
Abstract
This paper reports the self-assembly of two new tetrathiafulvalene (TTF) derivatives that contain one or two urethane groups. The formation of nanoribbons was evidenced by scanning electron microscopy (SEM) and X-ray diffraction (XRD), which showed that the self-assembly ability of T1 was better than that of T2. The results revealed that more urethane groups in a molecule did not necessarily instigate self-assembly. UV–vis and FTIR spectra were measured to explore noncovalent interactions. The driving forces for self-assembly of TTF derivatives were mainly hydrogen bond interactions and π–π stacking interactions. The electronic conductivity of the T1 and T2 films was tested by a four-probe method.
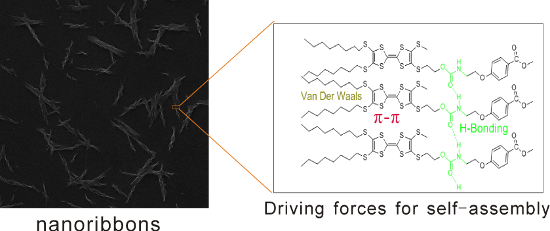
Graphical Abstract
Introduction
In recent years, there has been an enormous increase of interest in functional organic nanomaterials, given that they are promising materials with a variety of applications including optoelectronic and bioelectronic devices [1,2]. The mechanism behind the formation of functional organic nanomaterials is generally accepted to be the self-assembly of supermolecules, which is constructed through weak noncovalent interactions such as π–π stacking, van der Waals interactions, charge transfer and H-bonding interactions [3-6]. Generally speaking, H-bonding interactions are the key intermolecular interactions in molecular self-assembly systems. Therefore, molecules containing urea, amide and other similar groups have been investigated because these molecules can easily generate intermolecular hydrogen bonds [7-9].
Tetrathiafulvalene (TTF) derivatives have been widely investigated in the fields of supramolecular and materials chemistry due to their great potential application in molecular electronics, for example, as switches and conductors [10-14]. As we all know, TTF derivatives can form charge transfer (CT) complexes with electron acceptors such as tetracyanoquinodimethane (TCNQ), and the CT complexes of TTF derivatives and TCNQ exhibit high electrical conductivity [14-16]. Therefore, TTF derivatives are extensively used in the field of functional organic conductive nanomaterials.
Herein, we designed and synthesized two compounds, T1 and T2, which contain TTF units and urethane groups (Figure 1). The combination of the urethane group (forming hydrogen bonds) and the TTF unit (forming π–π stacking) may promote the formation of nanostructures. To the best of our knowledge, urethane groups have been rarely introduced into the molecular structure of TTF derivatives to generate an H-bonding chain.
![[1860-5397-11-255-1]](/bjoc/content/figures/1860-5397-11-255-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of TTF derivatives T1 and T2.
Figure 1: Molecular structure of TTF derivatives T1 and T2.
Results and Discussion
Synthesis and characterization
The synthetic routes for two newly designed TTF derivatives containing one or two urethane groups are shown in Scheme 1. Compounds 2 [17], 3 [18], 4 [19], 5 [19], 6 [18,20] and 7 [18,21] were synthesized from commercially available starting materials according to the reported methods. Compound 8 [18,21] was obtained by the reaction of 7 with 2-chloroethyl isocyanate in dry and degassed toluene. Finally, the TTF derivative T1 was obtained in acceptable yield (72%). For the synthesis of T2, urethane groups were introduced first, and then the coupling reaction was carried out. The new compounds T1 and T2 were characterized by 1H, 13C NMR, HRMS–ESI (for the spectra see Supporting Information File 1) and elemental analysis. In addition, other intermediates previously reported in the literature were also characterized by 1H NMR, 13C NMR, and EIMS.
![[1860-5397-11-255-i1]](/bjoc/content/inline/1860-5397-11-255-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthetic routes of compounds T1 and T2.
Scheme 1: The synthetic routes of compounds T1 and T2.
