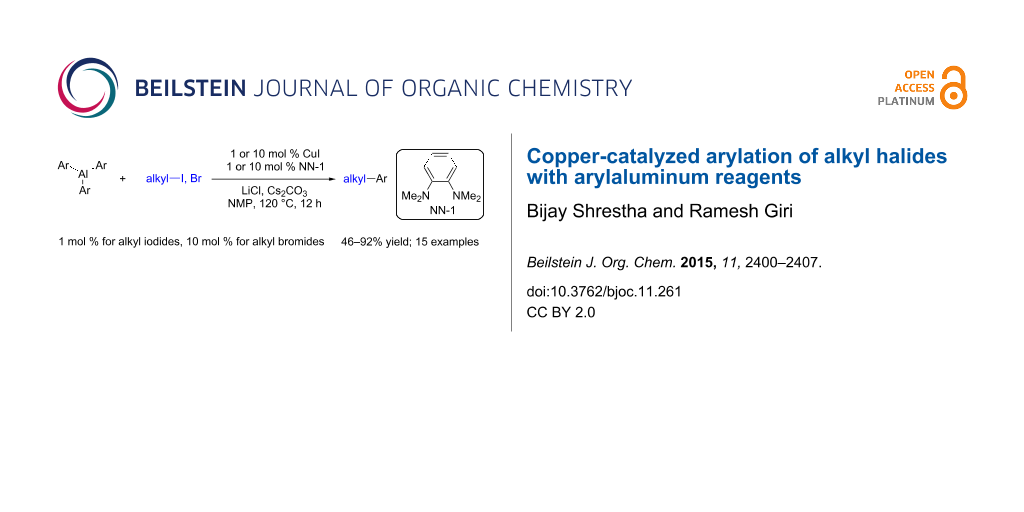
Abstract
We report a Cu-catalyzed coupling between triarylaluminum reagents and alkyl halides to form arylalkanes. The reaction proceeds in the presence of N,N,N’,N’-tetramethyl-o-phenylenediamine (NN-1) as a ligand in combination with CuI as a catalyst. This catalyst system enables the coupling of primary alkyl iodides and bromides with electron-neutral and electron-rich triarylaluminum reagents and affords the cross-coupled products in good to excellent yields.

Graphical Abstract
Introduction
Cross-coupling reactions represent one of the most important transformation for carbon–carbon (C–C) bond formation in organic synthesis [1-9]. These reactions, typically catalyzed by Pd and Ni, exploit a wide range of organometallic reagents of Mg, Zr, Zn, Sn, Al, B, Si and In as sources of nucleophiles. Among these metals/non-metals, Al offers a unique feature due to its high chemoselectivity and Lewis acidity [10-12]. In addition, Al also has low toxicity and is an inexpensive and earth-abundant metal. Organoaluminum reagents can be prepared directly from metallic aluminum [13-15], which further highlights the potential scope of these reagents in organic synthesis. However, despite extensive investigations and applications of organometallic reagents of Si, B, Mg, Zn and Sn in cross-coupling, the utility of organometallic complexes of Al are limited [13,14,16-21]. In many cases, direct transmetalation of organoalanes to Pd are sluggish and require ZnCl2 or CdCl2 to facilitate reactions through sequential transmetalations [19]. In some cases, intramolecular coordination to Al also enables the couplings of alkylalanes with organo halides [21]. Knochel [15] and Hoveyda [22] have also shown that organoaluminum reagents are capable of transmetalating to Cu-salts. Inspired by these literature reports and our recent investigations, we envisioned that organoaluminum reagents could participate as nucleophile sources in Cu-catalyzed cross-coupling reactions. In this artcle, we show that triarylaluminum reagents are excellent coupling partners for Cu-catalyzed cross-coupling reactions. The reaction proceeds for the coupling with primary alkyl iodides and bromides in good to excellent yields.
