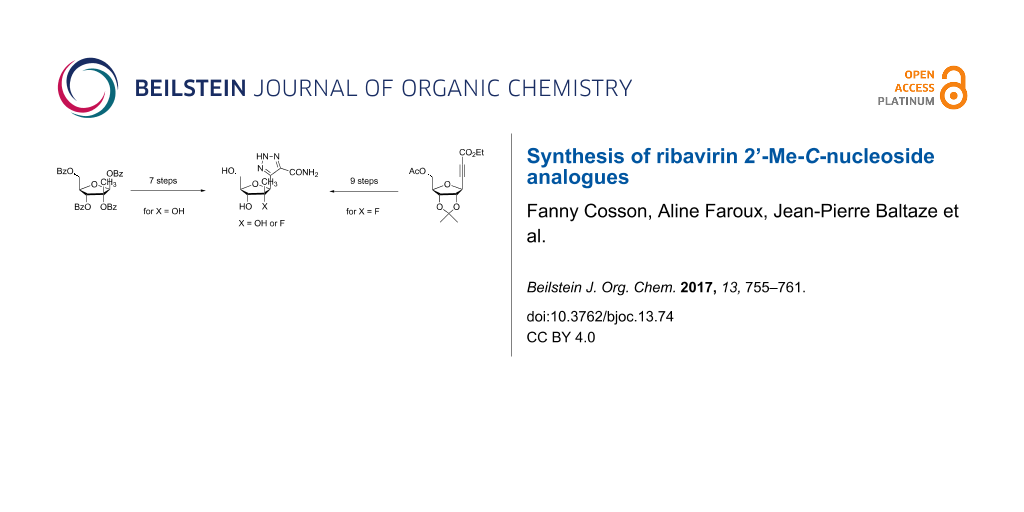
Abstract
An efficient synthetic pathway leading to two carbonated analogues of ribavirin is described. The key-steps in the synthesis of these ribosyltriazoles bearing a quaternary carbon atom in the 2’-position are an indium-mediated alkynylation and a 1,3-dipolar cyclization.

Graphical Abstract
Introduction
The triazole nucleoside ribavirin (RBV, Figure 1) is used for the treatment of a number of viral infections and may be promising as an anticancer drug [1-3]. The antiviral activity of ribavirin is ascribed to a combination of different mechanisms [4]. Although RBV causes some side effects [5-7] essentially due to its accumulation in red blood cells, it is indispensable in the treatment against hepatitis C virus (HCV). The current standard-of-care for hepatitis C involves taking a combination [8] of an antipolymerase compound (sofosbuvir [9,10]) and an antiprotease compound (simeprevir [11,12]), both associated to ribavirin. If the presence of interferon is not required for the therapy, ribavirin is mandatory in the combination, due to its particular role.
![[1860-5397-13-74-1]](/bjoc/content/figures/1860-5397-13-74-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Recently, we developed an alkynyl glycosylation protocol allowing us to obtain C-nucleoside derivatives and we turned our attention to ribavirin C-nucleoside analogues. Moreover, recently De Clerq [13] outlined the potential of C-nucleosides in the arsenal of antivirals due to their stability in biological fluids and their bioavailability.
Some years ago, we described an approach leading to the C-ribosylated analogue 1 of ribavirin (Figure 2) with the key-steps of the synthesis being an indium-mediated alkynylation of a ribose derivative followed by the Huisgen cycloaddition reaction onto the C-alkynyl riboside intermediately obtained [14].
![[1860-5397-13-74-2]](/bjoc/content/figures/1860-5397-13-74-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Herein we describe the synthesis of two new carbonated analogues 2 and 3 of RBV modified at the 2’-position (Figure 1). In fact, a quaternization in 2’-position of various nucleosides led to a higher efficacy against HCV as in the case of 2’-C-methylcytidine and 2’-C-methyladenosine [15]. The structure–activity studies of 2’-C-methylnucleosides showed that the methyl substituent must be in 2’-position and on the β face for an optimal efficacy that drops when the methyl is on the α face or in the 3’-position or if a bulkier ethyl group is used [16]. On the other hand, currently 2’-deoxy-2’-C-methyl-2’-C-fluoronucleosides are developed because a fluoro substituent in the 2’-position increases the antiviral activity and specificity due to a higher tolerance of viral polymerases with respect to incorporation of such compounds [17]. In clinical studies (phase I and II), the fluorinated compound mericitabine in combination with PEG-IFN and RBV was better tolerated and more effective in genotype 1 or 4 patients compared to the standard combination of Peg-IFN and RBV [18,19].
