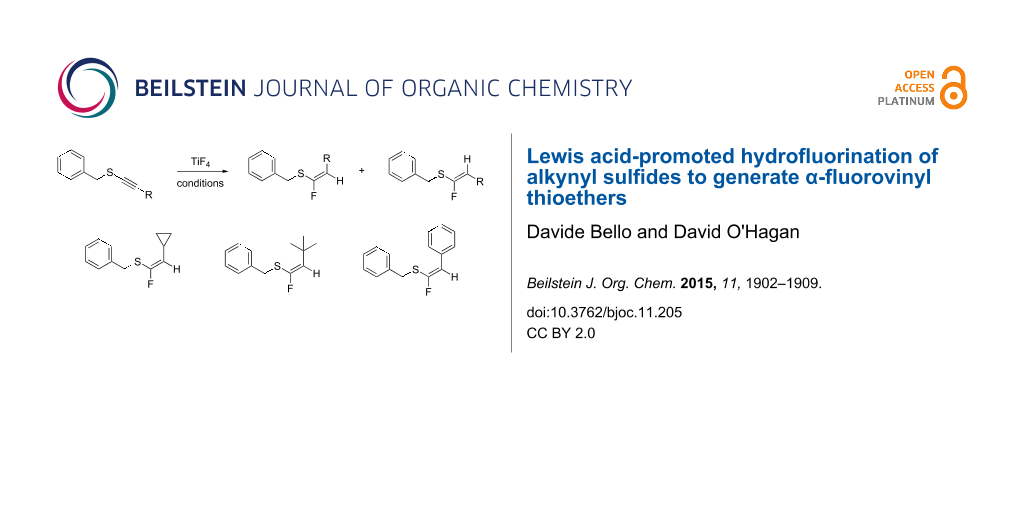
Abstract
A new method for the preparation of α-fluorovinyl thioethers is reported which involves the hydrofluorination of alkynyl sulfides with 3HF·Et3N, a process that requires Lewis acid activation using BF3·Et2O and TiF4. The method gives access to a range of α-fluorovinyl thioethers, some in high stereoselectivity with the Z-isomer predominating over the E-isomer. The α-fluorovinyl thioether motif has prospects as a steric and electronic mimetic of thioester enols and enolates, important intermediates in enzymatic C–C bond forming reactions. The method opens access to appropriate analogues for investigations in this direction.

Graphical Abstract
Introduction
Organofluorine compounds have found wide use in tuning the properties of performance compounds in medicinal and materials chemistry [1,2]. Also the electronegativity of fluorine has been used to design and tune steric and electronic mimetics of functional groups for applications in biomolecular chemistry. For example as illustrated in Figure 1, CF2-phosphonates became popular mimetics of the phosphate group [3,4], and vinyl fluorides were developed as analogues of the amide bond [5]. Difluorotoluene has proved to be a good spacial mimetic of the thymine base in thymidine, and has been shown to act as a functional and complementary template in enzymatic DNA synthesis [6].
![[1860-5397-11-205-1]](/bjoc/content/figures/1860-5397-11-205-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some spacial and electronic mimetics with fluorine as a design feature [3-6].
Figure 1: Some spacial and electronic mimetics with fluorine as a design feature [3-6].
We have recently begun to explore synthesis methods to prepare α-fluorovinyl thioethers, to open up the possibility of exploring this motif as a mimetic for enols and enolates of biochemically relevant thioesters. Thioesters of low molecular weight carboxylic acids are found widely in metabolism, often as their co-enzyme A esters, and they then undergo condensation reactions through enols or enolates to generate C–C bonds typified by the processes of long chain fatty acid biosynthesis. α-Fluorovinyl thioethers, illustrated in Figure 2, have a spatial and electrostatic profile consistent with the potential to mimic these enzyme intermediates.
![[1860-5397-11-205-2]](/bjoc/content/figures/1860-5397-11-205-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: α-Fluorovinyl thioesters offer prospects as thioester enol/ate mimetics [7].