Results and Discussion
Recently, we [23-27] and others [10,28-39] reported efficient cross-couplings of oganometallic reagents of Si, B, In, Zr, Zn, Mg and Sn with organo halides [40,41]. Under our reaction conditions, a catalyst derived from the combination of CuI and 2-(diphenylphosphino)-N,N-dimethylaniline (PN-1) remains highly effective for coupling many of these organometallic reagents with aryl halides. In order to expand the scope of our coupling reactions, we utilized the standard condtions for the reaction of commercially available Ph3Al with 1-iodooctane using 1 mol % each of CuI and PN-1. However, the product, 1-phenyloctane (3), was formed only in 34% yield (Table 1, entry 1). Further optimization of the reaction conditions revealed that the coupling proceeded in 66% GC yield when the reaction was performed in NMP using 1 equivalent of Cs2CO3 as a base and 3 equivlents of LiCl as an additive in the absence of a ligand (Table 1, entry 2). We then screened a variety of ligands (Scheme 1) and found that N,N,N’,N’-tetramethyl-o-phenylenediamine (NN-1) was an efficient ligand for CuI that enabled us to increase the product yield to 81% GC yields (76% isolated, Table 1, entry 3) [10,35,42-44]. Reactions containing other PN- and NN-based ligands that are analogous to PN-1 and NN-1 (Scheme 1) afforded cross-coupled product 3 in lower yields than the reaction performed in the absence of NN-1. Reactions containing the bisphosphine ligand, o-bis(diphenylphosphine)benzene (PP) and anionic ligands such as 8-hydroxyquinoline (NO) and 2,2,6,6-tetramethyl-3,5-heptanedione (OO, Scheme 1) also formed the product 3 in lower yields and the reaction performed in the absence of NN-1. The reaction does not proceed in the absence of CuI (Table 1, entry 4). The cross-coupled product 3 is formed in 50% and 54% yield, respectively, in the absence of LiCl and Cs2CO3 (Table 1, entries 5 and 6). The reacton with 2 and 4 equivalents of LiCl also afforded product 3 in comparable yields (78% and 76%, respectively) to that of the standard reaction (Table 1, entries 7 and 8). However, excess of LiCl was found to be detrimental to the reaction (Table 1, entry 9). The reaction could also be performed at a temperature as low as 80 °C affording the coupled product 3 only in slightly lower yields than that of the standard reaction (Table 1, entries 10 and 11).
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-261-i3.svg?max-width=637&scale=1.0)
|
||
| Entry | Variation from the standard conditions | Yield (%)b |
|---|---|---|
| 1 | PN-1 instead of NN-1 in DMF, no Cs2CO3 | 34 |
| 2 | No NN-1 | 66 |
| 3 | none | 81 (76) |
| 4 | without CuI | 0 |
| 5 | without LiCl | 50 |
| 6 | without Cs2CO3 | 54 |
| 7 | 2 equiv LiCl | 78 |
| 8 | 4 equiv LiCl | 76 |
| 9 | 6 equiv LiCl | 35 |
| 10 | 100 °C | 78 |
| 11 | 80 °C | 75 |
aReactions were run in 0.5 mL DMF. Commercially available Ph3Al was used. bGC yields (average of at least two parallel runs) calibrated against 2-nitrobiphenyl as an internal standard. Value in parenthesis is the isolated yield (1.0 mmol).
![[1860-5397-11-261-i1]](/bjoc/content/inline/1860-5397-11-261-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Ligands used for reaction optimization.
Scheme 1: Ligands used for reaction optimization.
