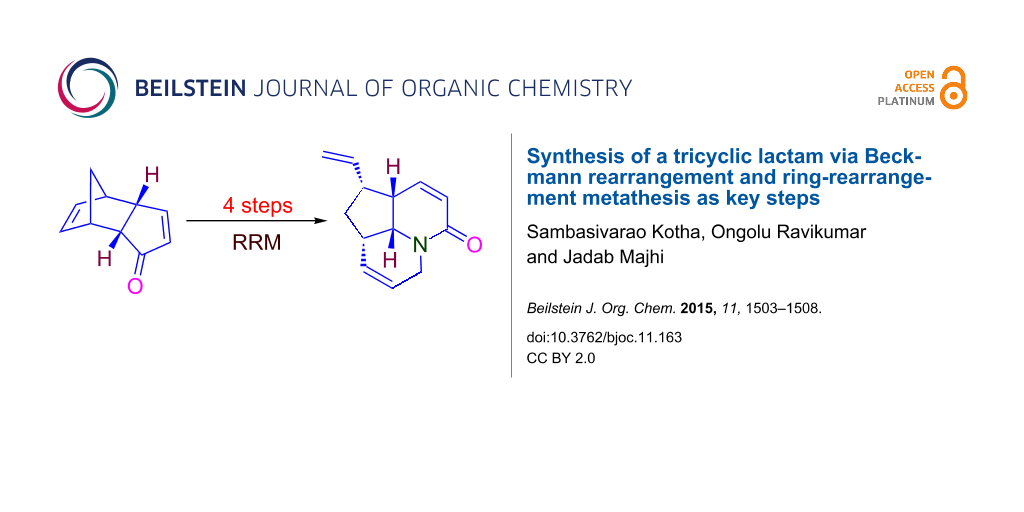
Abstract
A tricyclic lactam is reported in a four step synthesis sequence via Beckmann rearrangement and ring-rearrangement metathesis as key steps. Here, we used a simple starting material such as dicyclopentadiene.

Graphical Abstract
Introduction
The Beckmann rearrangement (BR), a well-known protocol for the conversion of ketoxime to an amide in the presence of acid was discovered in 1886. This rearrangement involves the migration of a group anti to the leaving group on the nitrogen atom. The BR has widely been used in synthetic organic chemistry, for example, a large-scale production of Nylon-6 is based on the synthesis of ε-caprolactam from cyclohexanone oxime involving the BR. The activation energy for the BR is almost the same as that of the nucleophilic substitution at sp2 nitrogen. To synthesize various aza-arenes and cyclic imines, such as quinolines, aza-spiro compounds and dihydropyrroles, the intramolecular SN2-type reaction at the oxime nitrogen is useful [1-6]. Here, we plan to use the BR in combination with a ring-rearrangement metathesis (RRM) [7-24] to generate lactam derivative 1. The RRM protocol involves a tandem process with several metathetic transformations such as ring-closing metathesis (RCM) and ring-opening metathesis (ROM). The RRM has emerged as a powerful tool in organic synthesis because of its ability to transform simple starting materials into complex targets involving an ingenious design. The retrosynthetic strategy to the target molecule 1 is shown in Figure 1. RRM of the tricyclic allylated compound 2 can deliver the target lactam 1. The key synthon 2 can be derived by allylation of lactam 3, which in turn can be prepared via BR starting with the known enone 4 [25-27], derived from dicyclopentadiene (5) [28-30].
![[1860-5397-11-163-1]](/bjoc/content/figures/1860-5397-11-163-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Retrosynthetic analysis of tricyclic amide 1.
Figure 1: Retrosynthetic analysis of tricyclic amide 1.
Results and Discussion
To begin with, the oxidation of dicyclopentadiene (5) in the presence of SeO2 gave 1α-dicyclopentadienol (6), which on treatment with pyridinium chlorochromate (PCC) [31] delivered the known tricyclic enone 4. Selective reduction of enone 4 with Zn in AcOH/EtOH under reflux conditions gave the saturated ketone 7 [32] (Scheme 1).
