Abstract
Here, we describe a new and simple synthetic strategy to various polycyclic sulfones via Diels–Alder reaction and ring-rearrangement metathesis (RRM) as the key steps. This approach delivers tri- and tetracyclic sulfones with six (n = 1), seven (n = 2) or eight-membered (n = 3) fused-ring systems containing trans-ring junctions unlike the conventional all cis-ring junctions generally obtained during the RRM sequence. Interestingly the starting materials used are simple and commercially available.

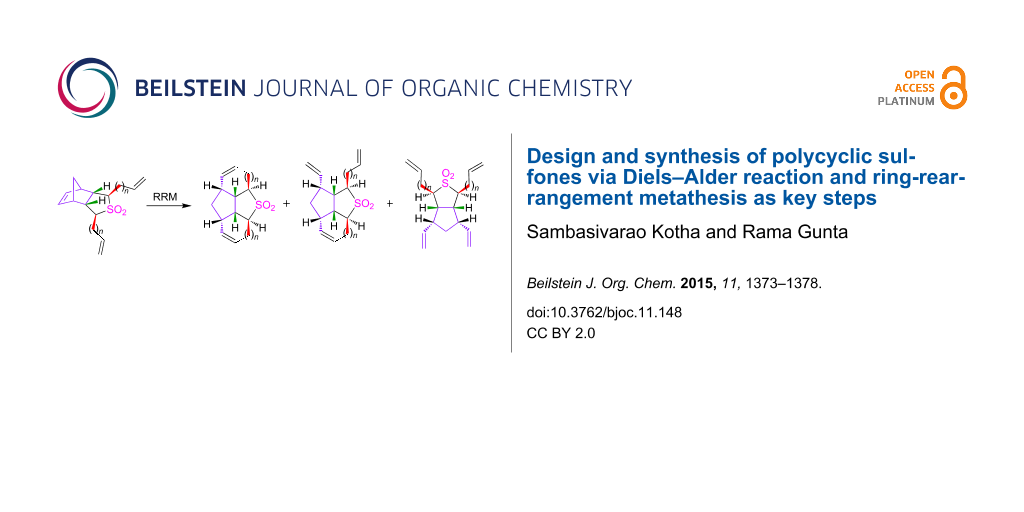
Graphical Abstract
Introduction
Sulfones [1-8] are popular building blocks [9] in organic synthesis. They are also useful substrates for the Ramberg–Bäcklund reaction [10] and they can be alkylated via carbanion chemistry. Moreover, they are suitable synthons in Diels–Alder (DA) reactions [11-14]. In view of various applications of sulfone derivatives, we envisioned a new synthetic strategy based on ring-rearrangement metathesis (RRM) as a key step. It is worth mentioning that the RRM strategy [15-23] with a variety of substrates affords intricate products that are inaccessible by conventional retrosynthetic routes. Several bicyclo[2.2.1]heptane systems [24-26] are known to undergo RRM. However, in almost all instances the products produced are cis-configured at the ring junctions. The main driving force for the RRM of these systems is the release of ring strain. The configuration is transferred from the starting material to the product. In connection with our interest to design new polycycles by RRM [27,28] as a key step, here we conceive unique examples where cis and trans ring junctions are produced in the RRM reactions.
Results and Discussion
Strategy
Our retrosynthetic strategy to diverse sulfone derivatives is shown in Figure 1. The target sulfone derivatives 1 could be synthesized from the functionalized tricyclic sulfone 2 by RRM sequence. The sulfone 2 may be prepared from the dimesylate 3, which in turn, can be assembled from the known anhydride 4 via reduction followed by mesylation of the resulting diol. Compound 4 could be prepared via DA reaction starting with freshly cracked cyclopentadiene and maleic anhydride (Figure 1).
![[1860-5397-11-148-1]](/bjoc/content/figures/1860-5397-11-148-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Retrosynthetic approach to polycyclic sulfones.
Figure 1: Retrosynthetic approach to polycyclic sulfones.
To realize the strategy shown in Figure 1, we started with the preparation of the known compound 4 [29,30]. Later, the DA adduct 4 was reduced with LiAlH4 to deliver the corresponding diol (95%) [31], which was subsequently treated with methanesulfonyl chloride in the presence of triethylamine as a base to obtain the dimesylate 3 (89%). Next, compound 3 was subjected to a cyclization reaction by treating with sodium sulfide nonahydrate (Na2S·9H2O) using 20% Aliquat® 336 as a phase–transfer catalyst (PTC) to produce the known sulfide 5 (83%) [31].