After establishing the combination of NN-1 and CuI as the best catalyst, we began to explore the substrate scope of the reaction. While the reaction proceeded in good yields with alkyl iodides (Table 2, entries 1–3) by using 1 mol % of the catalyst, reactions with alkyl bromides, which are more readily available and less expensive than alkyl iodides, required 10 mol % of NN-1/CuI (Table 2, entries 4–15). The reaction can be performed with electron-neutral and electron-rich triarylaluminum reagents [45]. The reaction tolerates a variety of functional groups on alkyl halides including highly sensitive esters (Table 2, entries, 5, 9 and 11), nitriles (Table 2, entries 6 and 7) and olefins (Table 2, entries 4, 8, 10, 13 and 15) [46]. With 10 mol % catalyst loading, the reaction can also be extended to the coupling of triarylaluminum reagents with benzyl bromides (Table 2, entries 12 and 14) [43].
Table 2: Coupling of triarylaluminum reagents with alkyl iodides and bromidesa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-261-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Ar in Ar3Al | Alkyl−I,Br | Alkyl−Ar | yield (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-261-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-261-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-261-i7.svg?max-width=637&scale=1.0)
|
76 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-261-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-261-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-261-i10.svg?max-width=637&scale=1.0)
|
61 |
| 3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-261-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-261-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-261-i13.svg?max-width=637&scale=1.0)
|
49 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-261-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-261-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-261-i16.svg?max-width=637&scale=1.0)
|
60 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-261-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-261-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-261-i19.svg?max-width=637&scale=1.0)
|
58 |
| 6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-261-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-261-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-261-i22.svg?max-width=637&scale=1.0)
|
71 |
| 7 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-261-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-261-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-261-i25.svg?max-width=637&scale=1.0)
|
88 |
| 8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-261-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-261-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-261-i28.svg?max-width=637&scale=1.0)
|
53 |
| 9 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-261-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-261-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-261-i31.svg?max-width=637&scale=1.0)
|
59 |
| 10 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-261-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-261-i33.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-261-i34.svg?max-width=637&scale=1.0)
|
46 |
| 11 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-261-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 34]](/bjoc/content/inline/1860-5397-11-261-i36.svg?max-width=637&scale=1.0)
|
![[Graphic 35]](/bjoc/content/inline/1860-5397-11-261-i37.svg?max-width=637&scale=1.0)
|
92 |
| 12 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-11-261-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-11-261-i39.svg?max-width=637&scale=1.0)
|
![[Graphic 38]](/bjoc/content/inline/1860-5397-11-261-i40.svg?max-width=637&scale=1.0)
|
53 |
| 13 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-11-261-i41.svg?max-width=637&scale=1.0)
|
![[Graphic 40]](/bjoc/content/inline/1860-5397-11-261-i42.svg?max-width=637&scale=1.0)
|
![[Graphic 41]](/bjoc/content/inline/1860-5397-11-261-i43.svg?max-width=637&scale=1.0)
|
52 |
| 14 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-11-261-i44.svg?max-width=637&scale=1.0)
|
![[Graphic 43]](/bjoc/content/inline/1860-5397-11-261-i45.svg?max-width=637&scale=1.0)
|
![[Graphic 44]](/bjoc/content/inline/1860-5397-11-261-i46.svg?max-width=637&scale=1.0)
|
68 |
| 15 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-11-261-i47.svg?max-width=637&scale=1.0)
|
![[Graphic 46]](/bjoc/content/inline/1860-5397-11-261-i48.svg?max-width=637&scale=1.0)
|
![[Graphic 47]](/bjoc/content/inline/1860-5397-11-261-i49.svg?max-width=637&scale=1.0)
|
47 |
aReactions were run in 5 mL DMF. Reactions for entries 1–3 were run with 1 mol % NN-1/CuI. Reactions for entries 4–15 were run with 10 mol % NN-1/CuI. Triarylaluminum reagents, except the commercially available Ph3Al, were prepared from the reaction of 3 equivalents of ArLi reagents with AlCl3 (99.99% purity) in THF at room temperature and were used without further purification. Each reaction contains 3 equivalents of LiCl, written in parenthesis below the reaction arrow, which is generated during the preparation of triarylaluminum reagents. bYields are for products isolated by column chromatography from a 1.0 mmol scale reaction.