![[1860-5397-11-163-i1]](/bjoc/content/inline/1860-5397-11-163-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of tricyclic ketone 4.
Scheme 1: Synthesis of tricyclic ketone 4.
Later, tricyclic ketone 7 was reacted with NH2OH·HCl in the presence of NaOAc in dry MeOH at rt to give a mixture of oximes 8a and 8b and this mixture was subjected to a BR under different reaction conditions, like (a) p-TsCl, rt, 15 h, CH3CN (b) p-TsCl, reflux, 15 h, CH3CN (c) PPA, reflux for 20 min. Surprisingly, in all these instances no rearrangement product was observed. Interestingly, when the mixture of oximes 8a and 8b was treated with TsCl in the presence of NaOH at rt lactams 9a and 9b were obtained in 66% combined yield for two steps (9a:9b = 2:1) (Scheme 2) but the products were inseparable by column chromatography. Next, we attempted to separate the mixture of these isomers (9a and 9b) by selective crystallization using different solvent systems. Finally, one of the lactam derivative 9a (δ = 3.86, dd, J = 5.8, 2.9 Hz, 1H) was isolated in pure form from ethanol in 20% yield over two steps.
![[1860-5397-11-163-i2]](/bjoc/content/inline/1860-5397-11-163-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Beckmann rearrangement of oximes 8a and 8b.
Scheme 2: Beckmann rearrangement of oximes 8a and 8b.
Subsequently, we attempted to synthesize the desired lactam 9a via Schmidt reaction or BR of the keto derivative 7 in a single step. In this regard, the tricyclic ketone 7 was treated under different reaction conditions. These include: (a) NaN3, heat 1 day in TFA (b) NaN3, FeCl3 in DCE at rt and reflux, 1 day and (c) TMSN3, FeCl3 in DCE, 1 day. Surprisingly, the desired lactam 9a was not formed. Interestingly, when the tricyclic ketone 7 was treated with hydroxylamine-O-sulfonic acid (NH2OSO3H) in glacial AcOH under reflux conditions, the lactams 9a and 9b were obtained in 48% yield (9a:9b = 2:1) the ratio of oximes 9a and 9b was calculated based on 1H NMR spectral data (Scheme 3).
![[1860-5397-11-163-i3]](/bjoc/content/inline/1860-5397-11-163-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Beckmann rearrangement reaction in a single step.
Scheme 3: Beckmann rearrangement reaction in a single step.
Having prepared the lactams 9a and 9b, the allylation reaction was attempted with the lactam mixture in the presence of NaH/allyl bromide in dry DMF to generate allyl derivatives 10a and 10b in 84% yield. Later, without separation of allyl lactams 10a and 10b, RRM was attempted with the lactam mixture under different catalyst conditions. For example, reaction conditions such as: (a) G-I in dry CH2Cl2, under ethylene atmosphere at rt; (b) G-II in dry CH2Cl2, under ethylene atmosphere at rt and (c) G-I and G-II in dry toluene under ethylene atmosphere did not deliver the desired RRM product 1a (Scheme 4).
![[1860-5397-11-163-i4]](/bjoc/content/inline/1860-5397-11-163-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of ring-rearrangement precursors.
Scheme 4: Synthesis of ring-rearrangement precursors.
Separation of the required isomer from the mixture of oximes 8a and 8b or the lactams 9a and 9b was not possible by column chromatography because of the same Rf value of the individual compounds. Finally, isolation of the required lactam 9a from the mixture was accomplished by using crystallization. Since this method is cumbersome, an alternate method was attempted. To this end, we changed our synthetic route and tried to use the unsaturated ketone 4 and hoped for a different outcome during the BR. In this content, oximation of the enone 4 was carried out with NH2OH·HCl in the presence of NaOAc in dry MeOH. The stereoisomeric oximes, i.e., (E)-11b and (Z)-11a were separated by silica gel column chromatography to deliver 47% and 23% yields, respectively (Scheme 5).
![[1860-5397-11-163-i5]](/bjoc/content/inline/1860-5397-11-163-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of Beckmann rearrangement precursors.
Scheme 5: Synthesis of Beckmann rearrangement precursors.