Further, the therapy of untreated patients with HCV genotype 1, 2, or 3 infections with a combination of sofosbuvir (Gilead) and ribavirin for 12 weeks is considered as the most effective treatment at the moment [20].
Results and Discussion
2’-C-Methylnucleoside 2 was synthesized according to a seven step pathway starting from the commercially available 2-C-methyl-1,2,3,5-tetra-O-benzoyl-β-D-ribofuranose (4) as shown in Scheme 1. Debenzoylation of 4 followed by selective protection led to derivative 5, which was submitted to the indium-mediated alkynylation reaction affording the alkynyl riboside 6 with the same β-anomeric selectivity as for the non-methylated derivative [21]. Then, the 1,3-dipolar cycloaddition reaction of 6 with benzyl azide in toluene at 70 °C led to a mixture of regioisomeric triazoles 7 in a 42:58 ratio. The removal of all protecting groups was achieved by treatment of compounds 7 with ammonia followed by catalytic hydrogenolysis. The latter reaction simultaneously cleaves the benzyl and isopropylidene groups affording compound 2 as a single isomer [22].
![[1860-5397-13-74-i1]](/bjoc/content/inline/1860-5397-13-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 5-(2’-C-methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (2).
Scheme 1: Synthesis of 5-(2’-C-methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (2).
In the case of 5-(2’-deoxy-2’-methyl-2’-fluoro-β-D-ribosyl)-1,2,3-triazole-4-carboxamide (3) the synthesis was more delicate as it is necessary to differentiate the 2’ position.
After the indium-mediated alkynylation, the obtained alkynyl riboside 8 was submitted to a Huisgen cycloaddition reaction with benzyl azide, under the same conditions as in the previous case, affording the mixture of regioisomeric triazoles 9a and 9b in a 37:63 ratio [14] (Scheme 2).
![[1860-5397-13-74-i2]](/bjoc/content/inline/1860-5397-13-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the 2’-keto derivatives 12a/12b.
Scheme 2: Synthesis of the 2’-keto derivatives 12a/12b.
The selective protection of the 3’ and 5’-positions requires the full deprotection of the ribose. This was performed by the treatment with Dowex 50Wx8 (H+) in methanol. However, carrying out this step with the mixture of 9a/9b the reaction led to only a moderate yield (52%) and with formation of a partially deprotected compound (only acetal deprotected), stemming from 9b, demonstrating the different hydrolysis rate of each regioisomer. An HPLC analysis of the disappearance of 9a and 9b and the formation of 10a and 10b showed a ratio of 2:1 in favor of regioisomer 10a. The hydrolysis was more efficient when performed with the isolated 9a or 9b isomers. In this case, the fully deprotected compounds 10a and 10b were obtained in 89% (after 36 h) and 61% (after 2 weeks) yields, respectively.
This rate difference was also observed in the subsequent protection of the 3’,5’ positions with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2). While this reaction proceeded with a very poor yield in the case of the mixture 10a/10b (~10% yield), compound 11a was obtained in 79% yield from pure 10a (24% yield for 11b starting from 10b). Thereafter, the oxidation of 11a to the corresponding ketone with Dess–Martin periodinane afforded 12a in 87% yield whereas the reaction of the less reactive isomer 11b led to 12b in 44% yield.
With the aim to get some explanations for the different reactivities observed for the two isomers 10a and 10b, we investigated the structure of the less reactive compound 10b. As depicted in Figure 3, compound 10b displayed an S-type conformation and an anti arrangement of the atoms O(1’)-C(1’)-C(1)-N(2) according to the dihedral angle of 0° lower than 90° (43°). Moreover, the benzyl group appeared to be present in two different positions covering the furan ring.
![[1860-5397-13-74-3]](/bjoc/content/figures/1860-5397-13-74-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray spectrum of compound 10b.
Figure 3: X-ray spectrum of compound 10b.
For the attempted methylation of the 2’-position different conditions were tested (Table 1). As described for other nucleosides [23], the use of MeLi led to compound 13b obtained by an attack from the α-face even if this proceeded with a very low yield (7%). The use of MeMgBr gave an almost 1:1 mixture by α- and β-attack; this second one can be explained by a magnesium complexation with the base. More interestingly, the methylation proceeded stereoselectively leading to 13b in 87% yield when trimethylaluminium was used [24].