Self-assembly and SEM investigation of T1 and T2
The studies showed that T1 and T2 gels were not formed in several common solvents such as hexane, chloroform, dichloromethane, tetrahydrofuran, toluene, diethyl ether, acetone, dimethylformamide, ethanol, methanol and acetonitrile when they were heated and cooled by the methods reported in the literature [2-4]. A loose gel of T1 was observed in ethyl acetate when the concentration was increased to 20 mg/mL. However, the precipitate of T2 was obtained under the same conditions. Moreover, their micromorphology was recorded with SEM images (Figure 2). The samples were prepared by different methods (drop-coating, spin-coating). The experiments were performed as follows: the solid compounds were completely dissolved in ethyl acetate while heating, then cooled to room temperature. The studies showed that drop-coating was better than direct spin-coating, likely because slow solvent evaporation is more conducive to the formation of regular structure. The SEM images of the T1 films (Figure 2a, drop-coated from a diluted T1 solution) showed that regular helical nanoribbons were observed. The diameter of the nanoribbons was approximately 500 nm with a length of >20 μm. Although nanoribbons were observed in the SEM images of T2 (Figure 2b), they showed no similar ordered structure.
![[1860-5397-11-255-2]](/bjoc/content/figures/1860-5397-11-255-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: SEM images of T1 (a) and T2 (b) films on glass substrates (drop-coated from diluted T1 or T2 solution).
Figure 2: SEM images of T1 (a) and T2 (b) films on glass substrates (drop-coated from diluted T1 or T2 soluti...
In addition, the X-ray diffraction (XRD) patterns of T1 and T2 nanoribbons were taken (Supporting Information File 1, Figure S7). The XRD pattern of T1 showed three sharp peaks at 7.4°, 14.9° and 22.1°, which suggested that a lamellar stacking organization was formed [4]. This was not the case for the XRD pattern of T2. In general, intermolecular hydrogen bonding is the main driving force behind self-assembly. Although T2 contains two urethane groups and T1 contains one urethane group, the self-assembly ability of T2 is not better than that of T1. We concluded that more intramolecular hydrogen bonds were formed in molecules of T2 instead of intermolecular hydrogen bonds in ethyl acetate, which was not conducive to form regular nanoribbons.
UV–vis and FTIR spectroscopy
To study the intermolecular interactions, the UV–vis absorption spectra of T1 and T2 in ethyl acetate at different concentrations were measured (Figure 3a,b). Figure 3a shows that the two absorption peaks of T1 are blue-shifted from 314 nm and 338 nm (1 × 10−6 M) to 294 nm and 315 nm (aggregated solid state). This was also observed for T2, which illustrated that π–π interactions and H-aggregation occurred with the increase in concentration [22-24]. To further study the driving forces for the self-assembly of T1 and T2, FTIR spectra were also measured (Figure 4a,b). The FTIR spectra of T1 showed an absorption peak at 3352 cm−1 for the N–H stretching vibration, 1706 cm−1 for amide I and 1519 cm−1 for amide II related to the urethane groups. The same situation was observed for T2. The absence of a free N–H stretching vibration (around 3400 cm−1) and a free C=O stretching vibration (around 1720 cm−1) suggested that strong hydrogen bonds between urethane groups were formed [25,26]. These results indicated that π–π interactions and hydrogen bonding were the main driving forces behind the self-assembly.
![[1860-5397-11-255-3]](/bjoc/content/figures/1860-5397-11-255-3.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 3: The UV–vis spectra of T1 (a) and T2 (b) at different concentrations in ethyl acetate.
Figure 3: The UV–vis spectra of T1 (a) and T2 (b) at different concentrations in ethyl acetate.
![[1860-5397-11-255-4]](/bjoc/content/figures/1860-5397-11-255-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: IR spectra of (a) T1, (b) T2, (c) TCNQ, (d) T2/TCNQ, and (e) T1/TCNQ.
Figure 4: IR spectra of (a) T1, (b) T2, (c) TCNQ, (d) T2/TCNQ, and (e) T1/TCNQ.
In addition, UV−vis and FTIR spectra were measured to explore the formation of the charge-transfer complexes. TTF derivates are representative electron donors, while TCNQ is a typical electron acceptor. When one equivalent of TCNQ was added to the solution of T1 in ethyl acetate, TCNQ radical anion species (TCNQ•−) and TTF radical cation species (TTF•+) were formed, which was possibly supported by the increase of the absorption bands around 600–900 nm (Figure 5a) [2,4]. Moreover, the UV–vis spectra of self-assembled nanoribbons doped with iodine were collected. It was concluded that the assembled solid structures were maintained. Figure 5b shows the UV–vis spectrum of T1 (thin film on glass) before and after iodine doping. Upon exposure to iodine vapor for 30 min in a sealed container, a new absorption band was observed at approximately 850 nm, which suggested the formation of the CT complex [27].