Having the sulfide 5 in hand, our next task was to prepare sulfone 6. In this regard, Trost and Curran [32] have reported the conversion of sulfides to sulfones in the presence of other common functional groups such as olefins by reacting with the oxidizing agent, potassium hydrogen persulfate (KHSO5, commercially available as Oxone®) in aqueous methanol. Equipped with this information, oxidation of compound 5 was attempted under similar reaction conditions to get the desired sulfone 6 [33] (Scheme 1, Table 1).
![[1860-5397-11-148-i1]](/bjoc/content/inline/1860-5397-11-148-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the sulfone 6 via oxidation.
Scheme 1: Preparation of the sulfone 6 via oxidation.
Table 1: Different reaction conditions used to improve the yield of the sulfone 6.
| Entry | Reaction conditions | 6 yield [%] | 7 yield [%] |
|---|---|---|---|
| 1 | Oxone® (3 equiv), MeOH, H2O, 0 °C, 22 h | 29 | 40 |
| 2 | Oxone® (2.5 equiv), MeOH, H2O, −5 °C, 6 h | 89 | 8 |
| 3 | Oxone® (2.5 equiv), MeOH, H2O, −5 °C, 5.5 h | 83 | 15 |
| 4 | Oxone® (2.2 equiv), MeOH, H2O, −8 °C, 4.5 h | 82 | 5 |
| 5 | Oxone® (2 equiv), MeOH, H2O, −20 °C, 5 h | 71 | 5 |
Initially, when the reaction was carried out at 0 °C, the epoxy sulfone 7 was the major product (Table 1, entry 1). However, after a considerable amount of experimentation (Table 1), the desired sulfone 6 has been produced in 89% yield (Table 1, entry 2) but it was not possible to eliminate the formation of the epoxy sulfone 7.
Next, our efforts were directed towards the synthesis of various alkenylated sulfone derivatives. In this regard, Bloch and co-workers reported a useful preparation of monoallylated sulfone 8a [34]. To this end, we carried out the allylation of sulfone 6 with allyl bromide (1.2 equiv) and n-BuLi (2.7 equiv) at −75 °C to rt. The monoallylated sulfone 8a was obtained in 22% yield and the diallylated sulfone 2a in 5% yield. Also, 25% of the starting material was recovered. To optimize the yield of diallylated sulfone 2a various conditions were studied (e.g., NaH and LDA). In this regard, increasing the equivalents of allyl bromide and n-BuLi produced the diallylated sulfone 2a in 80% yield and the monoallylated compound 8a in 10% yield (Table 2, entry 1a) [35] along with a minor amount (3%) of triallylated sulfone 9 (Scheme 2). However, with an excess amount of base (5 equiv) and allyl bromide the diallylated sulfone 2a was isolated as a major product and the triallylated sulfone 9 in 6% yield (Table 2, entry 1b). Later, the monoallylated sulfone 8a has been converted to the desired diallyl compound 2a (88%) under similar reaction conditions. The structures of the diallyl (2a) and triallyl (9) sulfones have been confirmed by 1H and 13C NMR spectral data and further supported by HRMS data. In addition, the structure and stereochemistry of the allyl groups present in compound 2a have been confirmed by single-crystal X-ray diffraction studies and this data clearly indicated that the allylation had occurred at α-position of the sulfone moiety and the two allyl groups are in cis-arrangement with each other [35-37].
![[1860-5397-11-148-i2]](/bjoc/content/inline/1860-5397-11-148-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of alkenylated sulfone derivatives.
Scheme 2: Synthesis of alkenylated sulfone derivatives.
Table 2: Optimized reaction conditions to realize mono and dialkenylated sulfones.
| Entry | n | Reaction conditions |
Monoalkenylated product
yield [%] |
Dialkenylated product
yield [%] |
|---|---|---|---|---|
| 1a | 1 |
allyl bromide (3 equiv), n-BuLi
THF, −75 °C to rt, 25 h |
8a (10) | 2a (80) & 9a (3) |
| 1b |
allyl bromide (10 equiv), n-BuLi
THF, −58 °C to rt, 26 h |
8a (0) | 2a (80) & 9a (6) | |
| 2 | 2 |
4-bromo-1-butene (3 equiv), n-BuLi
HMPA, THF, −74 °C to rt, 20 h |
8b (75b) | 2b (21b) |
| 3 | 3 |
5-bromo-1-pentene (2.5 equiv), n-BuLi
HMPA, THF, −78 °C to rt, 17.5 h |
8c (5) | 2c (57) |
| 4 | 4 |
6-bromo-1-hexene (2.8 equiv), n-BuLi
HMPA, THF, −78 °C to rt, 17 h |
8d (9) | 2d (75) |
aTriallylated product, bisolated yield based on starting material recovered.