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-74-i5.svg?max-width=637&scale=1.0)
The stereochemical outcome of this reaction was determined by selective 1D NOESY experiments (Figure 4). First, the hydrogen H3’ in 13a and 13b was selectively excited. The nOes observed for compound 13a are in the following order of decreasing intensity: CH3 > H5’a > H5’b-H4’ > H1’ confirming that CH3 and H3’ are spatially close. In the case of 13b the nOe intensities decrease in the order: H5’b-H4’ > CH3 > H1’. The selective excitation of H1’ in 13a led to nOes with decreasing intensities in the order: H5’b-H4’ > CH3 > H3’, whereas for 13b the order was CH3 > H5’b-H4’. This second series of nOes confirms that CH3 is closer to H1’ in 13b than in 13a.
![[1860-5397-13-74-4]](/bjoc/content/figures/1860-5397-13-74-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Structural study of isomeric compounds 13.
Figure 4: Structural study of isomeric compounds 13.
The fluorination of 13b with DAST was performed at −20 °C and afforded the desired fluorinated derivative 14 (24%) along with two elimination products, the exocyclic olefin 15 (16%) and the corresponding endocyclic one 16 (20%). As it was impossible to separate 15 from 14 at this stage, the mixture 14/15 was deprotected with tetrabutylammonium fluoride leading to the mixture of diols 17 and 18 in quantitative yield which were easily separated (Scheme 3).
![[1860-5397-13-74-i3]](/bjoc/content/inline/1860-5397-13-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Fluorination of ethyl 1-benzyl-4-(2’-C-methyl-3’,5’-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl)-1,2,3-triazole-5-carboxylate (13b).
Scheme 3: Fluorination of ethyl 1-benzyl-4-(2’-C-methyl-3’,5’-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-ribof...
Finally, the aminolysis of compound 17 followed by catalytic hydrogenolysis in the presence of palladium chloride led to the desired compound 3 [22] (Scheme 4).
![[1860-5397-13-74-i4]](/bjoc/content/inline/1860-5397-13-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 5-(2’-deoxy-2’-fluoro-2’-methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (3).
Scheme 4: Synthesis of 5-(2’-deoxy-2’-fluoro-2’-methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (3).
Conclusion
The indium-mediated alkynylation of a ribose derivative followed by a Huisgen cyclization allowed the access to 2’-quaternized carbonated analogues of ribavirin. While the synthesis of compound 2 starting from 2-methylated ribose derivative 4 was quite easy to perform, the preparation of the fluorinated analogue 3 required a more complicated pathway. This included the selective protection of the 3’,5’ positions, the stereoselective methylation and the fluorination of the 2’ position. The synthesized compounds are currently investigated for their antiviral activities.
Experimental
Experimental procedures for compounds 2, 5–7, and 10–18 are covered by the Ph.D. thesis of Fanny Cosson [22].
5-(2’-C-Methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (2) [22]: Through the solution of compound 7 (mixture of 7a and 7b, 0.49 g, 1.1 mmol) in anhydrous methanol (14 mL) was bubbled ammonia gas for 2 h at 0 °C. Then the mixture was stirred for 12 h at rt and concentrated in vacuum. The residue was purified by flash chromatography (EtOAc/cyclohexane, 3:7 to 1:0) affording the mixture of the corresponding 1-benzyl-4-(2’,3’-O-isopropylidene-2’-C-methyl-β-D-ribofuranosyl)-1,2,3-triazole-5-carboxamide and 1-benzyl-5-(2’,3’-O-isopropylidene-2’-C-methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (0.25 g, 60%) in a 27:73 ratio as an oil. IR (cm−1) νmax: 3336, 2360, 2342, 1635; 1H NMR (400 MHz, CDCl3) δ 7.32–7.25 (m, 5H, Ph), 6.05 (d, J = 14.6 Hz, 1H, CH2Ph), 5.59 (d, J = 14.6 Hz, 1H, CH2Ph), 5.37 (s, 1H, H1), 4.54 (d, J = 5.0 Hz, 1H, H3), 4.35–4.32 (m, 1H, H4), 3.82–3.75 (m, 2H, H5, H5’), 1.60 (s, 3H, CH3), 1.37 (s, 3H, C(CH3)2), 1.34 (s, 3H, C(CH3)2) and δ 7.32-7.25 (m, 5H, Ph), 6.12 (d, J = 14 Hz, 1H, CH2Ph), 5.80 (d, J = 14 Hz, 1H, CH2Ph), 5.33 (s, 1H, H1), 4.84–4.81 (m, 1H, H4), 4.45 (d, J = 2.7 Hz, 1H, H3), 3.91 (dd, J = 3.7 Hz, 11.9 Hz, 1H, H5), 3.82–3.75 (m, 1H, H5’), 1.64 (s, 3H, CH3), 1.24 (s, 3H, C(CH3)2), 1.23 (s, 3H, C(CH3)2); 13C NMR (100 MHz, CDCl3) δ 160.3 (CONH2), 149.7 (triazole), 129.6 (Cq Ph), 129–128 (Ph), 125.0 (triazole), 112.9 (C(CH3)2), 90.0 (C2), 87.1 (C3), 85.1 (C4), 84.1 (C1), 62.0 (C5), 53.8 (CH2Ph), 26.6 (C(CH3)2), 26.5 (C(CH3)2), 14.3 (CH3) and δ 159.1 (CONH2), 141.7 (triazole), 135.6 (Cq Ph), 135.1 (triazole), 129–128 (Ph), 114.5 (C(CH3)2), 90.7 (C2), 87.8 (C3), 84.8 (C4), 82.4 (C1), 62.8 (C5), 54.0 (CH2Ph), 26.0 (C(CH3)2), 25.3 (C(CH3)2); HRMS calcd for C19H22O5, 330.1467; found, 330.1478.
