 55, 5635–5638. doi:10.1016/j.tetlet.2014.08.054 |
| 24. | Georgescu, E.; Nicolescu, A.; Georgescu, F.; Teodorescu, F.; Marinescu, D.; Macsim, A.-M.; Deleanu, C. Beilstein J. Org. Chem. 2014, 10, 2377–2387. doi:10.3762/bjoc.10.248 |
| 25. | Dumitraşcu, F.; Vasilescu, M.; Drăghici, C.; Căproiu, M. T.; Barbu, L.; Dumitrescu, D. G. ARKIVOC 2011, No. x, 338–350. doi:10.3998/ark.5550190.0012.a28 |
| 2. | Koszarna, B.; Matczak, R.; Krzeszewwski, M.; Vakuliuk, O.; Klajn, J.; Tasior, M.; Nowicki, J. T.; Gryko, D. T. Tetrahedron 2014, 70, 225–231. doi:10.1016/j.tet.2013.11.088 |
| 3. | Matczak, R.; Koszarna, B.; Gryko, D. T. Tetrahedron 2014, 70, 7006–7009. doi:10.1016/j.tet.2014.07.060 |
| 4. | Akula, M.; El-Khoury, P. Z.; Nag, A.; Bhattacharya, A. RSC Adv. 2014, 4, 25605–25608. doi:10.1039/c4ra00922c |
| 15. | Dinica, R. M.; Druta, I. I.; Pettinari, C. Synlett 2000, 1013–1015. doi:10.1055/s-2000-6657 |
| 17. | Singh, G. S.; Mmatli, E. E. Eur. J. Med. Chem. 2011, 46, 5237–5257. doi:10.1016/j.ejmech.2011.08.042 |
| 18. | Rise, F.; Wikstroem, H.; Ugland, S.; Dijkstra, D.; Gundersen, L. L.; De Boer, P.; Bast, A.; Haenen, G.; Antonsen, O. G. The use of heterocyclic compounds as antioxidants, radical scavangers, Fe2+ complexing agents, tissue and/or neuroprotectants. WO 9621662 A1 July 18, 1996. |
| 19. | Ono, M.; Sun, L.; Xia, Z. Q.; Kostik, E.; Koya, K.; Wu, Y.; Nagai, M. Fused pyrrole compounds. WO 2004082606 A2 Sept 30, 2004. |
| 20. | Mangalagiu, G.; Ungureanu, M.; Grosu, G.; Mangalagiu, I. I.; Petrovanu, M. Ann. Pharm. Fr. 2001, 59, 139–140. |
| 33. | Etter, M. C.; Macdonald, J. C.; Bernstein, J. Acta Crystallogr., Sect. B 1990, 46, 256–262. doi:10.1107/S0108768189012929 |
| 14. | Georgescu, E.; Dumitrascu, F.; Georgescu, F.; Draghici, C.; Barbu, L. J. Heterocycl. Chem. 2013, 50, 78–82. doi:10.1002/jhet.997 |
| 15. | Dinica, R. M.; Druta, I. I.; Pettinari, C. Synlett 2000, 1013–1015. doi:10.1055/s-2000-6657 |
| 14. | Georgescu, E.; Dumitrascu, F.; Georgescu, F.; Draghici, C.; Barbu, L. J. Heterocycl. Chem. 2013, 50, 78–82. doi:10.1002/jhet.997 |
| 15. | Dinica, R. M.; Druta, I. I.; Pettinari, C. Synlett 2000, 1013–1015. doi:10.1055/s-2000-6657 |
| 33. | Etter, M. C.; Macdonald, J. C.; Bernstein, J. Acta Crystallogr., Sect. B 1990, 46, 256–262. doi:10.1107/S0108768189012929 |
| 13. | Yu, D.-G.; de Azambuja, F.; Gensch, T.; Daniliuc, C. G.; Glorius, F. Angew. Chem., Int. Ed. 2014, 53, 9650–9654. doi:10.1002/anie.201403782 |
| 29. | Bennett, G. B.; Mason, R. B.; Alden, L. J.; Roach, J. B., Jr. J. Med. Chem. 1978, 21, 623–628. doi:10.1021/jm00205a006 |
| 1. | Zhao, D.; You, J.; Hu, C. Chem. – Eur. J. 2011, 17, 5466–5492. doi:10.1002/chem.201003039 |
| 11. | Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310. doi:10.1016/j.tet.2009.10.015 |
| 12. | Qin, X.; Liu, H.; Qion, D.; Wu, Q.; You, J.; Zhao, D.; Guo, Q.; Huang, X.; Lan, J. Chem. Sci. 2013, 4, 1964–1969. doi:10.1039/c3sc22241a |
| 16. | Chaichi, M. J.; Ehsani, M.; Asghari, S.; Behboodi, V. Luminescence 2014, 29, 1169–1176. doi:10.1002/bio.2678 |
| 31. | Georgescu, E.; Georgescu, F.; Draghici, C.; Cristian, L.; Popa, M. M.; Dumitrascu, F. Comb. Chem. High Throughput Screening 2013, 16, 851–857. doi:10.2174/13862073113169990050 |
| 32. | Georgescu, E.; Georgescu, F.; Popa, M. M.; Draghici, C.; Tarko, L.; Dumitrascu, F. ACS Comb. Sci. 2012, 14, 101–107. doi:10.1021/co2002125 |
© 2015 Popa et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)