![[1860-5397-11-255-5]](/bjoc/content/figures/1860-5397-11-255-5.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 5: (a) UV–vis spectra of T1 solutions TCNQ and T1/TCNQ in ethyl acetate (1 × 10−3 M). (b) UV–vis spectra of T1 before and after iodine doping for 30 min.
Figure 5: (a) UV–vis spectra of T1 solutions TCNQ and T1/TCNQ in ethyl acetate (1 × 10−3 M). (b) UV–vis spect...
IR spectra of TCNQ, T1/TCNQ, and T2/TCNQ are shown in Figure 4c–e. In contrast to those of T1 and T2, the N–H and C=O stretching bands of the amide groups were not obviously shifted after doping with TCNQ. This indicated that the doping did not change the hydrogen-bonded structures.
Cyclic voltammetry (CV)
The cyclic voltammetry experiments were carried out to explore the electrochemical properties of the TTF compounds. The cyclic voltammograms of T1 and T2 were measured in dry and degassed dichloromethane solution [28]. Both T1 and T2 displayed two, reversible, one-electron redox couples, in which the first oxidation at ![[Graphic 1]](/bjoc/content/inline/1860-5397-11-255-i2.svg?max-width=637&scale=1.18182) = +0.628 V (T1) and +0.643 V (T2) (vs Ag/AgCl) was in the anodic window. This indicated the successive reversible oxidation of neutral TTF (TTF0) to the radical cation (TTF•+). The second oxidation at
= +0.628 V (T1) and +0.643 V (T2) (vs Ag/AgCl) was in the anodic window. This indicated the successive reversible oxidation of neutral TTF (TTF0) to the radical cation (TTF•+). The second oxidation at ![[Graphic 2]](/bjoc/content/inline/1860-5397-11-255-i3.svg?max-width=637&scale=1.18182) = +0.958 V (T1) and +0.973 V (T2) (vs Ag/AgCl) corresponded to the reversible oxidation of the radical cation (TTF•+) to the dication (TTF2+) (Figure 6). Both the first-wave and the second-wave oxidation potentials of T2 were higher (15 mV) than those of T1, which indicated that introduction of another urethane group resulted in a decrease of the electron-donating ability.
= +0.958 V (T1) and +0.973 V (T2) (vs Ag/AgCl) corresponded to the reversible oxidation of the radical cation (TTF•+) to the dication (TTF2+) (Figure 6). Both the first-wave and the second-wave oxidation potentials of T2 were higher (15 mV) than those of T1, which indicated that introduction of another urethane group resulted in a decrease of the electron-donating ability.
![[1860-5397-11-255-6]](/bjoc/content/figures/1860-5397-11-255-6.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 6: Cyclic voltammograms of T1 and T2 in DCM. Conditions: 0.1 M tetrabutylammonium hexafluorophosphate, 100 mV s−1, Ag/AgCl as the reference electrode, Pt wire as the counter electrode, and glassy carbon as the working electrode; measured under argon at 20 °C. Concentration: 1 mM for T1 and 1 mM for T2.
Figure 6: Cyclic voltammograms of T1 and T2 in DCM. Conditions: 0.1 M tetrabutylammonium hexafluorophosphate,...