Figure 2: α-Fluorovinyl thioesters offer prospects as thioester enol/ate mimetics [7].
There is limited literature for preparing such analogues. We have previously described the preparation of α-fluorovinyl thioethers by hydrofluorination of the corresponding alkynyl sulfides using HF·Py [7]; in this article we wish to report an improved synthesis of α-fluoroalkenyl thioethers via Lewis acid-mediated hydrofluorination of alkynyl sulfides, a method which brings us closer to being able to prepare analogues of particular design for enzyme inhibition studies.
Results and Discussion
Several methods for the synthesis of vinyl thioethers have been reported, including Wittig reactions [8], ionic and radical additions of thiols to alkynes [9] and coupling of 1-alkenyl halides with thiols, among others [10,11]. However, the literature for the preparation of α-fluorovinyl thioethers is somewhat scarce. The only account we are aware of involves the AIBN-promoted thiodesulfonylation of aromatic fluorovinyl sulfones as reported by Wnuk [12], a reaction which works in varying yields and stereoselectivities.
Following from our previous experience [7] with terminal acetylene thioethers, we now explore this reaction with alkynyl sulfides. In this regard 1a [13] was used as a model substrate and was treated with 50% HF·Py in dichloromethane. This, however, resulted in a very poor conversion (~10%) and gave a 4:1 product mixture of the fluorinated products 2a and 3a as illustrated in Scheme 1. When 70% HF·Py was employed, up to 70% conversion was achieved, but with over-fluorination to generate only the difluoromethylene thioether 4a (not isolated).
![[1860-5397-11-205-i1]](/bjoc/content/inline/1860-5397-11-205-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: HF·Py mediated hydrofluorinations of 1a.
Scheme 1: HF·Py mediated hydrofluorinations of 1a.
In view of the lack of control with HF·Py attention turned to triethylamine trihydrogen fluoride (3HF·Et3N). This proved unsuccessful presumably as it is a less acidic reagent compared to HF·Py, and thus activation of alkynyl sulfide 1a was explored by addition of a Lewis acid.
At this stage we were pleased to find that the use of BF3·Et2O allowed for a conversion of over 90% of 1a (16 h at room temperature). However, products 2a and 3a were obtained as a 4:1 mixture of Z/E-isomers, and they could only be isolated in a modest yield (35%) as shown in Scheme 2 and Table 1 (entry 7).
![[1860-5397-11-205-i2]](/bjoc/content/inline/1860-5397-11-205-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: BF3·Et2O/3HF·Et3N mediated hydrofluorination of 1a.
Scheme 2: BF3·Et2O/3HF·Et3N mediated hydrofluorination of 1a.
Encouraged by this result, a number of Lewis acids were tested, including SnCl2, ZnCl2, Sc(OTf)3, AuCl·SMe2 and B(C6F5)3 (Table 1). The Lewis acids (1.5 equivalents) were added to a mixture of sulfide 1a and 3HF·Et3N (3.0 equivalents) at 0 ºC, but no reactions took place under these conditions. The HBF4·SiO2 reagent was chosen as a solid phase-supported HBF4 equivalent [14]; carrying out the reaction in the presence of this reactant and 3HF·Et3N led to complete decomposition of sulfide 1a.
Table 1: Lewis acid screening.
| Entry | Lewis acid | HF source | Time | Temp | Conversion | Yield | Z/E |
|---|---|---|---|---|---|---|---|
| 1 | SnCl2 | 3HF·Et3N | 16 h | 0 °C to rt | 0% | – | – |
| 2 | ZnCl2 | 3HF·Et3N | 16 h | 0 °C to rt | 0% | – | – |
| 3 | Sc(OTf)3 | 3HF·Et3N | 16 h | 0 °C to rt | 0% | – | – |
| 4 | SiO2·HBF4 | 3HF·Et3N | 16 h | 0 °C to rt | n.a.a | – | – |
| 5 | AuCl·SMe2 | 3HF·Et3N | 16 h | 0 °C to rt | 0% | – | – |
| 6 | B(C6F5)3 | 3HF·Et3N | 16 h | 0 °C to rt | 0% | – | – |
| 7 | BF3·Et2O | 3HF·Et3N | 16 h | 0 °C to rt | >90% | 35% | 4:1 |
| 8 | TiF4 | 3HF·Et3N | 16 h | 0 °C to rt | 70% | 42% | 4:1 |
aSubstrate decomposed.