When the oxime 11a was treated with TsCl in the presence of NaOH in dioxane/H2O (3:4 v/v) at rt lactam 12 was formed in 34% yield. However, the oxime 11b did not give the rearranged product under the same reaction conditions, which clearly indicates that the oxime 11b is unreactive towards BR (Scheme 6). The stereostructure of the oxime 11b has been determined by single crystal X-ray diffraction data (Figure 2) [33].
![[1860-5397-11-163-i6]](/bjoc/content/inline/1860-5397-11-163-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Beckmann rearrangement of oxime isomers 11a and 11b.
Scheme 6: Beckmann rearrangement of oxime isomers 11a and 11b.
![[1860-5397-11-163-2]](/bjoc/content/figures/1860-5397-11-163-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular crystal structure of compound 11b.
Figure 2: Molecular crystal structure of compound 11b.
Allylation of lactam 12 in the presence of NaH/allyl bromide in dry DMF gave the allyl derivative 2 in 80% yield. Finally, the RRM of compound 2 was accomplished with G-II catalyst in dry CH2Cl2, under ethylene atmosphere at rt in the presence of Ti(OiPr)4 to deliver the tricyclic compound 1 in 90% yield (Scheme 7). Its structure has been established on the basis of 1H NMR and 13C NMR spectral data and further supported by HRMS data.
![[1860-5397-11-163-i7]](/bjoc/content/inline/1860-5397-11-163-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of aza tricyclic compound 1 by RRM.
Scheme 7: Synthesis of aza tricyclic compound 1 by RRM.
Conclusion
In summary, we have demonstrated the RRM strategy with the norbornene derivative 2 to deliver the tricyclic compound 1 involving a short synthetic sequence. However, a similar compound 10a did not deliver the RRM product. For the first time, we have demonstrated that BR in combination with RRM is a useful strategy to generate azacyclic compounds. Here we have used an inexpensive starting material such as dicyclopentadiene (5).
Experimental
Synthesis of compounds 9a and 9b
Method 1: Analogously as described in [4], a mixture of 7 (2 g, 13.51 mmol), hydroxylamine hydrochloride (1.41 g, 20.27 mmol), NaOAc (1.66 g, 20.27 mmol) and methanol (50 mL) was stirred at rt for 1 h. The residue after evaporation of the solvent was diluted with water and extracted with ether. Removal of ether furnished the crude oxime (2.4 g). p-Toluenesulfonyl chloride (6.15 g, 32.28 mmol) was added portion-wise over 15 min to a stirred solution of the crude oxime (2.4 g) and NaOH (2.97 g. 74.44 mmol) in 150 mL dioxane/water 3:4 at 5 °C. The mixture was stirred at rt for 15 h and dioxane was removed in vacuo. The residue was dissolved in CH2Cl2 and washed with brine. Removal of the solvent and column chromatography gave a mixture of amide isomers (9a, 9b) (1.45 g, 66%). The amide mixture was crystalized in different solvents and finally one of isomer 9a was isolated from ethanol 20%.
Method 2: A mixture of 7 (100 mg, 0.68 mmol) and hydroxylamine-O-sulfonic acid (113 mg, 1.0 mmol) in AcOH (5 mL) was heated at reflux conditions for 4 h under nitrogen. After completion of the reaction (TLC monitoring), the reaction mixture was basified with 3 N NaOH solution and the organic layer was extracted with CH2Cl2, washed with water, brine and dried by Na2SO4. The combined organic layer was concentrated under reduced pressure and column chromatography gave a mixture of amide isomers 9a and 9b (1.06 g, 48%). The amide mixture was crystalized in different solvents and finally isomer 9a was isolated from ethanol. White solid 9a; mp = 150–155 °C; yield 15%: Rf = 0.30 (EtOAc/pentane 1:1 v/v); IR (neat): 3195 (m), 3067 (w), 2938 (s), 2868 (m), 1674 (s), 1627 (m), 1452 (w), 1434 (w), 1410 (m), 1333 (m), 1252 (w), 1201 (m), 1031 (w), 783 (m), 541 (m) cm−1; 1H NMR (400 MHz, CDCl3) δ 6.31 (s, 1H), 6.22 (dd, J = 5.8, 3.0 Hz, 1H), 6.10 (dd, J = 5.8, 3.0 Hz, 1H), 3.86, (dd, J = 5.8, 2.9 Hz, 1H), 2.97 (s, 1H), 2.88 (s, 2H), 2.48–2.40 (m, 1H), 2.13–2.05 (m,1H), 1.94–1.87 (m, 1H), 1.56 (dt, J = 8.8, 1.8 Hz, 1H), 1.42 (d, J = 8.8 Hz, 1H), 1.23–1.13 (m, 1H) ppm; 13C NMR (100 Hz, CDCl3) δ 175.5, 137.6, 134.2, 54.8, 48.0, 47.8, 46.5, 39.49, 31.4, 23.2 ppm.