Cyclic voltammograms were also measured to explore the formation of the charge-transfer complex. For the mixture of T1 and TCNQ, five oxidation potentials at ![[Graphic 3]](/bjoc/content/inline/1860-5397-11-255-i4.svg?max-width=637&scale=1.18182) = −0.956 V (I),
= −0.956 V (I), ![[Graphic 4]](/bjoc/content/inline/1860-5397-11-255-i5.svg?max-width=637&scale=1.18182) = −0.368 V (II),
= −0.368 V (II), ![[Graphic 5]](/bjoc/content/inline/1860-5397-11-255-i6.svg?max-width=637&scale=1.18182) = +0.221 V (III),
= +0.221 V (III), ![[Graphic 6]](/bjoc/content/inline/1860-5397-11-255-i7.svg?max-width=637&scale=1.18182) = +0.527 V (IV), and
= +0.527 V (IV), and ![[Graphic 7]](/bjoc/content/inline/1860-5397-11-255-i8.svg?max-width=637&scale=1.18182) = +0.852 V (V) (vs saturated calomel electrode, SCE) were clearly discernible (Figure 7). The first three oxidation potentials belonged to TCNQ2−/TCNQ− (I), TCNQ−/TCNQ0 (II) and TCNQ0/TCNQ+ (III), which were all lower than those of TCNQ (
= +0.852 V (V) (vs saturated calomel electrode, SCE) were clearly discernible (Figure 7). The first three oxidation potentials belonged to TCNQ2−/TCNQ− (I), TCNQ−/TCNQ0 (II) and TCNQ0/TCNQ+ (III), which were all lower than those of TCNQ (![[Graphic 8]](/bjoc/content/inline/1860-5397-11-255-i9.svg?max-width=637&scale=1.18182) = −0.954 V(I),
= −0.954 V(I), ![[Graphic 9]](/bjoc/content/inline/1860-5397-11-255-i10.svg?max-width=637&scale=1.18182) = −0.341 V(II),
= −0.341 V(II), ![[Graphic 10]](/bjoc/content/inline/1860-5397-11-255-i11.svg?max-width=637&scale=1.18182) = +0.224 V(III)). The (IV) and (V) processes could be assigned to TTF•+/TTF0 (IV) and TTF•2+/ TTF•+ (V), which were all higher than those of T1 (
= +0.224 V(III)). The (IV) and (V) processes could be assigned to TTF•+/TTF0 (IV) and TTF•2+/ TTF•+ (V), which were all higher than those of T1 (![[Graphic 11]](/bjoc/content/inline/1860-5397-11-255-i12.svg?max-width=637&scale=1.18182) = +0.514 V (I),
= +0.514 V (I), ![[Graphic 12]](/bjoc/content/inline/1860-5397-11-255-i13.svg?max-width=637&scale=1.18182) = +0.841 V (II)). These changes indicated the formation of the CT complex.
= +0.841 V (II)). These changes indicated the formation of the CT complex.
![[1860-5397-11-255-7]](/bjoc/content/figures/1860-5397-11-255-7.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 7: Cyclic voltammograms of T1 and TCNQ in DCM. Conditions: 0.1 M tetrabutylammonium hexafluorophosphate, 100 mV s−1, saturated calomel electrode (SCE) as the reference electrode, Pt wire as the counter electrode, and glassy carbon as the working electrode; measured under argon at 20 °C. Concentration: 1 mM for T1 and 1 mM for TCNQ.
Figure 7: Cyclic voltammograms of T1 and TCNQ in DCM. Conditions: 0.1 M tetrabutylammonium hexafluorophosphat...
Electrical conductivity measurements
The electrical conductivity of thin films obtained from the T1 and T2 samples with TCNQ (1:1 molar)/I2 (30 min) were further evaluated. To eliminate the influence of contact resistance, the four-probe method was carried out instead of the two-probe method [29,30]. To prepare the thin films, a diluted ethyl acetate solution was dropcasted onto a glass substrates (20 mm × 20 mm) and dried overnight at 40 °C under vacuum. The T1 and T2 films in the neutral state before doping behaved as typical, undoped semiconductors (σ < 10−9 S cm−1) at room temperature. Nevertheless, for T1, the conductivity increased to 5.8 × 10−6 S cm−1 when doped with TCNQ and to 3.0 × 10−6 S cm−1 when exposed to iodine vapor. As for T2, the results were 6.3 × 10−7 S cm−1 when doped with TCNQ and 1.8 × 10−7 S cm−1 when exposed to iodine vapor. These results indicated their CT complexes can function as semiconducting materials.
Conclusion
In summary, we demonstrated that T2 (containing two urethane groups) formed amorphous structures while T1 (possessing one urethane group) formed nanoribbons. The self-assembly ability of T1 was better than that of T2, and the results revealed that more urethane groups in a molecule did not necessarily lead to more efficient self-assembly. This may be associated with the formation of intramolecular hydrogen bonds in the T2 molecule. The formation of hydrogen bonds between urethane groups and the π–π stacking interaction from TTF units were regarded as the main driving forces behind the self-assembly process. Cyclic voltammetry showed that the TTF derivatives underwent two reversible oxidation processes. In addition, the doping of nanoribbons by TCNQ/iodine resulted in the formation of charge transfer states exhibiting semiconducting properties. There is significant potential for the application of the conducting nanoribbons in molecular electronics devices.