Analogously, the alkenylation of sulfone 6 was optimized with other electrophiles and the results are summarized in Table 2 (entries 2–4). In this regard, sulfone 6 was butenylated with 4-bromo-1-butene and n-BuLi in the presence of HMPA at −74 °C to rt to deliver the monobutenylated sulfone 8b in 75% yield. Surprisingly, here a minor amount of the desired dibutenylated sulfone 2b (21%) was isolated (Table 2, entry 2). However, the monobutenylated sulfone 8b can be converted to the dibutenylated sulfone 2b under similar conditions. Next, the same synthetic sequence has been extended to the dipentenyl and the dihexenyl sulfone derivatives. Thus, treatment of sulfone 6 with 5-bromo-1-pentene and n-BuLi using HMPA at −78 °C to rt (Table 2, entry 3) gave the desired dipentenylated sulfone 2c (57%) and a minor amount of monopentenylated sulfone 8c (5%).
Similarly, we synthesized the hexenyl sulfone derivatives 8d and 2d by treating compound 6 with 6-bromo-1-hexene using HMPA and n-BuLi at −78 °C. The desired dihexenylated sulfone 2d has been furnished in 75% yield along with monohexenyl sulfone derivative 8d (9%, Table 2, entry 4). Based on these optimization studies, it was concluded that it is necessary to use the appropriate number of equivalents of the alkenyl bromide and the suitable base to generate the dialkenylated products (Table 2 and Scheme 2).
After the successful synthesis of various dialkenyl sulfone derivatives 2a–d, we focussed our attention towards the RRM step. Initially, the diallyl sulfone 2a (~0.0141 M solution in dry CH2Cl2) was subjected to RRM using G-I catalyst in the presence of ethylene gas in refluxing CH2Cl2 to get the tetracyclic sulfone 1a, however, we isolated the tricyclic sulfone 10 in 48% yield. When the G-I catalyst was replaced with G-II a complex mixture of products was observed as indicated by 1H and 13C NMR spectral data. Later, compound 10 was treated with conventional Grubbs catalysts under different reaction conditions (Table 3) to obtain the RRM product 1a (Scheme 3). Unfortunately, the expected compound 1a was not obtained. The strain present in the trans-fused compound 1a may be responsible for its absence in the RRM sequence.
![[1860-5397-11-148-i3]](/bjoc/content/inline/1860-5397-11-148-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 3: Toluene (~0.004 M) reflux conditions to convert 10 to 1a.
| Entry | Conditions | Result |
|---|---|---|
| 1 | G-I (10 mol %), C2H4, 19 h | SMa recovered |
| 2 | G-II (10 mol %), Ti(OiPr)4, C2H4, 24 h | No productb |
| 3 | HG-IIc (10 mol %), Ti(OiPr)4, C2H4, 24 h | No productb |
aStarting material. bSM not recovered, cHoveyda–Blechert–Grubbs catalyst.
Interestingly, dibutenyl sulfone 2b (~0.0034 M solution in toluene) smoothly underwent RRM with Grubbs 2nd generation (G-II) catalyst in the presence of ethylene in refluxing toluene to produce the anticipated tetracyclic sulfone 1b (97%) (Scheme 4). The sulfone 1b has been characterized by 1H and 13C NMR and DEPT-135 spectral data including HRMS data.
![[1860-5397-11-148-i4]](/bjoc/content/inline/1860-5397-11-148-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, the RRM of dipentenyl sulfone 2c (~0.0031 M solution in toluene) was carried out under similar reaction conditions to furnish 1c. Interestingly, the tricyclic sulfone 11 was isolated in 60% along with the expected tetracyclic sulfone 1c (32%) and a minor amount of ring-opened product 12 (6%, Scheme 5). A complex mixture of products was obtained when compound 2c was exposed to the metathesis catalyst for a longer period of time as indicated by 1H and 13C NMR spectral data.