With TiF4 the overall conversion was around 70%, and the hydrofluorinated product could be isolated in an improved yield (42%, 4:1 Z:E).
In order to improve the reaction yields, reactions with the BF3·Et2O/3HF·Et3N and TiF4/3HF·Et3N systems were optimised and the outcomes described in Table 2 and Table 3, respectively. Shorter reaction times (5 h) led to reduced conversions (Table 2, entry 2) and BF3·Et2O or TiF4 are required to be stoichiometric, otherwise the reaction does not occur (Table 2, entry 4) and an excess of BF3·Et2O over the alkynyl sulfide is required for an improved outcome (Table 2, entry 1).
Table 2: Optimisation of BF3·Et2O/3HF·Et3N mediated hydrofluorination.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-205-i4.svg?max-width=637&scale=1.0)
|
|||||||
| Entry |
BF3·Et2O
(equiv) |
3HF·Et3N
(equiv) |
Time | Temp. | Solvent | Conversion | Yield |
|---|---|---|---|---|---|---|---|
| 1 | 1.5 | 3.0 | 16 h | 0 °C to rt | DCM | >90% | 35% |
| 2 | 1.5 | 3.0 | 5 h | 0 °C to rt | DCM | 39% | 28% |
| 3 | 1.0 | 2.0 | 16 h | 0 °C to rt | DCM | >80% | 30% |
| 4 | 0.5 | 3.0 | 16 h | 0 °C to rt | DCM | – | – |
| 5 | 1.5 | 3.0 | 5 days | 0 °C | DCM | 20% | – |
| 6 | 1.5 × 2 | 3.0 | 7 h | 0 °C | DCM | 20% | – |
| 7 | 1.5 | 3.0 | 5 h | 40 °C | DCM | >95% | 30% |
| 8a | 1.5 | 3.0 | 16 h | 0 °C to rt | DCM | 70% | 28% |
| 9 | 1.5 × 2 | 3.0 × 2 | 21 h | b | THF | 25% | – |
| 10 | 1.5 | 3.0 | 16 h | 0 °C to rt | DCE | <5% | – |
| 11 | 1.5 | 3.0 | 21 h | c | DCE | 10% | – |
| 12 | 1.5 x 2 | 3.0 x 2 | 21 h | d | DCE | n.a.e | – |
aBF3·Et2O and 3HF·Et3N were pre-mixed at 0 °C prior to adding starting material 1a. bMixture stirred for 16 hours at room temperature, then heated to 50 °C for 5 hours. cMixture stirred for 16 hours at room temperature, then stirred under reflux for 5 hours. dMixture stirred for 5 hours at room temperature, then stirred under reflux for 16 hours. eSubstrate decomposed.
The high conversion of 1a but low product (2a and 3a) isolation is attributed to substrate decomposition. When the reaction is followed by 19F NMR (vide infra), the presence of the hydrofluorinated products 2a and 3a is obvious and the anion BF4−, when using BF3·Et2O, or TiF62− when using TiF4 are clearly identifiable. No other fluorinated species are detected, thus it does not appear that products 2a and 3a decompose.