Supporting Information
| Supporting Information File 1: Experimental section and copies of 1H, 13C NMR spectra, MS and XRD pattern of T1 and T2. | ||
| Format: PDF | Size: 1017.7 KB | Download |
References
-
Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022
Return to citation in text: [1] -
Nalluri, S. K. M.; Shivarova, N.; Kanibolotsky, A. L.; Zelzer, M.; Gupta, S.; Frederix, P. W. J. M.; Skabara, P. J.; Gleskova, H.; Ulijn, R. V. Langmuir 2014, 30, 12429–12437. doi:10.1021/la503459y
Return to citation in text: [1] [2] [3] -
Giansante, C.; Raffy, G.; Schäfer, C.; Rahma, H.; Kao, M.-T.; Olive, A. G. L.; Del Guerzo, A. J. Am. Chem. Soc. 2011, 133, 316–325. doi:10.1021/ja106807u
Return to citation in text: [1] [2] -
Liu, Y.; Zheng, N.; Li, H.; Yin, B. Soft Matter 2013, 9, 5261–5269. doi:10.1039/c3sm50614b
Return to citation in text: [1] [2] [3] [4] -
Pratihar, P.; Gosh, S.; Stepanenko, V.; Patwardhan, S.; Grozema, F. C.; Siebbeles, L. D. A.; Würthner, F. Beilstein J. Org. Chem. 2010, 6, 1070–1078. doi:10.3762/bjoc.6.122
Return to citation in text: [1] -
Banerjee, S.; Das, R. K.; Terech, P.; de Geyer, A.; Aymonier, C.; Loppinet-Serani, A.; Raffy, G.; Maitra, U.; Del Guerzo, A.; Desvergne, J. P. J. Mater. Chem. C 2013, 1, 3305–3316. doi:10.1039/c3tc30104d
Return to citation in text: [1] -
George, M.; Tan, G.; John, V. T.; Weiss, R. G. Chem. – Eur. J. 2005, 11, 3243–3254. doi:10.1002/chem.200401066
Return to citation in text: [1] -
Goyal, N.; Mangunuru, H. P. R.; Parikh, B.; Shrestha, S.; Wang, G. Beilstein J. Org. Chem. 2014, 10, 3111–3121. doi:10.3762/bjoc.10.328
Return to citation in text: [1] -
Skilling, K. J.; Citossi, F.; Bradshaw, T. D.; Ashford, M.; Kellam, B.; Marlow, M. Soft Matter 2014, 10, 237–256. doi:10.1039/C3SM52244J
Return to citation in text: [1] -
Wang, C.; Zhang, D.; Zhu, D. J. Am. Chem. Soc. 2005, 127, 16372–16373. doi:10.1021/ja055800u
Return to citation in text: [1] -
Jeppesen, J. O.; Becher, J. Eur. J. Org. Chem. 2003, 3245–3266. doi:10.1002/ejoc.200300078
Return to citation in text: [1] -
Yang, X.; Zhang, G.; Zhang, D.; Zhu, D. Langmuir 2010, 26, 11720–11725. doi:10.1021/la101193z
Return to citation in text: [1] -
Gomar-Nadal, E.; Veciana, J.; Rovira, C.; Amabilino, D. B. Adv. Mater. 2005, 17, 2095–2098. doi:10.1002/adma.200500348
Return to citation in text: [1] -
Bryce, M. R. Chem. Soc. Rev. 1991, 20, 355–390. doi:10.1039/cs9912000355
Return to citation in text: [1] [2] -
PuigmartÍ-Luis, J.; Laukhin, V.; Pérez del Pino, Á.; Vidal-Gancedo, J.; Rovira, C.; Laukhina, E.; Amabilino, D. B. Angew. Chem., Int. Ed. 2007, 46, 238–241. doi:10.1002/anie.200602483
Return to citation in text: [1] -
Puigmartı-Luis, J.; Pérez del Pino, Á.; Laukhina, E.; Esquena, J.; Laukhin, V.; Rovira, C.; Vidal-Gancedo, J.; Kanaras, A. G.; Nichols, R. J.; Brust, M.; Amabilino, D. B. Angew. Chem., Int. Ed. 2008, 47, 1861–1865. doi:10.1002/anie.200704864