The mixture of carboxamides and palladium chloride (23 mg, 0.13 mmol) in ethanol (10 mL) was hydrogenated with H2 at 4 bar for 48 h. After filtration over Celite and concentration in vacuum, the crude was purified by flash chromatography (cyclohexane/EtOAc 2:8 to EtOAc/MeOH 1:1) to afford compound 2 as a white solid (0.12 g, 75%) that was lyophilized. [α]D24 −47 (c 1, MeOH); mp 185–190 °C; IR (cm−1) νmax: 3202, 1663, 1604, 1382, 1014; 1H NMR (400 MHz, MeOD) δ 5.48 (s, 1H, H1), 4.11–4.04 (m, 1H, H4), 3.99 (d, J = 8.3 Hz, 1H, H3), 3.83 (dd, J = 2.7 Hz, 12.4 Hz, 1H, H5), 3.64 (dd, J = 3.6 Hz, 11.9 Hz, 1H, H5’), 1.22 ppm (s, 3H, CH3); 13C NMR (100 MHz, MeOD) δ 164.1 (CONH2), 137.8 (triazole), 82.4 (C4), 78.5 (C1), 78.1 (C2), 76.2 (C3), 61.6 (C5), 19.9 (CH3); HRMS calcd for C9H15N4O5, 259.1042; found, 259.1055.
5-(2’-Deoxy-2’-fluoro-2’-methyl-β-D-ribofuranosyl)-1,2,3-triazole-4-carboxamide (3) [22]: Through the solution of compound 17 (110 mg, 0.29 mmol) in anhydrous methanol (4 mL) was bubbled ammonia for 2 h at 0 °C. After stirring at rt overnight, the solution was concentrated in vacuum affording 1-benzyl-4-(2’-deoxy-2’-fluoro-2’-methyl-β-D-ribofuranosyl)-1,2,3-triazole-5-carboxamide (90 mg, 89%) as a white powder. [α]D25 −18.6 (c 0.9, MeOH); mp 176 °C; IR (cm−1) νmax: 3397, 3304, 2944, 2883, 1668, 1598, 1458, 1218, 1126, 1048; 1H NMR (400 MHz, MeOD) δ 7.25–7.18 (m, 5H, Ph), 5.85 (d, J = 14.6 Hz, 1H, CH2Ph), 5.73 (d, J = 14.6 Hz, 1H, CH2Ph), 5.42 (d, J = 24.8 Hz, 1H, H1), 4.14 (dd, J = 8.7 Hz, 18.8 Hz, 1H, H3), 3.97–3.93 (m, 1H, H4), 3.90 (dd, J = 2.8 Hz, 12.4 Hz, 1H, H5), 3.72 (dd, J = 5.0 Hz, 12.4 Hz, 1H, H5’), 1.18 (d, J = 22.5 Hz, 3H, CH3); 13C NMR (100 MHz, MeOD) δ 160.8 (CONH2), 143.7 (d, J = 11.4 Hz, triazole), 135.3 (Cq Ph), 130.4 (triazole), 128.6, 128.2, 127.7 (Ph), 100.8 (d, J = 190 Hz, C2), 82.2 (C4), 79.7 (d, J = 39 Hz, C1), 73.9 (d, J = 23 Hz, C3), 61.3 (C5), 53.0 (-CH2Ph), 17.2 (d, J = 26 Hz, CH3); 19F NMR (376.2 MHz, MeOD) −158.2 (m); HRMS calcd for C16H20FN4O4, 351.1469; found, 351.1468.