Synthesis of compounds 11a and 11b
Analogously as described in [4], a mixture of 4 (9 g, 61.64 mmol), hydroxylamine hydrochloride (6.41 g, 92.34 mmol), NaOAc (7.58 g, 92.49 mmol) and methanol (225 mL) were stirred at rt for 1 h. The residue after evaporation of the solvent was diluted with water and extracted with diethyl ether. Removal of ether furnished the crude oxime which was purified by silica gel column chromatography by eluting appropriate mixture of ethyl acetate/petroleum ether to afford compounds 11a (2.29 g, 23%) and 11b (4.61 g, 47%) as colourless solids.
11a: Rf = 0.29 (EtOAc/pentane 2:8 v/v); IR (neat): 3325 (m), 3013 (m), 2400 (w), 1725 (w), 1337 (w), 1216 (m), 927 (m), 759 (s) cm−1; 1H NMR (500 MHz, CDCl3) δ 9.15 (s, 1H), 6.54 (dd, J = 5.8, 1.3 Hz, 1H), 6.38 (dd, J = 5.8, 2.5 Hz, 1H), 5.97 (dd, J = 5.6, 2.8 Hz, 1H), 5.77 (dd, J = 5.6, 2.9 Hz, 1H), 3.32 (m, 1H), 3.18 (dd, J = 10.7, 4.5 Hz, 1H), 3.16 (s, 1H), 2.28 (s, 1H), 2.90 (s, 1H), 1.61 (d, J = 8.3 Hz, 1H), 1.47 (d, J = 8.3 Hz, 1H) ppm; 13C NMR (125 Hz, CDCl3) δ 165.1, 149.1, 133.3, 133.1, 126.4, 51.0, 50.5, 46.1, 45.9, 44.1 ppm; HRMS (Q-Tof) m/z: [M + Na]+ calcd for C10H11NNaO, 184.0733; found, 184.0734.
11b: mp = 89–91 °C; Rf = 0.30 (EtOAc/petroleum ether 2:8 v/v); IR (neat): 3322 (m), 3020 (m), 2396 (w), 2125 (w), 1705 (m), 1217 (m), 926 (m), 759 (s) cm−1; 1H NMR (500 MHz, CDCl3) δ 8.71 (s, 1H), 6.30 (dd, J = 5.8, 2.5 Hz, 1H), 6.00 (dd, J = 5.7, 1.3 Hz, 1H), 5.90 (dd, J = 5.6, 3.0 Hz, 1H), 5.76 (dd, J = 5.6, 2.9 Hz, 1H), 3.43 (s, 1H), 3.35 (dd, J = 6.1, 4.2 Hz, 1H), 3.30 (m, 1H), 2.90 (s, 1H), 1.64 (d, J = 8.3 Hz, 1H), 1.50 (d, J = 8.3, 1H) ppm; 13C NMR (125 Hz, CDCl3) δ 168.1, 147.0, 133.1, 132.9, 131.1, 51.9, 50.8, 45.1, 45.0, 44.1 ppm; HRMS (Q-Tof) m/z: [M + Na]+ calcd for C10H11NNaO, 184.0733; found, 184.0737.