Return to citation in text: [1] -
Massue, J.; Bellec, N.; Chopin, S.; Levillain, E.; Roisnel, T.; Clérac, R.; Lorcy, D. Inorg. Chem. 2005, 44, 8740–8748. doi:10.1021/ic051017r
Return to citation in text: [1] -
Lyskawa, J.; Oçafrain, M.; Trippé, G.; Le Derf, F.; Sallé, M.; Viel, P.; Palacin, S. Tetrahedron 2006, 62, 4419–4425. doi:10.1016/j.tet.2006.02.054
Return to citation in text: [1] [2] [3] [4] -
Zhang, X.; Wang, C.; Lai, G.; Zhang, L.; Shen, Y. New J. Chem. 2010, 34, 318–324. doi:10.1039/B9NJ00520J
Return to citation in text: [1] [2] -
Benbellat, N.; Le Gal, Y.; Golhen, S.; Gouasmia, A.; Ouahab, L. Synth. Met. 2012, 162, 1789–1797. doi:10.1016/j.synthmet.2012.08.018
Return to citation in text: [1] -
Tatewaki, Y.; Watanabe, T.; Watanabe, K.; Kikuchi, K.; Okada, S. Dalton Trans. 2013, 42, 16121–16127. doi:10.1039/c3dt51464a
Return to citation in text: [1] [2] -
Su, L.; Bao, C.; Lu, R.; Chen, Y.; Xu, T.; Song, D.; Tan, C.; Shi, T.; Zhao, Y. Org. Biomol. Chem. 2006, 4, 2591–2594. doi:10.1039/b602520j
Return to citation in text: [1] -
Kitamura, T.; Nakaso, S.; Mizoshita, N.; Tochigi, Y.; Shimomura, T.; Moriyama, M.; Ito, K.; Kato, T. J. Am. Chem. Soc. 2005, 127, 14769–14775. doi:10.1021/ja053496z
Return to citation in text: [1] -
Ding, Z.; Zhao, Q.; Xing, R.; Wang, X.; Ding, J.; Wang, L.; Han, Y. J. Mater. Chem. C 2013, 1, 786–792. doi:10.1039/C2TC00125J
Return to citation in text: [1] -
Demir-Ordu, Ő.; Şimşir, H.; Alper, K. Tetrahedron 2015, 71, 1529–1539. doi:10.1016/j.tet.2015.01.042
Return to citation in text: [1] -
Zhang, Y.; Liang, C.; Shang, H.; Ma, Y.; Jiang, S. J. Mater. Chem. C 2013, 1, 4472–4480. doi:10.1039/c3tc30545g
Return to citation in text: [1] -
Le Gall, T.; Pearson, C.; Bryce, M. R.; Petty, M. C.; Dahlgaard, H.; Becher, J. Eur. J. Org. Chem. 2003, 3562–3568. doi:10.1002/ejoc.200300286
Return to citation in text: [1] -
Vilela, F.; Skabara, P. J.; Mason, C. R.; Westgate, T. D. J.; Luquin, A.; Coles, S. J.; Hursthouse, M. B. Beilstein J. Org. Chem. 2010, 6, 1002–1014. doi:10.3762/bjoc.6.113
Return to citation in text: [1] -
Akutagawa, T.; Kakiuchi, K.; Hasegawa, T.; Nakamura, T.; Christensen, C. A.; Becher, J. Langmuir 2004, 20, 4187–4195. doi:10.1021/la049950e
Return to citation in text: [1] -
Skabara, P. J.; Berridge, R.; McInnes, E. J. L.; West, D. P.; Coles, S. J.; Hursthouse, M. B.; Müllen, K. J. Mater. Chem. 2004, 14, 1964–1969. doi:10.1039/b400809j
Return to citation in text: [1]
| 28. | Vilela, F.; Skabara, P. J.; Mason, C. R.; Westgate, T. D. J.; Luquin, A.; Coles, S. J.; Hursthouse, M. B. Beilstein J. Org. Chem. 2010, 6, 1002–1014. doi:10.3762/bjoc.6.113 |
| 2. | Nalluri, S. K. M.; Shivarova, N.; Kanibolotsky, A. L.; Zelzer, M.; Gupta, S.; Frederix, P. W. J. M.; Skabara, P. J.; Gleskova, H.; Ulijn, R. V. Langmuir 2014, 30, 12429–12437. doi:10.1021/la503459y |