Debenzylation proceeded in ethanol (5 mL) with palladium chloride (9.1 mg, 0.05 mmol) under 4 bar pressure of hydrogen for 24 h. After filtration over Celite and concentration in vacuum, the crude was purified by flash chromatography (pure EtOAc to EtOAc/MeOH 1:1) to afford 3 as a white solid (0.03 g, 45%) that was lyophilized. [α]D25 +18.5 (c 0.7, MeOH); mp 52 °C; IR (cm−1) νmax 3357, 2361, 2341, 1635, 1455, 1072, 1021; 1H NMR (400 MHz, MeOD) δ 5.80 (d, J = 22 Hz, 1H, H1), 4.06–3.98 (m, 3H, H3, H4, H5), 3.84 (dd, J = 2.8 Hz, 11.9 Hz, 1H, H5’), 1.13 (d, J = 24.8 Hz, 3H, CH3); 13C NMR (100 MHz, MeOD) δ 163.8 (-CONH2), 137.9 (triazole), 101.2 (d, J = 174 Hz, C2), 81.5 (C4), 78.5 (d, J = 39 Hz, C1), 72.6 (d, J = 23 Hz, C3), 60.6 (C5), 16.3 (d, J = 26 Hz, CH3); 19F NMR (376.2 MHz, MeOD) −160.8 (m); HRMS calcd for C9H14FN4O4, 261.0999; found, 261.1006.
Supporting Information
| Supporting Information File 1: Experimental details, copies of 1H NMR, 13C NMR, HRMS and X-ray spectra. | ||
| Format: PDF | Size: 4.6 MB | Download |
References
-
Borden, K. L. B.; Culjkovic-Kraljacic, B. Leuk. Lymphoma 2010, 51, 1805–1815. doi:10.3109/10428194.2010.496506
Return to citation in text: [1] -
Zahreddine, H. A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A. A.; Morris, S. J.; Cormack, G.; Jaquith, J. B.; Cerchietti, L.; Cocolakis, E.; Amri, A.; Bergeron, J.; Leber, B.; Becker, M. W.; Pei, S.; Jordan, C. T.; Miller, W. H.; Borden, K. L. B. Nature 2014, 511, 90–93. doi:10.1038/nature13283
Return to citation in text: [1] -
Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2016, 116, 14379–14455. doi:10.1021/acs.chemrev.6b00209
Return to citation in text: [1] -
Paeshuyse, J.; Dallmeier, K.; Neyts, J. Curr. Opin. Virol. 2011, 1, 590–598. doi:10.1016/j.coviro.2011.10.030
Return to citation in text: [1] -
Manns, M. P.; Wedemeyer, H.; Cornberg, M. Gut 2006, 55, 1350–1359. doi:10.1136/gut.2005.076646
Return to citation in text: [1] -
Fried, M. W.; Shiffman, M. L.; Reddy, K. R.; Smith, C.; Marinos, G.; Gonçales, F. L., Jr.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; Craxi, A.; Lin, A.; Hoffman, J.; Yu, J. N. Engl. J. Med. 2002, 347, 975–982. doi:10.1056/NEJMoa020047
Return to citation in text: [1] -
Manns, M. P.; McHutchison, J. G.; Gordon, S. C.; Rustgi, V. K.; Shiffman, M.; Reindollar, R.; Goodman, Z. D.; Koury, K.; Ling, M.-H.; Albrecht, J. K. Lancet 2001, 358, 958–965. doi:10.1016/S0140-6736(01)06102-5
Return to citation in text: [1] -
Miller, M. H.; Agarwal, K.; Austin, A.; Brown, A.; Barclay, S. T.; Dundas, P.; Dusheiko, G. M.; Foster, G. R.; Fox, R.; Hayes, P. C.; Leen, C.; Millson, C.; Ryder, S. D.; Tait, J.; Ustianowski, A.; Dillon, J. F. Aliment. Pharmacol. Ther. 2014, 39, 1363–1375. doi:10.1111/apt.12764
Return to citation in text: [1] -
Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S.; Wang, P.; Zhang, H.-R.; Bansal, S.; Espiritu, C.; Keilman, M.; Lam, A. M.; Micolochick Steuer, H. M.; Niu, C.; Otto, M. J.; Furman, P. A. J. Med. Chem. 2010, 53, 7202–7218. doi:10.1021/jm100863x
Return to citation in text: [1] -