Synthesis of compound 12
Analogously as described in [4], p-toluenesulfonyl chloride (2.36 g, 12.42 mmol) was added portionwise over 15 min to a stirred solution of oxime 11a (1.0 g, 6.21 mmol) and NaOH (1.24 g. 31.05 mmol) in 100 mL dioxane/water 3:4 at 5 °C. The mixture was stirred at rt for 15 h and the dioxane was removed in vacuo. The residue was dissolved in CH2Cl2 and washed with the brine. Removal of solvent and column chromatography using an appropriate mixture of ethyl acetate/petroleum ether gave the pure lactam 12 (0.33 g, 34%) as a semi solid. IR (neat): 3020 (m), 2400 (w), 2125 (w), 1678 (w), 1422 (w), 1216 (m), 1049 (w), 1022 (w), 929 (w), 759 (s) cm−1; 1H NMR (500 MHz, CDCl3) δ 6.36–6.34 (m, 1H), 6.15 (dd, J = 5.5, 3 Hz, 1H), 6.07 (dd, J = 5.5, 3 Hz, 1H), 5.96 (bs, 1H), 5.63 (dt, J = 8.5, 2 Hz, 1H), 4.12–4.08 (m, 1H), 3.10 (t, J = 0.5 Hz, 1H), 3.06 (d, J = 0.5 Hz, 1H), 2.99–2.95 (m, 1H), 1.44 (dt, J = 8.5, 2Hz, 1H), 1.25–1.22 (m, 1H) pmm; 13C NMR (125 Hz, CDCl3) δ 164.4, 142.4, 136.9, 134.5, 122.4, 54.9, 49.8, 47.8, 44.5, 39.3 ppm; HRMS (Q-Tof) m/z: [M + Na]+ calcd for C10H11NNaO, 184.0733; found, 184.0733.
Synthesis of compound 2
Analogously as described in [8], a suspension of NaH (20 mg, 0.83 mmol) in dry DMF (5mL), was added to compound 12 (70 mg, 0.43 mmol) in dry DMF (5 mL) and allyl bromide (57 mg, 0.47 mmol) at 0 °C under nitrogen and it was stirred for 20 minutes at 0 °C. After completion of the reaction (TLC monitoring) the reaction mixture was acidified with saturated ammonium chloride and extracted with ethyl acetate. The combined organic layer was washed with water and brine and then dried over sodium sulfate. Later, the organic layer was concentrated under reduced pressure and purified by silica gel column chromatography by eluting with an appropriate mixture of ethyl acetate/petroleum ether to afford compound 2 as a brown liquid (87 mg, 80%). IR (neat): 3370 (s), 2945 (m), 2832 (m), 2532 (w), 2044 (w), 1662 (w), 1450 (m), 1114 (m), 1030 (s), 770 (m) cm−1; 1H NMR (500 MHz, CDCl3) δ 6.25–6.23 (m, 1H), 6.05–6.01 (m, 2H), 5.85–5.77 (m, 1H), 5.67 (dd, J = 10, 2 Hz, 1H), 5.26–5.22 (m, 2H), 4.47–4.46 (m, 1H), 4.02 (dd, J = 10, 3.5 Hz, 1H), 3.65–3.60 (m, 1H), 3.29 (s, 1H), 3.08 (s, 1H), 3.01–2.97 (m, 1H), 1.45 (dt, J = 9, 2 Hz, 1H), 1.21–1.24 (m, 1H) ppm; 13C NMR (125 Hz, CDCl3) δ 162.5, 140.1, 137.1, 133.8, 133.6, 123.1, 117.7, 59.43, 48.4, 47.4, 47.3, 44.7, 40.0 ppm; HRMS (Q-Tof) m/z: [M + Na]+ calcd for C13H15NNaO, 224.1046; found, 224.1041.