A number of attempts were made to improve the yields and reduce starting material decomposition. At low temperatures the reaction is sluggish and conversions are low (~20%), even with prolonged reaction times (5 days, Table 2, entry 5). A second addition of 1.5 equivalents of BF3·Et2O after a few hours at 0 °C proved ineffective (Table 2, entry 6). On the other hand, warming the mixture to reflux (40 ºC for dichloromethane) allowed for complete conversion in just 5 hours (Table 2, entry 7) although the isolated yield (30%) was relatively modest. Thus heating promotes the reaction but also substrate decomposition. Pre-equilibration of BF3·Et2O and 3HF·Et3N at 0 °C prior to starting material 1a addition resulted in a 70% conversion and a modest 28% yield (Table 2, entry 8). When tetrahydrofuran or dichloroethane were explored as solvents the conversions were low, even when warming (tetrahydrofuran, Table 2, entry 9, dichloroethane, Table 2, entries 10–12).
For the TiF4/3HF·Et3N reactions (Table 3) shorter reaction times also afforded lower conversions, and sub-stoichiometric levels of TiF4 failed to initiate the reaction. Tetrahydrofuran and dichloroethane at different temperatures were again not useful solvents.
Table 3: Optimisation of TiF4/3HF·Et3N mediated hydrofluorination.
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-205-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry |
TiF4
(equiv) |
3HF·TEA
(equiv) |
Time | Temp. | Solvent | Conversion | Yield |
|---|---|---|---|---|---|---|---|
| 1 | 1.5 | 3.0 | 5 h | 0 °C to rt | DCM | 39% | – |
| 2 | 1.5 | 3.0 | 16 h | 0 °C to rt | DCM | >90% | 42% |
| 3 | 0.5 | 3.0 | 16 h | 0 °C to rt | DCM | – | – |
| 4 | 1.5 | 3.0 | 16 h | 0 °C to rt or reflux | THF | – | – |
| 5 | 1.5 | 3.0 | 16 h | 0 °C to rt, then reflux | DCE | 10% | – |
Having optimised the reaction to some extent with substrate 1a, a range of alkynyl sulfides [15] were now prepared and each individually treated with both hydrofluorination protocols using BF3·Et2O/3HF·Et3N and TiF4/3HF·Et3N. The results are summarised in Table 4. Cyclohexylethynyl(benzyl)sulfane (1b) gave an improved outcome relative to 1a with higher yields and better stereoselectivity. The BF3·Et2O reaction furnished an inseparable 9:1 mixture of Z-2b and E-3b isomers in 48% yield. When TiF4 was used, the reaction showed complete stereoselectivity, affording the Z-isomer of 2b in 55% yield.
Table 4: Scope of BF3·Et2O and TiF4-mediated hydrofluorination reaction.
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-205-i6.svg?max-width=637&scale=1.0)
|
|||
| Substrate | Conversion and yield | Productsa | |
|---|---|---|---|
|
1a
R = Bn R1 = n-Bu |
BF3·Et2O |
>90%, 35%
Z/E 4:1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-205-i7.svg?max-width=637&scale=1.0)
|
| TiF4 |
70%, 42%
Z/E 4:1 |
||
|
1b
R = Bn R1 = Cy |
BF3·Et2O |
>90%, 48%
Z/E 9:1 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-205-i8.svg?max-width=637&scale=1.0)
|
| TiF4 |
80%, 55%
Z only (2b) |
||
|
1c
R = Bn R1 = Ph |
BF3·Et2O |
60%, 45%
Z only |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-205-i9.svg?max-width=637&scale=1.0)
|
| TiF4 |
>90%, 57%
Z only |
||
|
1d
R = Cy R1 = Ph |
BF3·Et2O |
complete, 47%
Z only |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-205-i10.svg?max-width=637&scale=1.0)
|
| TiF4 |
>90%, 68%
Z only |
||
|
1e
R = Ph R1 = cyclopropyl |
BF3·Et2O |
>80%, 47%
Z/E 3:2 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-205-i11.svg?max-width=637&scale=1.0)
|
| TiF4 |
90%, 69%
Z/E 7:3 |
||
|
1f
R = Ph R1 = t-Bu |
BF3·Et2O |
80%, 40%
Z only (contains 2% 4f) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-205-i12.svg?max-width=637&scale=1.0)