Zeuzem, S.; Dusheiko, G. M.; Salupere, R.; Mangia, A.; Flisiak, R.; Hyland, R. H.; Illeperuma, A.; Svarovskaia, E.; Brainard, D. M.; Symonds, W. T.; Subramanian, G. M.; McHutchison, J. G.; Weiland, O.; Reesink, H. W.; Ferenci, P.; Hézode, C.; Esteban, R. N. Engl. J. Med. 2014, 370, 1993–2001. doi:10.1056/NEJMoa1316145
Return to citation in text: [1] -
Lin, T.-I.; Lenz, O.; Fanning, G.; Verbinnen, T.; Delouvroy, F.; Scholliers, A.; Vermeiren, K.; Rosenquist, Å.; Edlund, M.; Samuelsson, B.; Vrang, L.; de Kock, H.; Wigerinck, P.; Raboisson, P.; Simmen, K. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. doi:10.1128/AAC.01058-08
Return to citation in text: [1] -
Reesink, H. W.; Fanning, G. C.; Farha, K. A.; Weegink, C.; Van Vliet, A.; van 't Klooster, G.; Lenz, O.; Aharchi, F.; Mariën, K.; Van Remoortere, P.; de Kock, H.; Broeckaert, F.; Meyvisch, P.; Van Beirendonck, E.; Simmen, K.; Verloes, R. Gastroenterology 2010, 138, 913–921. doi:10.1053/j.gastro.2009.10.033
Return to citation in text: [1] -
De Clercq, E. J. Med. Chem. 2016, 59, 2301–2311. doi:10.1021/acs.jmedchem.5b01157
Return to citation in text: [1] -
Aït Youcef, R.; Dos Santos, M.; Roussel, S.; Baltaze, J.-P.; Lubin-Germain, N.; Uziel, J. J. Org. Chem. 2009, 74, 4318–4323. doi:10.1021/jo900594x
Return to citation in text: [1] [2] -
Stuyver, L. J.; McBrayer, T. R.; Tharnish, P. M.; Hassan, A. E. A.; Chu, C. K.; Pankiewicz, K. W.; Watanabe, K. A.; Schinazi, R. F.; Otto, M. J. J. Virol. 2003, 77, 10689–10694. doi:10.1128/JVI.77.19.10689-10694.2003
Return to citation in text: [1] -
Carroll, S. S.; Olsen, D. B. Infect. Disord.: Drug Targets 2006, 6, 17–29. doi:10.2174/187152606776056698
Return to citation in text: [1] -
Stuyver, L. J.; McBrayer, T. R.; Whitaker, T.; Tharnish, P. M.; Ramesh, M.; Lostia, S.; Cartee, L.; Shi, J.; Hobbs, A.; Schinazi, R. F.; Watanabe, K. A.; Otto, M. J. Antimicrob. Agents Chemother. 2004, 48, 651–654. doi:10.1128/AAC.48.2.651-654.2004
Return to citation in text: [1] -
Pockros, P. J.; Jensen, D.; Tsai, N.; Taylor, R.; Ramji, A.; Cooper, C.; Dickson, R.; Tice, A.; Kulkarni, R.; Vierling, J. M.; Munson, M. L.; Chen, Y.-C.; Najera, I.; Thommes, J. Hepatology 2013, 58, 514–523. doi:10.1002/hep.26275
Return to citation in text: [1] -
Wedemeyer, H.; Jensen, D.; Herring, R., Jr..; Ferenci, P.; Ma, M.-M.; Zeuzem, S.; Rodriguez-Torres, M.; Bzowej, N.; Pockros, P.; Vierling, J.; Ipe, D.; Munson, M. L.; Chen, Y.-C.; Najera, I.; Thommes, J. Hepatology 2013, 58, 524–537. doi:10.1002/hep.26274
Return to citation in text: [1] -
Gane, E. J.; Stedman, C. A.; Hyland, R. H.; Ding, X.; Svarovskaia, E.; Symonds, W. T.; Hindes, R. G.; Berrey, M. M. N. Engl. J. Med. 2013, 368, 34–44. doi:10.1056/NEJMoa1208953
Return to citation in text: [1] -
Lubin-Germain, N.; Baltaze, J.-P.; Coste, A.; Hallonet, A.; Lauréano, H.; Legrave, G.; Uziel, J.; Augé, J. Org. Lett. 2008, 10, 725–728. doi:10.1021/ol7029257
Return to citation in text: [1] -
Cosson, F. Synthèse d’analogues carbonés de la Ribavirine pour leurs activités antivirals. Ph.D. Thesis, University of Cergy-Pontoise, France, 2014.