Based on literature reports and our recent mechanistic work on Cu-catalyzed cross-couplings [23-25], we propose a catalytic cycle for the current reaction (Scheme 2). It is evident from the optimization of reaction conditions that both NN-1 and LiCl improve product yields for the current coupling of triarylaluminum reagents with alkyl halides (Table 1). As such, we believe that organoaluminate complexes such as 18, generated from the binding of LiCl to three-coordinate triarylaluminum reagents, are the actual species in solution that undergo transmetalation with NN-bound CuX (X = I, Br) to generate (NN)CuAr complexes as the reaction intermediates. Catalytically competent CuI-complexes that contain nitrogen-based ligands have previously been synthesized and fully characterized structurally [47-51]. Triorganoaluminum complexes are also known to form triorganoaluminate species in the presence of anions in solution [52-57]. In addition, organoaluminum reagents have been demonstrated to undergo transmetalation with Cu salts based on their participation in allylic and conjugate addition reactions [11,15,43]. Similar Cu-catalyzed couplings of organometallic reagents with alkyl electrophiles have previously been shown to proceed via an SN2 process [34,35]. Therefore, we believe that a similar mechanistic scenario can also be envisioned in the current Cu-catalyzed cross-coupling of triarylaluminum reagents with primary alkyl halides that involves (NN)CuAr as the reaction intermediates.
![[1860-5397-11-261-i2]](/bjoc/content/inline/1860-5397-11-261-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have developed a Cu-catalyzed coupling of triarylaluminum reagents with primary alkyl iodides and bromides. The reaction proceeds in the presence of NN-1/CuI as an effective catalyst. Electron-neutral and electron-rich triarylaluminum reagents can be coupled with a variety of alkyl iodides and bromides containing a range of sensitive functional groups such as olefins, nitriles and esters, affording the alkylated arenes in good to excellent yields.
Experimental
General information. All the reactions and handling of chemicals were done inside a nitrogen-filled glovebox unless stated otherwise. All glassware were dried in an oven before use. All commercial reagents were used as received without further purification. Anhydrous solvents and triphenylaluminum were purchased from Sigma-Aldrich. Pure triarylaluminum reagents other than Ph3Al were synthesized following the reported procedure [56]. Ligands PN-5, PN-6, NN-2, NN-3, PP, NO and OO were purchased from commercial sources. Ligands PN-1, PN-2, PN-3, PN-4, PN-7 [58], PN-8, NN-1 [59], and NN-4 [60] were synthesized following the reported procedures [61]. 1H and 13C NMR spectra were recorded on a Bruker instrument (300 and 75 MHz, respectively) and internally referenced to the residual solvent signals of CDCl3 at 7.26 and at 77.16 ppm, respectively.
General procedure for cross-coupling. To a suspension of AlCl3 (133.3 mg, 1.0 mmol) in THF (2 mL) was added dropwise a solution of aryllithium (3.0 mmol, generated from the lithiation of aryl iodides with 1 equiv of n-BuLi in THF) at room temperature. After 45 minutes, the solvent was removed to obtain a triarylaluminum reagent containing 3 equivalents of LiCl, which was then dissolved in NMP (5 mL). Alkyl halide (1.0 mmol), CuI (1.9 mg, 0.010 mmol, for alkyl iodides; 19.0 mg, 0.10 mmol, for alkyl bromides) and NN-1 (1.6 mg, 0.010 mmol, for alkyl iodides; 16.4 mg, 0.10 mmol, for alkyl bromides) were then added to the solution of the triarylaluminum reagent. The reaction mixture was then tightly capped, taken out of the glovebox, placed in an oil bath pre-heated to 120 °C and vigorously stirred. After 12 h, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (15 mL) and washed with H2O (5 mL × 3). The aqueous fraction was extracted back with ethyl acetate (5 mL × 3) and combined with the first ethyl acetate fraction. The combined ethyl acetate fractions were dried over Na2SO4 and the solvent was removed on a rotary evaporator. The product was purified by silica gel column chromatography using 0–5% ethyl acetate in hexanes.