Synthesis of compound 1
Analogously as described in [8], to a stirred solution of compound 2 (20 mg, 0.099 mmol) in dry CH2Cl2 (20 mL) degassed with nitrogen for 10 minutes, purged with ethylene gas for 10 minutes was then added Ti(OiPr)4 and Grubbs-II catalyst (8.4 mg, 10 mol %) and stirred for 5 h at reflux conditions under ethylene atmosphere. After completion of the reaction (TLC monitoring) the solvent was removed on a rotavapor under reduced pressure and purified by silica gel column chromatography by eluting with an appropriate mixture of ethyl acetate/petroleum ether to afford 1 as a brown coloured semi solid (18 mg, 90%). IR (neat): 3020 (m), 2927 (m), 2861 (m), 2396 (w), 1727 (w), 1608 (w), 1461 (w), 1216 (m), 929 (w), 762 (s) cm−1; 1H NMR (500 MHz, CDCl3) δ 6.35–6.27 (m, 1H), 6.05–5.89 (m, 1H), 5.88–5.83 (m, 1H), 5.75–5.72 (m, 1H), 5.63 (dt, J = 16.0, 9.7 Hz, 1H), 5.02–4.91 (m, 2H), 4.64–4.57 (m, 1H), 4.07–4.03 (m , 1H), 3.50–3.42 (m, 1H), 3.19–3.14 (m, 1H), 3.12–2.94 (m, 1H), 2.62–2.55 (m, 1H), 2.21–2.03 (m, 1H), 1.62–1.53 (m, 1H) ppm; 13C NMR (125 Hz, CDCl3) δ 164.2, 139.6, 139.6, 125.7, 123.5, 123.2, 115.5, 59.0, 58.8, 49.1, 42.3, 40.9, 39.6 ppm; HRMS (Q-Tof) m/z: [M + Na]+ calcd for C13H15NNaO, 224.1046; found, 224.1041.
Supporting Information
| Supporting Information File 1: NMR spectra of synthesized compounds and X-ray data of compound 11b. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
We thank the Department of Science and Technology (DST), New Delhi for the financial support and the Sophisticated Analytical Instrument Facility (SAIF), IIT-Bombay for recording spectral data and also thank Gaddamedi Sreevani and Darshan Mhatre for their help in collecting the X-ray data and structure refinement. S. K. thanks the Department of Science and Technology for the award of a J. C. Bose fellowship. O. R. thanks the University Grants Commission, New Delhi for the award of a research fellowship. J. M thanks DST for the award of inspire fellowship.
References
-
Beckmann, E. Ber. Dtsch. Chem. Ges. 1886, 19, 988. doi:10.1002/cber.188601901222
Return to citation in text: [1] -
Blatt, A. H. Chem. Rev. 1933, 12, 215. doi:10.1021/cr60042a002
Return to citation in text: [1] -
Chandrasekhar, S. The Beckmann and Related Reactions. In Comprehensive Organic Synthesis II; Knochel, P.; Molander, G. A., Eds.; Elsevier, 2014; Vol. 7, pp 770 ff. doi:10.1016/B978-0-08-097742-3.00730-8
Return to citation in text: [1] -
Mehta, G.; Praveen, M. J. Org. Chem. 1995, 60, 279. doi:10.1021/jo00106a052
Return to citation in text: [1] [2] [3] [4] -
Kaur, N.; Sharma, P.; Kishore, D. J. Chem. Pharm. Res. 2012, 4, 1938.
Return to citation in text: [1] -
Narasaka, K.; Kitamura, M. Eur. J. Org. Chem. 2005, 4505. doi:10.1002/ejoc.200500389
Return to citation in text: [1] -
Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397. doi:10.1016/j.tet.2011.10.018
Return to citation in text: [1] -
Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582. doi:10.1002/ejoc.201402273
Return to citation in text: [1] [2] [3] -
Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781. doi:10.1016/j.tetlet.2014.08.108
Return to citation in text: [1] -
Zuercher, W. J.; Hashimoto, M.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 6634. doi:10.1021/ja9606743
Return to citation in text: [1] -
Beligny, S.; Eibauer, S.; Maechling, S.; Blechert, S. Angew. Chem., Int. Ed. 2006, 45, 1900. doi:10.1002/anie.200503552
Return to citation in text: [1] -
Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064. doi:10.1002/asia.200700072
Return to citation in text: [1] -
Li, J.; Lee, D. Eur. J. Org. Chem. 2011, 4269. doi:10.1002/ejoc.201100438
Return to citation in text: [1] -
Kotha, S.; Singh, K. Eur. J. Org. Chem. 2007, 5909. doi:10.1002/ejoc.200700744
Return to citation in text: [1] -
Carreras, J.; Avenoza, A.; Busto, J. H.; Peregrina, J. M. J. Org. Chem. 2011, 76, 3381. doi:10.1021/jo200321t
Return to citation in text: [1] -
North, M.; Banti, D. Adv. Synth. Catal. 2002, 344, 694.