Return to citation in text: [1] [2] [3] [4] [5] -
Takenuki, K.; Itoh, H.; Matsuda, A.; Ueda, T. Chem. Pharm. Bull. 1990, 38, 2947–2952. doi:10.1248/cpb.38.2947
Return to citation in text: [1] -
Li, N.-S.; Lu, J.; Piccirilli, J. A. J. Org. Chem. 2009, 74, 2227–2230. doi:10.1021/jo802666k
Return to citation in text: [1]
| 22. | Cosson, F. Synthèse d’analogues carbonés de la Ribavirine pour leurs activités antivirals. Ph.D. Thesis, University of Cergy-Pontoise, France, 2014. |
| 23. | Takenuki, K.; Itoh, H.; Matsuda, A.; Ueda, T. Chem. Pharm. Bull. 1990, 38, 2947–2952. doi:10.1248/cpb.38.2947 |
| 24. | Li, N.-S.; Lu, J.; Piccirilli, J. A. J. Org. Chem. 2009, 74, 2227–2230. doi:10.1021/jo802666k |
| 1. | Borden, K. L. B.; Culjkovic-Kraljacic, B. Leuk. Lymphoma 2010, 51, 1805–1815. doi:10.3109/10428194.2010.496506 |
| 2. | Zahreddine, H. A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A. A.; Morris, S. J.; Cormack, G.; Jaquith, J. B.; Cerchietti, L.; Cocolakis, E.; Amri, A.; Bergeron, J.; Leber, B.; Becker, M. W.; Pei, S.; Jordan, C. T.; Miller, W. H.; Borden, K. L. B. Nature 2014, 511, 90–93. doi:10.1038/nature13283 |
| 3. | Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2016, 116, 14379–14455. doi:10.1021/acs.chemrev.6b00209 |
| 9. | Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S.; Wang, P.; Zhang, H.-R.; Bansal, S.; Espiritu, C.; Keilman, M.; Lam, A. M.; Micolochick Steuer, H. M.; Niu, C.; Otto, M. J.; Furman, P. A. J. Med. Chem. 2010, 53, 7202–7218. doi:10.1021/jm100863x |
| 10. | Zeuzem, S.; Dusheiko, G. M.; Salupere, R.; Mangia, A.; Flisiak, R.; Hyland, R. H.; Illeperuma, A.; Svarovskaia, E.; Brainard, D. M.; Symonds, W. T.; Subramanian, G. M.; McHutchison, J. G.; Weiland, O.; Reesink, H. W.; Ferenci, P.; Hézode, C.; Esteban, R. N. Engl. J. Med. 2014, 370, 1993–2001. doi:10.1056/NEJMoa1316145 |
| 22. | Cosson, F. Synthèse d’analogues carbonés de la Ribavirine pour leurs activités antivirals. Ph.D. Thesis, University of Cergy-Pontoise, France, 2014. |
| 8. | Miller, M. H.; Agarwal, K.; Austin, A.; Brown, A.; Barclay, S. T.; Dundas, P.; Dusheiko, G. M.; Foster, G. R.; Fox, R.; Hayes, P. C.; Leen, C.; Millson, C.; Ryder, S. D.; Tait, J.; Ustianowski, A.; Dillon, J. F. Aliment. Pharmacol. Ther. 2014, 39, 1363–1375. doi:10.1111/apt.12764 |
| 14. | Aït Youcef, R.; Dos Santos, M.; Roussel, S.; Baltaze, J.-P.; Lubin-Germain, N.; Uziel, J. J. Org. Chem. 2009, 74, 4318–4323. doi:10.1021/jo900594x |
| 5. | Manns, M. P.; Wedemeyer, H.; Cornberg, M. Gut 2006, 55, 1350–1359. doi:10.1136/gut.2005.076646 |
| 6. | Fried, M. W.; Shiffman, M. L.; Reddy, K. R.; Smith, C.; Marinos, G.; Gonçales, F. L., Jr.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; Craxi, A.; Lin, A.; Hoffman, J.; Yu, J. N. Engl. J. Med. 2002, 347, 975–982. doi:10.1056/NEJMoa020047 |
| 7. | Manns, M. P.; McHutchison, J. G.; Gordon, S. C.; Rustgi, V. K.; Shiffman, M.; Reindollar, R.; Goodman, Z. D.; Koury, K.; Ling, M.-H.; Albrecht, J. K. Lancet 2001, 358, 958–965. doi:10.1016/S0140-6736(01)06102-5 |