n-Octylbenzene (3) [62]: The title compound 3 was obtained as a colorless oil (144 mg, 76% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 0.89 (t, J = 6.6 Hz, 3H), 1.29–1.32 (m, 10H), 1.58–1.68 (m, 2H), 2.62 (m, J = 8.1 Hz, 2H), 7.16–7.21 (m, 3H), 7.27–7.32 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 14.2, 22.8, 29.4, 29.5, 29.6, 31.7, 32.0, 36.1, 125.7, 128.3, 128.5, 143.1; GC–MS (m/z) 190.1.
1-Dodecyl-3-methylbenzene (4): The title compound 4 was obtained as yellow oil (159 mg, 61% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 0.90 (t, J = 6.3 Hz, 3H), 1.28–1.31 (m, 18H), 1.53–1.64 (m, 2H), 2.35 (s, 3H), 2.55–2.60 (m, 2H), 6.99–7.02 (m, 3H), 7.18 (t, J = 6.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.3, 21.6, 22.9, 27.1, 29.5, 29.6, 29.7, 29.8, 31.7, 32.1, 36.1, 45.3, 125.5, 126.4, 128.3, 129.4, 137.9, 143.1; GC–MS (m/z) 260.1.
1-Isopentyl-3-methoxybenzene (5): The title compound 5 was obtained as colorless oil (87 mg, 49% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 0.93 (s, 6H), 1.46–1.64 (m, 3H), 2.6 (t, J = 7.8 Hz, 2H), 3.80 (s, 3H), 6.71–6.80 (m, 2H), 7.12–7.38 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 22.7, 27.8, 33.9, 40.8, 55.2, 110.9, 114.3, 120.9, 129.3, 144.9, 159.7; GC–MS (m/z) 178.1.
7-Octen-1-ylbenzene (6) [63]: The title compound 6 was obtained as a colorless oil (113 mg, 60% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.33–1.41 (m, 6H), 1.60–1.65 (m, 2H), 2.00–2.08 (m, 2H), 2.61 (t, J = 7.5 Hz, 2H), 4.91–5.03 (m, 2H), 5.75–5.88 (m, 1H), 7.17–7.20 (m, 3H), 7.25–7.31 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 28.9, 29.1, 29.2, 31.5, 33.9, 36.0, 114.3, 125.6, 128.3, 128.5, 139.2, 142.9; GC–MS (m/z) 188.1.
Ethyl 5-phenylvalerate (7) [64]: The title compound 7 was obtained as yellow oil (120 mg, 58% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.26 (t, J = 9.0 Hz, 3H), 1.66–1.70 (m, 4H), 2.31–2.35 (m, 2H), 2.62–2.67 (m, 2H), 4.13 (q, J = 6.9 Hz, 2H), 7.17–7.22 (m, 3H), 7.26–7.32 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 14.3, 24.7, 33.5, 34.2, 35.6, 60.3, 125.8, 128.4, 128.4, 142.2, 173.7; GC–MS (m/z) 206.1.
2,2-Dimethyl-6-phenylhexanenitrile (8) [65]: The title compound 8 was obtained as yellow oil (143 mg, 71% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.34 (s, 6H), 1.54–1.58 (m, 4H), 1.62–1.72 (m, 2H), 2.63–2.68 (m, 2H), 7.17–7.23 (m, 3H), 7.26–7.33 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 25.0, 26.7, 31.5, 32.4, 35.7, 40.9, 125.2, 125.8, 128.4, 142.2; GC–MS (m/z) 201.1.