Return to citation in text: [1] -
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953. doi:10.1021/ol990909q
Return to citation in text: [1] -
Nguyen, N. N. M.; Leclère, M.; Stogaitis, N.; Fallis, A. G. Org. Lett. 2010, 12, 1684. doi:10.1021/ol100150f
Return to citation in text: [1] -
Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 248. doi:10.1021/ol9025606
Return to citation in text: [1] -
Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592. doi:10.1002/anie.200300576
Return to citation in text: [1] -
Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18. doi:10.1021/ar000114f
Return to citation in text: [1] -
Vincent, G.; Kouklovsky, C. Chem. – Eur. J. 2011, 17, 2972. doi:10.1002/chem.201002558
Return to citation in text: [1] -
Gao, F.; Stamp, C. T. M.; Thoenton, P. D.; Cameron, T. S.; Doyle, L. E.; Miller, D. O.; Burnell, D. J. Chem. Commun. 2012, 48, 233. doi:10.1039/C1CC15452D
Return to citation in text: [1] -
Malik, C. K.; Hossain, M. F.; Ghosh, S. Tetrahedron Lett. 2009, 50, 3063. doi:10.1016/j.tetlet.2009.04.033
Return to citation in text: [1] -
Woodward, R. B.; Katz, T. J. Tetrahedron 1959, 5, 70. doi:10.1016/0040-4020(59)80072-7
Return to citation in text: [1] -
Rosenblum, M. J. Am. Chem. Soc. 1957, 79, 3179. doi:10.1021/ja01569a050
Return to citation in text: [1] -
Lin, H.-C.; Wu, H.-J. J. Chin. Chem. Soc. 2009, 56, 1072. doi:10.1002/jccs.200900155
Return to citation in text: [1] -
Álvarez, C.; Peláez, R.; Medarde, M. Tetrahedron 2007, 63, 2132. doi:10.1016/j.tet.2007.01.001
Return to citation in text: [1] -
Yamaguchi, T.; Ono, T. Chem. Ind. 1968, 769.
Return to citation in text: [1] -
Crivello, J. V.; Song, S. Chem. Mater. 2000, 12, 3674. doi:10.1021/cm000556l
Return to citation in text: [1] -
Corey, E. J.; Suggs, W. Tetrahedron Lett. 1975, 16, 2647. doi:10.1016/S0040-4039(00)75204-X
Return to citation in text: [1] -
Shao, C.; Yu, H.-J.; Wu, N.-Y.; Feng, C.-G.; Lin, G.-Q. Org. Lett. 2010, 12, 3820. doi:10.1021/ol101531r
Return to citation in text: [1] -
CCDC-1403298 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1]
| 1. | Beckmann, E. Ber. Dtsch. Chem. Ges. 1886, 19, 988. doi:10.1002/cber.188601901222 |
| 2. | Blatt, A. H. Chem. Rev. 1933, 12, 215. doi:10.1021/cr60042a002 |
| 3. | Chandrasekhar, S. The Beckmann and Related Reactions. In Comprehensive Organic Synthesis II; Knochel, P.; Molander, G. A., Eds.; Elsevier, 2014; Vol. 7, pp 770 ff. doi:10.1016/B978-0-08-097742-3.00730-8 |
| 4. | Mehta, G.; Praveen, M. J. Org. Chem. 1995, 60, 279. doi:10.1021/jo00106a052 |
| 5. | Kaur, N.; Sharma, P.; Kishore, D. J. Chem. Pharm. Res. 2012, 4, 1938. |
| 6. | Narasaka, K.; Kitamura, M. Eur. J. Org. Chem. 2005, 4505. doi:10.1002/ejoc.200500389 |
| 31. | Corey, E. J.; Suggs, W. Tetrahedron Lett. 1975, 16, 2647. doi:10.1016/S0040-4039(00)75204-X |