| 20. | Gane, E. J.; Stedman, C. A.; Hyland, R. H.; Ding, X.; Svarovskaia, E.; Symonds, W. T.; Hindes, R. G.; Berrey, M. M. N. Engl. J. Med. 2013, 368, 34–44. doi:10.1056/NEJMoa1208953 |
| 4. | Paeshuyse, J.; Dallmeier, K.; Neyts, J. Curr. Opin. Virol. 2011, 1, 590–598. doi:10.1016/j.coviro.2011.10.030 |
| 21. | Lubin-Germain, N.; Baltaze, J.-P.; Coste, A.; Hallonet, A.; Lauréano, H.; Legrave, G.; Uziel, J.; Augé, J. Org. Lett. 2008, 10, 725–728. doi:10.1021/ol7029257 |
| 15. | Stuyver, L. J.; McBrayer, T. R.; Tharnish, P. M.; Hassan, A. E. A.; Chu, C. K.; Pankiewicz, K. W.; Watanabe, K. A.; Schinazi, R. F.; Otto, M. J. J. Virol. 2003, 77, 10689–10694. doi:10.1128/JVI.77.19.10689-10694.2003 |
| 17. | Stuyver, L. J.; McBrayer, T. R.; Whitaker, T.; Tharnish, P. M.; Ramesh, M.; Lostia, S.; Cartee, L.; Shi, J.; Hobbs, A.; Schinazi, R. F.; Watanabe, K. A.; Otto, M. J. Antimicrob. Agents Chemother. 2004, 48, 651–654. doi:10.1128/AAC.48.2.651-654.2004 |
| 22. | Cosson, F. Synthèse d’analogues carbonés de la Ribavirine pour leurs activités antivirals. Ph.D. Thesis, University of Cergy-Pontoise, France, 2014. |
| 14. | Aït Youcef, R.; Dos Santos, M.; Roussel, S.; Baltaze, J.-P.; Lubin-Germain, N.; Uziel, J. J. Org. Chem. 2009, 74, 4318–4323. doi:10.1021/jo900594x |
| 18. | Pockros, P. J.; Jensen, D.; Tsai, N.; Taylor, R.; Ramji, A.; Cooper, C.; Dickson, R.; Tice, A.; Kulkarni, R.; Vierling, J. M.; Munson, M. L.; Chen, Y.-C.; Najera, I.; Thommes, J. Hepatology 2013, 58, 514–523. doi:10.1002/hep.26275 |
| 19. | Wedemeyer, H.; Jensen, D.; Herring, R., Jr..; Ferenci, P.; Ma, M.-M.; Zeuzem, S.; Rodriguez-Torres, M.; Bzowej, N.; Pockros, P.; Vierling, J.; Ipe, D.; Munson, M. L.; Chen, Y.-C.; Najera, I.; Thommes, J. Hepatology 2013, 58, 524–537. doi:10.1002/hep.26274 |
| 13. | De Clercq, E. J. Med. Chem. 2016, 59, 2301–2311. doi:10.1021/acs.jmedchem.5b01157 |
| 22. | Cosson, F. Synthèse d’analogues carbonés de la Ribavirine pour leurs activités antivirals. Ph.D. Thesis, University of Cergy-Pontoise, France, 2014. |
| 11. | Lin, T.-I.; Lenz, O.; Fanning, G.; Verbinnen, T.; Delouvroy, F.; Scholliers, A.; Vermeiren, K.; Rosenquist, Å.; Edlund, M.; Samuelsson, B.; Vrang, L.; de Kock, H.; Wigerinck, P.; Raboisson, P.; Simmen, K. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. doi:10.1128/AAC.01058-08 |
| 12. | Reesink, H. W.; Fanning, G. C.; Farha, K. A.; Weegink, C.; Van Vliet, A.; van 't Klooster, G.; Lenz, O.; Aharchi, F.; Mariën, K.; Van Remoortere, P.; de Kock, H.; Broeckaert, F.; Meyvisch, P.; Van Beirendonck, E.; Simmen, K.; Verloes, R. Gastroenterology 2010, 138, 913–921. doi:10.1053/j.gastro.2009.10.033 |
| 16. | Carroll, S. S.; Olsen, D. B. Infect. Disord.: Drug Targets 2006, 6, 17–29. doi:10.2174/187152606776056698 |
| 22. | Cosson, F. Synthèse d’analogues carbonés de la Ribavirine pour leurs activités antivirals. Ph.D. Thesis, University of Cergy-Pontoise, France, 2014. |
© 2017 Cosson et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)