![[1860-5397-11-148-i5]](/bjoc/content/inline/1860-5397-11-148-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: RRM of the dipentenyl sulfone 2c.
Scheme 5: RRM of the dipentenyl sulfone 2c.
Analogously, dihexenyl sulfone 2d (~0.0024 M solution in toluene) was treated with G-II catalyst to deliver the RRM product in the presence of ethylene in refluxing toluene. In this regard, only ring-opened sulfone 13 was produced in 88% yield (Scheme 6) and no cyclized product was observed. Presumably, this observation may be explained on the basis that the nine-membered ring product was not formed due to the unfavourable steric interactions involved.
![[1860-5397-11-148-i6]](/bjoc/content/inline/1860-5397-11-148-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: RRM of the dihexenyl sulfone 2d.
Scheme 6: RRM of the dihexenyl sulfone 2d.
Conclusion
Several interesting polycyclic sulfone derivatives were designed and assembled involving RRM. The RRM outcome of various sulfones (2a–d) depends on the length of the alkenyl chain. In this context, the dibutenyl sulfone derivative 2b is the most-promising candidate for the RRM protocol. In other instances, for example with propenyl analogue 2a the partial ring-closing product 10 was obtained. With substrate 2c, the eight-membered RRM compound 1c was formed as a minor product and partial ring-closing compound 11 as a major product. With substrate 2d, only ring-opened product 13 was produced. Interestingly, we demonstrated trans-ring junction products are possible in the RRM protocol. It is clear that RRM has a unique place in olefin metathesis [38-45] and further interesting examples are expected in future.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, characterization data and copies of 1H,13C NMR and HRMS spectra for all new compounds. | ||
| Format: PDF | Size: 3.6 MB | Download |
Acknowledgements
We thank the Department of Science and Technology (DST), New Delhi for the financial support and the Sophisticated Analytical Instrument Facility (SAIF), IIT-Bombay for recording spectral data. S.K. thanks the Department of Science and Technology for the award of a J. C. Bose fellowship. R.G. thanks the University Grants Commission (UGC), New Delhi for the award of a research fellowship.
References
-
Alba, A.-N. R.; Companyó, X.; Rios, R. Chem. Soc. Rev. 2010, 39, 2018–2033. doi:10.1039/B911852G
Return to citation in text: [1] -
Meadows, D. C.; Gervay-Hague, J. Med. Res. Rev. 2006, 26, 793–814. doi:10.1002/med.20074
Return to citation in text: [1] -
Bäckvall, J.-E.; Chinchilla, R.; Nájera, C.; Yus, M. Chem. Rev. 1998, 98, 2291–2312. doi:10.1021/cr970326z
Return to citation in text: [1] -
García Ruano, J. L.; Alemán, J.; Parra, A.; Marzo, L. Eur. J. Org. Chem. 2014, 1577–1588. doi:10.1002/ejoc.201301483
Return to citation in text: [1] -
Alonso, D. A.; Fuensanta, M.; Nájera, C.; Varea, M. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 1119–1131. doi:10.1080/10426500590910657
Return to citation in text: [1] -
Roy, K.-M. Sulfones and Sulfoxides. Ullmann's Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2000. doi:10.1002/14356007.a25_487
Return to citation in text: [1] -
Kotha, S.; Ali, R. Tetrahedron Lett. 2015, 56, 2172–2175. doi:10.1016/j.tetlet.2015.03.021
Return to citation in text: [1] -
Kotha, S.; Ali, R. Tetrahedron 2015, 71, 1597–1603. doi:10.1016/j.tet.2015.01.009
Return to citation in text: [1] -
Kotha, S. Acc. Chem. Res. 2003, 36, 342–351. doi:10.1021/Ar020147q
Return to citation in text: [1] -
Harvey, J. E.; Bartlett, M. J. Chem. N. Z. 2010, 74, 63–69.
And references cited therein.
Return to citation in text: [1] -
Metz, P.; Fleischer, M.; Fröhlich, R. Tetrahedron 1995, 51, 711–732. doi:10.1016/0040-4020(94)00969-2
Return to citation in text: [1] -
Barbarella, G.; Cinquini, M.; Colonna, S. J. Chem. Soc., Perkin Trans. 1 1980, 1646–1649. doi:10.1039/P19800001646
Return to citation in text: [1] -
Kotha, S.; Bandi, V. Heterocycles 2015, 90, 226–237. doi:10.3987/Com-14-S(K)9
Return to citation in text: [1] -
Kotha, S.; Khedkar, P. J. Org. Chem. 2009, 74, 5667–5670. doi:10.1021/jo900658z
Return to citation in text: [1] -
Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064–1082. doi:10.1002/asia.200700072
And references cited therein.