| 28. | Álvarez, C.; Peláez, R.; Medarde, M. Tetrahedron 2007, 63, 2132. doi:10.1016/j.tet.2007.01.001 |
| 29. | Yamaguchi, T.; Ono, T. Chem. Ind. 1968, 769. |
| 30. | Crivello, J. V.; Song, S. Chem. Mater. 2000, 12, 3674. doi:10.1021/cm000556l |
| 25. | Woodward, R. B.; Katz, T. J. Tetrahedron 1959, 5, 70. doi:10.1016/0040-4020(59)80072-7 |
| 26. | Rosenblum, M. J. Am. Chem. Soc. 1957, 79, 3179. doi:10.1021/ja01569a050 |
| 27. | Lin, H.-C.; Wu, H.-J. J. Chin. Chem. Soc. 2009, 56, 1072. doi:10.1002/jccs.200900155 |
| 7. | Kotha, S.; Dipak, M. K. Tetrahedron 2012, 68, 397. doi:10.1016/j.tet.2011.10.018 |
| 8. | Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582. doi:10.1002/ejoc.201402273 |
| 9. | Kotha, S.; Ravikumar, O. Tetrahedron Lett. 2014, 55, 5781. doi:10.1016/j.tetlet.2014.08.108 |
| 10. | Zuercher, W. J.; Hashimoto, M.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 6634. doi:10.1021/ja9606743 |
| 11. | Beligny, S.; Eibauer, S.; Maechling, S.; Blechert, S. Angew. Chem., Int. Ed. 2006, 45, 1900. doi:10.1002/anie.200503552 |
| 12. | Holub, N.; Blechert, S. Chem. – Asian J. 2007, 2, 1064. doi:10.1002/asia.200700072 |
| 13. | Li, J.; Lee, D. Eur. J. Org. Chem. 2011, 4269. doi:10.1002/ejoc.201100438 |
| 14. | Kotha, S.; Singh, K. Eur. J. Org. Chem. 2007, 5909. doi:10.1002/ejoc.200700744 |
| 15. | Carreras, J.; Avenoza, A.; Busto, J. H.; Peregrina, J. M. J. Org. Chem. 2011, 76, 3381. doi:10.1021/jo200321t |
| 16. | North, M.; Banti, D. Adv. Synth. Catal. 2002, 344, 694. |
| 17. | Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953. doi:10.1021/ol990909q |
| 18. | Nguyen, N. N. M.; Leclère, M.; Stogaitis, N.; Fallis, A. G. Org. Lett. 2010, 12, 1684. doi:10.1021/ol100150f |
| 19. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 248. doi:10.1021/ol9025606 |
| 20. | Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592. doi:10.1002/anie.200300576 |
| 21. | Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18. doi:10.1021/ar000114f |
| 22. | Vincent, G.; Kouklovsky, C. Chem. – Eur. J. 2011, 17, 2972. doi:10.1002/chem.201002558 |
| 23. | Gao, F.; Stamp, C. T. M.; Thoenton, P. D.; Cameron, T. S.; Doyle, L. E.; Miller, D. O.; Burnell, D. J. Chem. Commun. 2012, 48, 233. doi:10.1039/C1CC15452D |
| 24. | Malik, C. K.; Hossain, M. F.; Ghosh, S. Tetrahedron Lett. 2009, 50, 3063. doi:10.1016/j.tetlet.2009.04.033 |
| 8. | Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582. doi:10.1002/ejoc.201402273 |
| 8. | Kotha, S.; Ravikumar, O. Eur. J. Org. Chem. 2014, 5582. doi:10.1002/ejoc.201402273 |
| 33. | CCDC-1403298 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 32. | Shao, C.; Yu, H.-J.; Wu, N.-Y.; Feng, C.-G.; Lin, G.-Q. Org. Lett. 2010, 12, 3820. doi:10.1021/ol101531r |
© 2015 Kotha et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)