| 4. | Liu, Y.; Zheng, N.; Li, H.; Yin, B. Soft Matter 2013, 9, 5261–5269. doi:10.1039/c3sm50614b |
| 27. | Le Gall, T.; Pearson, C.; Bryce, M. R.; Petty, M. C.; Dahlgaard, H.; Becher, J. Eur. J. Org. Chem. 2003, 3562–3568. doi:10.1002/ejoc.200300286 |
| 1. | Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022 |
| 2. | Nalluri, S. K. M.; Shivarova, N.; Kanibolotsky, A. L.; Zelzer, M.; Gupta, S.; Frederix, P. W. J. M.; Skabara, P. J.; Gleskova, H.; Ulijn, R. V. Langmuir 2014, 30, 12429–12437. doi:10.1021/la503459y |
| 14. | Bryce, M. R. Chem. Soc. Rev. 1991, 20, 355–390. doi:10.1039/cs9912000355 |
| 15. | PuigmartÍ-Luis, J.; Laukhin, V.; Pérez del Pino, Á.; Vidal-Gancedo, J.; Rovira, C.; Laukhina, E.; Amabilino, D. B. Angew. Chem., Int. Ed. 2007, 46, 238–241. doi:10.1002/anie.200602483 |
| 16. | Puigmartı-Luis, J.; Pérez del Pino, Á.; Laukhina, E.; Esquena, J.; Laukhin, V.; Rovira, C.; Vidal-Gancedo, J.; Kanaras, A. G.; Nichols, R. J.; Brust, M.; Amabilino, D. B. Angew. Chem., Int. Ed. 2008, 47, 1861–1865. doi:10.1002/anie.200704864 |
| 22. | Su, L.; Bao, C.; Lu, R.; Chen, Y.; Xu, T.; Song, D.; Tan, C.; Shi, T.; Zhao, Y. Org. Biomol. Chem. 2006, 4, 2591–2594. doi:10.1039/b602520j |
| 23. | Kitamura, T.; Nakaso, S.; Mizoshita, N.; Tochigi, Y.; Shimomura, T.; Moriyama, M.; Ito, K.; Kato, T. J. Am. Chem. Soc. 2005, 127, 14769–14775. doi:10.1021/ja053496z |
| 24. | Ding, Z.; Zhao, Q.; Xing, R.; Wang, X.; Ding, J.; Wang, L.; Han, Y. J. Mater. Chem. C 2013, 1, 786–792. doi:10.1039/C2TC00125J |
| 10. | Wang, C.; Zhang, D.; Zhu, D. J. Am. Chem. Soc. 2005, 127, 16372–16373. doi:10.1021/ja055800u |
| 11. | Jeppesen, J. O.; Becher, J. Eur. J. Org. Chem. 2003, 3245–3266. doi:10.1002/ejoc.200300078 |
| 12. | Yang, X.; Zhang, G.; Zhang, D.; Zhu, D. Langmuir 2010, 26, 11720–11725. doi:10.1021/la101193z |
| 13. | Gomar-Nadal, E.; Veciana, J.; Rovira, C.; Amabilino, D. B. Adv. Mater. 2005, 17, 2095–2098. doi:10.1002/adma.200500348 |
| 14. | Bryce, M. R. Chem. Soc. Rev. 1991, 20, 355–390. doi:10.1039/cs9912000355 |
| 25. | Demir-Ordu, Ő.; Şimşir, H.; Alper, K. Tetrahedron 2015, 71, 1529–1539. doi:10.1016/j.tet.2015.01.042 |
| 26. | Zhang, Y.; Liang, C.; Shang, H.; Ma, Y.; Jiang, S. J. Mater. Chem. C 2013, 1, 4472–4480. doi:10.1039/c3tc30545g |
| 7. | George, M.; Tan, G.; John, V. T.; Weiss, R. G. Chem. – Eur. J. 2005, 11, 3243–3254. doi:10.1002/chem.200401066 |
| 8. | Goyal, N.; Mangunuru, H. P. R.; Parikh, B.; Shrestha, S.; Wang, G. Beilstein J. Org. Chem. 2014, 10, 3111–3121. doi:10.3762/bjoc.10.328 |
| 9. | Skilling, K. J.; Citossi, F.; Bradshaw, T. D.; Ashford, M.; Kellam, B.; Marlow, M. Soft Matter 2014, 10, 237–256. doi:10.1039/C3SM52244J |
| 2. | Nalluri, S. K. M.; Shivarova, N.; Kanibolotsky, A. L.; Zelzer, M.; Gupta, S.; Frederix, P. W. J. M.; Skabara, P. J.; Gleskova, H.; Ulijn, R. V. Langmuir 2014, 30, 12429–12437. doi:10.1021/la503459y |