|
| TiF4 |
>90%, 62%
Z only (contains 2% 4f) |
||
|
1g
R = Ph R1 = Ph |
BF3·Et2O |
75%, 32%
Z only |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-205-i13.svg?max-width=637&scale=1.0)
|
| TiF4 |
80%, 41%
Z only |
||
|
1h
R = Ph R1 = 4-MeOPh |
BF3·Et2O |
27%,b 9%
Z only |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-205-i14.svg?max-width=637&scale=1.0)
|
| TiF4 |
35%,b 17%
Z only |
||
|
1i
R = Ph R1 = 4-NO2Ph |
BF3·Et2O |
90% compound 5 [16], 45%
only traces of fluorinated products |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-205-i15.svg?max-width=637&scale=1.0)
|
| TiF4 |
15%,b 5%
Z only |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-205-i16.svg?max-width=637&scale=1.0)
|
|
|
1j
R = Ph R1 = 4-CF3Ph |
BF3·Et2O | <5%,b NO products isolated | – |
| TiF4 | <5%,b NO products isolated | ||
aThe regiochemistry of all products was determined by NMR analysis. The Z/E stereochemistry was determined by calculating the vinyl moieties H–F coupling constants. bReaction times were 16 hours for all entries except for substrates 1h, 1i, and 1j (7 days).
Replacement of the cyclohexyl moiety with a phenyl ring in 1c led to a fully stereoselective reaction both with BF3·Et2O and TiF4, giving the Z-stereoisomer 2c in 45% and 57% yields, respectively. We then maintained the phenyl moiety on the alkyne side of the sulfide, and replaced the benzyl group with a cyclohexyl fragment directly connected to the sulfur atom (compound 1d). This material allowed too for a stereoselective reaction, giving rise to the Z-stereoisomer of 2d in 47% and 68% yields, respectively. At this stage we decided to explore two simple variations of the groups directly connected to the ethynyl moiety, that are, a cyclopropyl group and the bulky tert-butyl group. Thus, we reacted cyclopropylethynyl(phenyl)sulfane (1e) with BF3·Et2O, obtaining an inseparable 3:2 mixture of Z-2e and E-3e isomers in 47% yield. The reaction with TiF4 showed a better stereoselectivity, furnishing a 7:3 Z/E mixture in 69% yield.
Interestingly, the reaction of tert-butylethynyl(phenyl)sulfane (1f) with BF3·Et2O and TiF4, while being completely stereoselective, furnished the Z-stereoisomer 2f in 40% and 62% yields, respectively, along with a 2% of difluorinated compound 4f. The formation of this byproduct could not be avoided; in fact lower temperatures or shorter reaction times did not change the outcome, and the contaminant 4f could always be detected (and not removed) from the desired product 2f.
We were also interested in exploring the electronic effects of para-substitution of the phenyl group directly attached to the ethynyl moiety on the reaction outcome; thus we selected compounds 1g–j and reacted them under our hydrofluorinating conditions. Phenylethynyl(phenyl)sulfane (1g) represented the “unactivated” compound in the series. Although the stereoselectivity was complete with the Z-isomer of 2g as the sole product, the yields were unexpectedly low both with BF3·Et2O and TiF4 (32% and 41%, respectively).
We thought that the electron-donating 4-methoxy group would release enough electron density towards the triple bond to increase the yields, and possibly shorten the reaction times. Thus, we prepared compound 1h and then reacted it with our hydrofluorinating systems; surprisingly, almost no reaction took place during 16 hours, and it was necessary to extend the reaction time to 7 days to obtain the desired product 2h, which was isolated in 9% yield from the BF3·Et2O reaction and in 17% when TiF4 was employed. It appears that the methoxy group is able to efficiently coordinate the Lewis acid reactants and thus almost prevent the reaction from occurring.