Return to citation in text: [1] -
Vincent, G.; Kouklovsky, C. Chem. – Eur. J. 2011, 17, 2972–2980. doi:10.1002/chem.201002558
Return to citation in text: [1] -
Nolan, S. P.; Clavier, H. Chem. Soc. Rev. 2010, 39, 3305–3316. doi:10.1039/B912410c
Return to citation in text: [1] -
Clavier, H.; Broggi, J.; Nolan, S. P. Eur. J. Org. Chem. 2010, 937–943. doi:10.1002/ejoc.200901316
Return to citation in text: [1] -
Tsao, K.-W.; Devendar, B.; Liao, C.-C. Tetrahedron Lett. 2013, 54, 3055–3059. doi:10.1016/j.tetlet.2013.03.142
Return to citation in text: [1] -
Standen, P. E.; Kimber, M. C. Tetrahedron Lett. 2013, 54, 4098–4101. doi:10.1016/j.tetlet.2013.05.112
Return to citation in text: [1] -
Li, J.; Lee, D. Chem. Sci. 2012, 3, 3296–3301. doi:10.1039/C2sc20812a
Return to citation in text: [1] -
Imhof, S.; Blechert, S. Synlett 2003, 609–614. doi:10.1055/s-2003-38367
Return to citation in text: [1] -
Arjona, O.; Csákÿ, A. G.; Plumet, J. Eur. J. Org. Chem. 2003, 611–622. doi:10.1002/ejoc.200390100
Return to citation in text: [1] -
Nguyen, N. N. M.; Leclère, M.; Stogaitis, N.; Fallis, A. G. Org. Lett. 2010, 12, 1684–1687. doi:10.1021/Ol100150f
Return to citation in text: [1] -
Datta, R.; Bose, S.; Viththlbhai, P. B.; Ghosh, S. Tetrahedron Lett. 2014, 55, 3538–3540. doi:10.1016/j.tetlet.2014.04.091
Return to citation in text: [1] -
Lam, J. K.; Pham, H. V.; Houk, K. N.; Vanderwal, C. D. J. Am. Chem. Soc. 2013, 135, 17585–17594. doi:10.1021/ja409618p
Return to citation in text: [1] -
Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781–5784. doi:10.1016/j.tetlet.2014.08.108
Return to citation in text: [1] -
Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582–5590. doi:10.1002/ejoc.201402273
Return to citation in text: [1] -
Goll, J. M.; Fillion, E. Organometallics 2008, 27, 3622–3625. doi:10.1021/Om800390w
Return to citation in text: [1] -
Wilcox, C. F., Jr.; Craig, R. R. J. Am. Chem. Soc. 1961, 83, 3866–3871. doi:10.1021/ja01479a030
Return to citation in text: [1] -
Zhang, M.; Flynn, D. L.; Hanson, P. R. J. Org. Chem. 2007, 72, 3194–3198. doi:10.1021/Jo0620260
Return to citation in text: [1] [2] -
Trost, B. M.; Curran, D. P. Tetrahedron Lett. 1981, 22, 1287–1290. doi:10.1016/S0040-4039(01)90298-9
Return to citation in text: [1] -
Bloch, R.; Abecassis, J. Tetrahedron Lett. 1982, 23, 3277–3280. doi:10.1016/S0040-4039(00)87591-7
Return to citation in text: [1] -
Bloch, R.; Abecassis, J.; Hassan, D. Can. J. Chem. 1984, 62, 2019–2024. doi:10.1139/V84-345
Return to citation in text: [1] -
Kotha, S.; Gunta, R. Acta Crystallogr., Sect. E 2014, 70, o1163–o1164. doi:10.1107/S1600536814022053
Return to citation in text: [1] [2] -
Bloch, R.; Abecassis, J. Synth. Commun. 1985, 15, 959–963. doi:10.1080/00397918508076826
Return to citation in text: [1] -
Bloch, R.; Abecassis, J. Tetrahedron Lett. 1983, 24, 1247–1250. doi:10.1016/S0040-4039(00)81626-3
Return to citation in text: [1] -
Grubbs, R. H. Handbook of Metathesis, 1st ed.; Wiley-VCH: Weinheim, 2003.