| 3. | Giansante, C.; Raffy, G.; Schäfer, C.; Rahma, H.; Kao, M.-T.; Olive, A. G. L.; Del Guerzo, A. J. Am. Chem. Soc. 2011, 133, 316–325. doi:10.1021/ja106807u |
| 4. | Liu, Y.; Zheng, N.; Li, H.; Yin, B. Soft Matter 2013, 9, 5261–5269. doi:10.1039/c3sm50614b |
| 3. | Giansante, C.; Raffy, G.; Schäfer, C.; Rahma, H.; Kao, M.-T.; Olive, A. G. L.; Del Guerzo, A. J. Am. Chem. Soc. 2011, 133, 316–325. doi:10.1021/ja106807u |
| 4. | Liu, Y.; Zheng, N.; Li, H.; Yin, B. Soft Matter 2013, 9, 5261–5269. doi:10.1039/c3sm50614b |
| 5. | Pratihar, P.; Gosh, S.; Stepanenko, V.; Patwardhan, S.; Grozema, F. C.; Siebbeles, L. D. A.; Würthner, F. Beilstein J. Org. Chem. 2010, 6, 1070–1078. doi:10.3762/bjoc.6.122 |
| 6. | Banerjee, S.; Das, R. K.; Terech, P.; de Geyer, A.; Aymonier, C.; Loppinet-Serani, A.; Raffy, G.; Maitra, U.; Del Guerzo, A.; Desvergne, J. P. J. Mater. Chem. C 2013, 1, 3305–3316. doi:10.1039/c3tc30104d |
| 4. | Liu, Y.; Zheng, N.; Li, H.; Yin, B. Soft Matter 2013, 9, 5261–5269. doi:10.1039/c3sm50614b |
| 19. | Zhang, X.; Wang, C.; Lai, G.; Zhang, L.; Shen, Y. New J. Chem. 2010, 34, 318–324. doi:10.1039/B9NJ00520J |
| 18. | Lyskawa, J.; Oçafrain, M.; Trippé, G.; Le Derf, F.; Sallé, M.; Viel, P.; Palacin, S. Tetrahedron 2006, 62, 4419–4425. doi:10.1016/j.tet.2006.02.054 |
| 21. | Tatewaki, Y.; Watanabe, T.; Watanabe, K.; Kikuchi, K.; Okada, S. Dalton Trans. 2013, 42, 16121–16127. doi:10.1039/c3dt51464a |
| 19. | Zhang, X.; Wang, C.; Lai, G.; Zhang, L.; Shen, Y. New J. Chem. 2010, 34, 318–324. doi:10.1039/B9NJ00520J |
| 18. | Lyskawa, J.; Oçafrain, M.; Trippé, G.; Le Derf, F.; Sallé, M.; Viel, P.; Palacin, S. Tetrahedron 2006, 62, 4419–4425. doi:10.1016/j.tet.2006.02.054 |
| 21. | Tatewaki, Y.; Watanabe, T.; Watanabe, K.; Kikuchi, K.; Okada, S. Dalton Trans. 2013, 42, 16121–16127. doi:10.1039/c3dt51464a |
| 18. | Lyskawa, J.; Oçafrain, M.; Trippé, G.; Le Derf, F.; Sallé, M.; Viel, P.; Palacin, S. Tetrahedron 2006, 62, 4419–4425. doi:10.1016/j.tet.2006.02.054 |
| 29. | Akutagawa, T.; Kakiuchi, K.; Hasegawa, T.; Nakamura, T.; Christensen, C. A.; Becher, J. Langmuir 2004, 20, 4187–4195. doi:10.1021/la049950e |
| 30. | Skabara, P. J.; Berridge, R.; McInnes, E. J. L.; West, D. P.; Coles, S. J.; Hursthouse, M. B.; Müllen, K. J. Mater. Chem. 2004, 14, 1964–1969. doi:10.1039/b400809j |
| 17. | Massue, J.; Bellec, N.; Chopin, S.; Levillain, E.; Roisnel, T.; Clérac, R.; Lorcy, D. Inorg. Chem. 2005, 44, 8740–8748. doi:10.1021/ic051017r |
| 18. | Lyskawa, J.; Oçafrain, M.; Trippé, G.; Le Derf, F.; Sallé, M.; Viel, P.; Palacin, S. Tetrahedron 2006, 62, 4419–4425. doi:10.1016/j.tet.2006.02.054 |
| 20. | Benbellat, N.; Le Gal, Y.; Golhen, S.; Gouasmia, A.; Ouahab, L. Synth. Met. 2012, 162, 1789–1797. doi:10.1016/j.synthmet.2012.08.018 |
© 2015 Sun et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