Conversely, and as expected, the 4-nitro group had a detrimental effect on the reaction outcome. When 4-nitrophenyl(ethynyl)sulfane (1i) was treated with TiF4, it took nearly 7 days to observe some reaction progress, and the desired Z-isomer of 2i could be isolated in only 5% yield. However, when 1i was reacted with the BF3·Et2O, the starting material was completely consumed in 16 hours, but only traces of the desired compound 2i could be detected, with thioester 5 being the main reaction product (45% yield). An explanation for this behaviour can be drawn from the fact that the 4-nitrophenyl group surely must increase the triple bond electrophilicity, hence any trace of water present in the reaction mixture could lead to an intermediate enol thioester which would in turn readily convert to the stable thioester 5. Nonetheless, ensuring rigorously anhydrous reaction conditions and using fresh BF3·Et2O could not prevent the formation of 5, while the same compound was never detected when TiF4 was used, even after extended reaction times.
Because of the peculiar reactivity of electron-poor alkynyl sulfide 1i with respect to BF3·Et2O and TiF4, we decided to carry out a further test with compound 1j, with the intention of having the 4-trifluoromethylphenyl group removing electron-density from the triple bond, thus possessing a reactivity similar to that of nitro compound 1i. Surprisingly, compound 1j was found mostly unreacted after 7 days, and NMR analysis of the crude reaction mixtures did indicate the presence of product 2j only in traces (<5% conversion). Since 1j behaved in a similar way both with BF3·Et2O and TiF4, we could only conclude that the formation of thioester 5 from sulfide 1i was due to some very specific side-reaction promoted by the nitro group, possibly with its participation in the reaction process.
19F NMR was used to probe changes in the Lewis acids in the reaction. Ratios of 1:2 Lewis acid:3HF·Et3N mixtures in CD2Cl2 were stirred at room temperature for 2 hours, and the aliquots (0.7 mL) were assayed in Teflon NMR tubes. 19F NMR indicated that for each Lewis acid, BF3 and TiF4, respectively had disappeared, forming the corresponding anions BF4− (−150.75 ppm) and [TiF6]2− (75.37 ppm), respectively. Broad peaks corresponding to the excess 3HF·Et3N reagent were present. BF4− is known to be an inherently inert, non-nucleophilic counter ion; in the case of TiF4, [TiF6]2− was the only species present in solution, and we were unable to detect any penta-coordinated [TiF5]− species. It has been reported that an excess of hydrofluoric acid positions the equilibrium between [TiF5]− and [TiF6]2− in favour of the latter [17]. Moreover [TiF6]2− is rather unreactive [18], similar to the BF4− anion. We then analysed both reaction mixtures by 19F NMR, separately in CD2Cl2, in the presence of sulfide 1a, after stirring at room temperature for 2 hours. This showed the presence of 2a and 3a, as well anions BF4− or [TiF6]2− and also an excess 3HF·Et3N.
In light of these observations, our working hypothesis is that the Lewis acid acts to increase the acidity of the 3HF·Et3N by sequestering fluoride ions as relatively unreactive metal fluorides; thus, the alkynyl sulfides are activated by protonation possibly through an intermediate such as A as illustrated in Scheme 3. Such an intermediate would then be susceptible to fluoride ion attack, and progress to the reaction products. The major cis stereoselectivity is consistent with the attack of an intermediate such as A from the less hindered face, opposite to the R1 substituent (Scheme 3).
![[1860-5397-11-205-i3]](/bjoc/content/inline/1860-5397-11-205-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed Lewis acid-mediated hydroflurination of sulfides 1.
Scheme 3: Proposed Lewis acid-mediated hydroflurination of sulfides 1.
Conclusion
In summary, we have developed a mild method for the synthesis of α-fluorovinyl thioethers. The method involves the hydrofluorination of alkynyl sulfides by 3HF·Et3N and requires activation using BF3·Et2O or TiF4. The reactions display moderate to good stereoselectivity in favour of the Z-hydrofluorination product, and this opens the way forward for making appropriate analogues as potential steric and electronic mimetics of thioester enols and enolates relevant to particular enzymatic transformations.