Return to citation in text: [1] -
Kotha, S.; Sreenivasachary, N. Indian J. Chem., Sect. B 2001, 40, 763–780.
Return to citation in text: [1] -
Kotha, S.; Lahiri, K. Synlett 2007, 2767–2784. doi:10.1055/s-2007-990954
Return to citation in text: [1] -
Kotha, S.; Mandal, K. Chem. – Asian J. 2009, 4, 354–362. doi:10.1002/asia.200800244
Return to citation in text: [1] -
Kotha, S.; Misra, S.; Sreevani, G.; Babu, B. V. Curr. Org. Chem. 2013, 17, 2776–2795. doi:10.2174/13852728113179990118
Return to citation in text: [1] -
Kotha, S.; Meshram, M.; Tiwari, A. Chem. Soc. Rev. 2009, 38, 2065–2092. doi:10.1039/B810094m
Return to citation in text: [1] -
Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397–421. doi:10.1016/j.tet.2011.10.018
Return to citation in text: [1] -
Kotha, S.; Krishna, N. G.; Halder, S.; Misra, S. Org. Biomol. Chem. 2011, 9, 5597–5624. doi:10.1039/c1ob05413a
Return to citation in text: [1]
| 1. | Alba, A.-N. R.; Companyó, X.; Rios, R. Chem. Soc. Rev. 2010, 39, 2018–2033. doi:10.1039/B911852G |
| 2. | Meadows, D. C.; Gervay-Hague, J. Med. Res. Rev. 2006, 26, 793–814. doi:10.1002/med.20074 |
| 3. | Bäckvall, J.-E.; Chinchilla, R.; Nájera, C.; Yus, M. Chem. Rev. 1998, 98, 2291–2312. doi:10.1021/cr970326z |
| 4. | García Ruano, J. L.; Alemán, J.; Parra, A.; Marzo, L. Eur. J. Org. Chem. 2014, 1577–1588. doi:10.1002/ejoc.201301483 |
| 5. | Alonso, D. A.; Fuensanta, M.; Nájera, C.; Varea, M. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 1119–1131. doi:10.1080/10426500590910657 |
| 6. | Roy, K.-M. Sulfones and Sulfoxides. Ullmann's Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2000. doi:10.1002/14356007.a25_487 |
| 7. | Kotha, S.; Ali, R. Tetrahedron Lett. 2015, 56, 2172–2175. doi:10.1016/j.tetlet.2015.03.021 |
| 8. | Kotha, S.; Ali, R. Tetrahedron 2015, 71, 1597–1603. doi:10.1016/j.tet.2015.01.009 |
| 15. |
Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064–1082. doi:10.1002/asia.200700072
And references cited therein. |
| 16. | Vincent, G.; Kouklovsky, C. Chem. – Eur. J. 2011, 17, 2972–2980. doi:10.1002/chem.201002558 |
| 17. | Nolan, S. P.; Clavier, H. Chem. Soc. Rev. 2010, 39, 3305–3316. doi:10.1039/B912410c |
| 18. | Clavier, H.; Broggi, J.; Nolan, S. P. Eur. J. Org. Chem. 2010, 937–943. doi:10.1002/ejoc.200901316 |
| 19. | Tsao, K.-W.; Devendar, B.; Liao, C.-C. Tetrahedron Lett. 2013, 54, 3055–3059. doi:10.1016/j.tetlet.2013.03.142 |
| 20. | Standen, P. E.; Kimber, M. C. Tetrahedron Lett. 2013, 54, 4098–4101. doi:10.1016/j.tetlet.2013.05.112 |
| 21. | Li, J.; Lee, D. Chem. Sci. 2012, 3, 3296–3301. doi:10.1039/C2sc20812a |
| 22. | Imhof, S.; Blechert, S. Synlett 2003, 609–614. doi:10.1055/s-2003-38367 |
| 23. | Arjona, O.; Csákÿ, A. G.; Plumet, J. Eur. J. Org. Chem. 2003, 611–622. doi:10.1002/ejoc.200390100 |
| 35. | Kotha, S.; Gunta, R. Acta Crystallogr., Sect. E 2014, 70, o1163–o1164. doi:10.1107/S1600536814022053 |
| 36. | Bloch, R.; Abecassis, J. Synth. Commun. 1985, 15, 959–963. doi:10.1080/00397918508076826 |