2,2-Dimethyl-6-(3-methylphenyl)hexanenitrile (9): The title compound 9 was obtained as yellow oil (189 mg, 88% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.35 (s, 6H), 1.55–1.59 (m, 4H), 1.63–1.72 (m, 2H), 2.36 (s, 3H), 2.61–2.67 (m, 2H), 6.99–7.02 (m, 3H), 7.19 (t, J = 3.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 21.4, 25.0, 26.6, 31.5, 32.3, 35.6, 41.3, 125.2, 125.3, 126.5, 128.2, 129.2, 137.8, 142.1; GC–MS (m/z) 215.1.
1-(6-Hepten-1-yl)-3-methylbenzene (10): The title compound 10 was obtained as colorless oil (100 mg, 53% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.34–1.45 (m, 4H), 1.59–1.66 (m, 2H), 2.02–2.09 (m, 2H), 2.33 (s, 3H), 2.57 (t, J = 7.8 Hz, 2H), 4.92–5.03 (m, 2H), 5.75–5.88 (m, 1H), 6.97–7.00 (m, 3H), 7.17 (t, J = 3.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 28.8, 29.7, 31.4, 33.7, 35.6, 114.2, 125.4, 126.3, 128.1, 129.2, 137.7, 139.1, 142.8; GC–MS (m/z) 188.2.
Ethyl 5-(3-methylphenyl)valerate (11) [66]: The title compound 11 was obtained as a yellow oil (130 mg, 59% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H), 1.66–1.72 (m, 4H), 2.33–2.37 (m, 5H), 2.60–2.65 (m, 2H), 4.15 (q, J = 7.2 Hz, 2H), 6.98–7.04 (m, 3H), 7.18 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.3, 21.5, 24.7, 31.0, 34.3, 35.6, 60.2, 125.4, 126.5, 128.3, 129.3, 137.9, 142.2, 173.7; GC–MS (m/z) 220.2.
1-(5-Hexen-1-yl)-2-methylbenzene (12) [67]: The title compound 12 was obtained as colorless oil (80 mg, 46% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.48–1.67 (m, 4H), 2.09–2.16 (m, 2H), 2.33 (s, 3H), 2.59–2.65 (m, 2H), 4.98–5.07 (m, 2H), 5.77–5.91 (m, 1H), 7.11–7.15 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 19.4, 29.1, 29.9, 33.3, 33.8, 114.6, 125.9, 125.9, 128.9, 130.2, 135.9, 139.0, 141.0; GC–MS (m/z) 174.1.
Ethyl 5-(2-methylphenyl)valerate (13) [68]: The title compound 13 was obtained as yellow oil (203 mg, 92% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.26 (t, J = 7.2 Hz, 3H), 1.58–1.76 (m, 4H), 2.31–2.37 (m, 5H), 2.6–2.65 (m, 2H), 4.13 (q, J = 7.2 Hz, 2H), 7.11–7.12 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 14.4, 19.4, 25.1, 29.8, 33.1, 34.4, 60.4, 126.0, 128.9, 130.3, 135.9, 140.5, 173.8; GC–MS (m/z) 220.2.
1-Benzyl-3-methoxybenzene (14) [69]: The title compound 14 was obtained as a light yellow oil (105 mg, 53% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 2.82 (s, 3H), 3.8 (s, 2H), 6.77–6.84 (m, 2H), 6.91 (t, J = 9.0 Hz, 1H), 7.20–7.34 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 42.0, 55.2, 111.4, 114.9, 121.4, 126.2, 128.3, 128.5, 129.5, 140.9, 142.7, 159.8; GC–MS (m/z) 198.1.
1-Methoxy-3-(oct-7-en-1-yl)benzene (15): The title compound 15 was obtained as colorless oil (113 mg, 52% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.27–1.42 (m, 4H), 1.55–1.65 (m, 4H), 2.01–2.05 (m, 2H), 2.59 (t, J = 7.5 Hz, 2H), 3.81 (s, 3H), 4.92–5.02 (m, 2H), 5.75–5.88 (m, 1H), 6.72–7.36 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 29.1, 29.3, 29.6, 31.4, 33.9, 35.8, 55.2, 110.9, 114.3, 121.0, 129.3, 129.8, 139.3, 144.7; GC–MS (m/z) 218.2.