Supporting Information
| Supporting Information File 1: Experimental part and NMR spectra of synthesised compounds. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley: Hoboken, NJ, 2009. doi:10.1002/9781444312096
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Berkowitz, D. B.; Bose, M.; Pfannenstiel, T. J.; Doukov, T. J. Org. Chem. 2000, 65, 4498–4508. doi:10.1021/jo000220v
Return to citation in text: [1] [2] -
Chambers, R. D.; Jaouhari, R.; O'Hagan, D. J. Chem. Soc., Chem. Commun. 1988, 1169–1170. doi:10.1039/c39880001169
Return to citation in text: [1] [2] -
Urban, J. J.; Tillman, B. G.; Cronin, W. A. J. Phys. Chem. A 2006, 110, 11120–11129. doi:10.1021/jp062881n
Return to citation in text: [1] [2] -
Jarchow-Choy, S. K.; Sjuvarsson, E.; Sintim, H. O.; Eriksson, S.; Kool, E. T. J. Am. Chem. Soc. 2009, 131, 5488–5494. doi:10.1021/ja808244t
Return to citation in text: [1] [2] -
Bello, D.; Cormanich, R. A.; O’Hagan, D. Aust. J. Chem. 2015, 68, 72–79. doi:10.1071/CH14298
Return to citation in text: [1] [2] [3] -
Aucagne, V.; Tatibouët, A.; Rollin, P. Tetrahedron 2004, 60, 1817–1826. doi:10.1016/j.tet.2003.12.040
Return to citation in text: [1] -
Oswald, A. A.; Griesbaum, K.; Hudson, B. E.; Bregman, J. M. J. Am. Chem. Soc. 1964, 86, 2877–2884. doi:10.1021/ja01068a023
Return to citation in text: [1] -
Kabir, M. S.; Van Linn, M. L.; Monte, A.; Cook, J. M. Org. Lett. 2008, 10, 3363–3366. doi:10.1021/ol801149n
Return to citation in text: [1] -
Kondo, T.; Mitsudo, T.-a. Chem. Rev. 2000, 100, 3205–3220. doi:10.1021/cr9902749
Return to citation in text: [1] -
Sacasa, P. R.; Zayas, R. J.; Wnuk, S. F. Tetrahedron Lett. 2009, 50, 5424–5427. doi:10.1016/j.tetlet.2009.07.063
Return to citation in text: [1] -
Zheng, W.; Zheng, F.; Hong, Y.; Hu, L. Heteroat. Chem. 2012, 23, 105–110. doi:10.1002/hc.20744
Return to citation in text: [1] -
Chakraborti, A. K.; Gulhane, R. Tetrahedron Lett. 2003, 44, 3521–3525. doi:10.1016/S0040-4039(03)00683-X
Return to citation in text: [1] -
The ethynyl sulfides were prepared using previously reported procedures; see Supporting Information File 1.