| 37. | Bloch, R.; Abecassis, J. Tetrahedron Lett. 1983, 24, 1247–1250. doi:10.1016/S0040-4039(00)81626-3 |
| 11. | Metz, P.; Fleischer, M.; Fröhlich, R. Tetrahedron 1995, 51, 711–732. doi:10.1016/0040-4020(94)00969-2 |
| 12. | Barbarella, G.; Cinquini, M.; Colonna, S. J. Chem. Soc., Perkin Trans. 1 1980, 1646–1649. doi:10.1039/P19800001646 |
| 13. | Kotha, S.; Bandi, V. Heterocycles 2015, 90, 226–237. doi:10.3987/Com-14-S(K)9 |
| 14. | Kotha, S.; Khedkar, P. J. Org. Chem. 2009, 74, 5667–5670. doi:10.1021/jo900658z |
| 38. | Grubbs, R. H. Handbook of Metathesis, 1st ed.; Wiley-VCH: Weinheim, 2003. |
| 39. | Kotha, S.; Sreenivasachary, N. Indian J. Chem., Sect. B 2001, 40, 763–780. |
| 40. | Kotha, S.; Lahiri, K. Synlett 2007, 2767–2784. doi:10.1055/s-2007-990954 |
| 41. | Kotha, S.; Mandal, K. Chem. – Asian J. 2009, 4, 354–362. doi:10.1002/asia.200800244 |
| 42. | Kotha, S.; Misra, S.; Sreevani, G.; Babu, B. V. Curr. Org. Chem. 2013, 17, 2776–2795. doi:10.2174/13852728113179990118 |
| 43. | Kotha, S.; Meshram, M.; Tiwari, A. Chem. Soc. Rev. 2009, 38, 2065–2092. doi:10.1039/B810094m |
| 44. | Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397–421. doi:10.1016/j.tet.2011.10.018 |
| 45. | Kotha, S.; Krishna, N. G.; Halder, S.; Misra, S. Org. Biomol. Chem. 2011, 9, 5597–5624. doi:10.1039/c1ob05413a |
| 10. |
Harvey, J. E.; Bartlett, M. J. Chem. N. Z. 2010, 74, 63–69.
And references cited therein. |
| 34. | Bloch, R.; Abecassis, J.; Hassan, D. Can. J. Chem. 1984, 62, 2019–2024. doi:10.1139/V84-345 |
| 35. | Kotha, S.; Gunta, R. Acta Crystallogr., Sect. E 2014, 70, o1163–o1164. doi:10.1107/S1600536814022053 |
| 31. | Zhang, M.; Flynn, D. L.; Hanson, P. R. J. Org. Chem. 2007, 72, 3194–3198. doi:10.1021/Jo0620260 |
| 32. | Trost, B. M.; Curran, D. P. Tetrahedron Lett. 1981, 22, 1287–1290. doi:10.1016/S0040-4039(01)90298-9 |
| 29. | Goll, J. M.; Fillion, E. Organometallics 2008, 27, 3622–3625. doi:10.1021/Om800390w |
| 30. | Wilcox, C. F., Jr.; Craig, R. R. J. Am. Chem. Soc. 1961, 83, 3866–3871. doi:10.1021/ja01479a030 |
| 33. | Bloch, R.; Abecassis, J. Tetrahedron Lett. 1982, 23, 3277–3280. doi:10.1016/S0040-4039(00)87591-7 |
| 27. | Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781–5784. doi:10.1016/j.tetlet.2014.08.108 |
| 28. | Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582–5590. doi:10.1002/ejoc.201402273 |
| 24. | Nguyen, N. N. M.; Leclère, M.; Stogaitis, N.; Fallis, A. G. Org. Lett. 2010, 12, 1684–1687. doi:10.1021/Ol100150f |
| 25. | Datta, R.; Bose, S.; Viththlbhai, P. B.; Ghosh, S. Tetrahedron Lett. 2014, 55, 3538–3540. doi:10.1016/j.tetlet.2014.04.091 |
| 26. | Lam, J. K.; Pham, H. V.; Houk, K. N.; Vanderwal, C. D. J. Am. Chem. Soc. 2013, 135, 17585–17594. doi:10.1021/ja409618p |
| 31. | Zhang, M.; Flynn, D. L.; Hanson, P. R. J. Org. Chem. 2007, 72, 3194–3198. doi:10.1021/Jo0620260 |
© 2015 Kotha and Gunta; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)