Return to citation in text: [1] -
Burhardt, M. N.; Ahlburg, A.; Skrydstrup, T. J. Org. Chem. 2014, 79, 11830–11840. doi:10.1021/jo5009965
Return to citation in text: [1] -
Itoh, Y.; Jang, S.; Ohba, S.; Mikami, K. Chem. Lett. 2004, 33, 776–777. doi:10.1246/cl.2004.776
Return to citation in text: [1] -
Nikiforov, G. B.; Roesky, H. W.; Koley, D. Coord. Chem. Rev. 2014, 258–259, 16–57. doi:10.1016/j.ccr.2013.09.002
Return to citation in text: [1]
| 17. | Itoh, Y.; Jang, S.; Ohba, S.; Mikami, K. Chem. Lett. 2004, 33, 776–777. doi:10.1246/cl.2004.776 |
| 18. | Nikiforov, G. B.; Roesky, H. W.; Koley, D. Coord. Chem. Rev. 2014, 258–259, 16–57. doi:10.1016/j.ccr.2013.09.002 |
| 1. | Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley: Hoboken, NJ, 2009. doi:10.1002/9781444312096 |
| 2. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2004. doi:10.1002/352760393X |
| 3. | Berkowitz, D. B.; Bose, M.; Pfannenstiel, T. J.; Doukov, T. J. Org. Chem. 2000, 65, 4498–4508. doi:10.1021/jo000220v |
| 4. | Chambers, R. D.; Jaouhari, R.; O'Hagan, D. J. Chem. Soc., Chem. Commun. 1988, 1169–1170. doi:10.1039/c39880001169 |
| 5. | Urban, J. J.; Tillman, B. G.; Cronin, W. A. J. Phys. Chem. A 2006, 110, 11120–11129. doi:10.1021/jp062881n |
| 6. | Jarchow-Choy, S. K.; Sjuvarsson, E.; Sintim, H. O.; Eriksson, S.; Kool, E. T. J. Am. Chem. Soc. 2009, 131, 5488–5494. doi:10.1021/ja808244t |
| 15. | The ethynyl sulfides were prepared using previously reported procedures; see Supporting Information File 1. |
| 6. | Jarchow-Choy, S. K.; Sjuvarsson, E.; Sintim, H. O.; Eriksson, S.; Kool, E. T. J. Am. Chem. Soc. 2009, 131, 5488–5494. doi:10.1021/ja808244t |
| 16. | Burhardt, M. N.; Ahlburg, A.; Skrydstrup, T. J. Org. Chem. 2014, 79, 11830–11840. doi:10.1021/jo5009965 |
| 5. | Urban, J. J.; Tillman, B. G.; Cronin, W. A. J. Phys. Chem. A 2006, 110, 11120–11129. doi:10.1021/jp062881n |
| 13. | Zheng, W.; Zheng, F.; Hong, Y.; Hu, L. Heteroat. Chem. 2012, 23, 105–110. doi:10.1002/hc.20744 |
| 3. | Berkowitz, D. B.; Bose, M.; Pfannenstiel, T. J.; Doukov, T. J. Org. Chem. 2000, 65, 4498–4508. doi:10.1021/jo000220v |
| 4. | Chambers, R. D.; Jaouhari, R.; O'Hagan, D. J. Chem. Soc., Chem. Commun. 1988, 1169–1170. doi:10.1039/c39880001169 |
| 14. | Chakraborti, A. K.; Gulhane, R. Tetrahedron Lett. 2003, 44, 3521–3525. doi:10.1016/S0040-4039(03)00683-X |
| 9. | Oswald, A. A.; Griesbaum, K.; Hudson, B. E.; Bregman, J. M. J. Am. Chem. Soc. 1964, 86, 2877–2884. doi:10.1021/ja01068a023 |
| 12. | Sacasa, P. R.; Zayas, R. J.; Wnuk, S. F. Tetrahedron Lett. 2009, 50, 5424–5427. doi:10.1016/j.tetlet.2009.07.063 |
| 8. | Aucagne, V.; Tatibouët, A.; Rollin, P. Tetrahedron 2004, 60, 1817–1826. doi:10.1016/j.tet.2003.12.040 |
| 7. | Bello, D.; Cormanich, R. A.; O’Hagan, D. Aust. J. Chem. 2015, 68, 72–79. doi:10.1071/CH14298 |
| 7. | Bello, D.; Cormanich, R. A.; O’Hagan, D. Aust. J. Chem. 2015, 68, 72–79. doi:10.1071/CH14298 |
| 7. | Bello, D.; Cormanich, R. A.; O’Hagan, D. Aust. J. Chem. 2015, 68, 72–79. doi:10.1071/CH14298 |
| 10. | Kabir, M. S.; Van Linn, M. L.; Monte, A.; Cook, J. M. Org. Lett. 2008, 10, 3363–3366. doi:10.1021/ol801149n |
| 11. | Kondo, T.; Mitsudo, T.-a. Chem. Rev. 2000, 100, 3205–3220. doi:10.1021/cr9902749 |
© 2015 Bello and O'Hagan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)