1-Benzyl-2-methoxybenzene (16) [70]: The title compound 16 was obtained as light yellow oil (135 mg, 68% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 3.86 (s, 3H), 4.04 (s, 2H), 6.90–6.96 (m, 2H), 7.11–7.14 (m, 1H), 7.21–7.36 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 35.9, 55.4, 110.5, 120.6, 125.8, 127.5, 128.3, 129.0, 129.7, 130.4, 141.1, 157.4; GC–MS (m/z) 198.2.
1-Methoxy-2-(pent-4-en-1-yl)benzene (17) [71]: The title compound 17 was obtained as yellow oil (83 mg, 47% yield) after purification by silica gel column chromatography. 1H NMR (300 MHz, CDCl3) δ 1.64–1.74 (m, 2H), 2.08–2.15 (m, 2H), 2.61–2.66 (m, 2H), 3.83 (s, 3H), 5.01–5.08 (m, 2H), 5.80–5.94 (m, 1H), 6.84–6.92 (m, 3H), 7.15 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz,CDCl3) δ 29.2, 33.8, 41.5, 55.4, 110.4, 114.5, 117.9, 120.4, 127.0, 139.1, 145.3, 157.2; GC–MS (m/z) 176.1.
References
-
Heck, R. F. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 833 ff.
Return to citation in text: [1] -
Diederich, F.; Stang, P. J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: New York, 1998.
Return to citation in text: [1] -
Negishi, E.-i.; Hu, Q.; Huang, Z.; Qian, M.; Wang, G. Aldrichimica Acta 2005, 38, 71.
Return to citation in text: [1] -
Fu, G. C. Acc. Chem. Res. 2008, 41, 1555–1564. doi:10.1021/ar800148f
Return to citation in text: [1] -
Knochel, P.; Jones, P., Eds. Organozinc Reagents: A Practical Approach; Oxford University Press: New York, 1999.
Return to citation in text: [1] -
Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417–1492. doi:10.1021/cr100327p
Return to citation in text: [1] -
Chang, W.-T. T.; Smith, R. C.; Regens, C. S.; Bailey, A. D.; Werner, N. S.; Denmark, S. E. Org. React. 2011, 75, 213–746.
Return to citation in text: [1] -
Nakao, Y.; Hiyama, T. Chem. Soc. Rev. 2011, 40, 4893–4901. doi:10.1039/c1cs15122c
Return to citation in text: [1] -
Sore, H. F.; Galloway, W. R. J. D.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 1845–1866. doi:10.1039/C1CS15181A
Return to citation in text: [1] -
Tsubouchi, A.; Muramatsu, D.; Takeda, T. Angew. Chem., Int. Ed. 2013, 52, 12719–12722. doi:10.1002/anie.201306882
Return to citation in text: [1] [2] [3] -
Thaler, T.; Knochel, P. Angew. Chem., Int. Ed. 2009, 48, 645–648. doi:10.1002/anie.200804446
Return to citation in text: [1] [2] -
Krasovskiy, A.; Malakhov, V.; Gavryushin, A.; Knochel, P. Angew. Chem., Int. Ed. 2006, 45, 6040–6044. doi:10.1002/anie.200601450
Return to citation in text: [1] -
Huo, S. Org. Lett. 2003, 5, 423–425. doi:10.1021/ol0272693
Return to citation in text: [1] [2] -
Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442–4489. doi:10.1002/anie.200500368
Return to citation in text: [1] [2] -
Blümke, T. D.; Groll, K.; Karaghiosoff, K.; Knochel, P. Org. Lett. 2011, 13, 6440–6443. doi:10.1021/ol202733v
Return to citation in text: [1] [2] [3] -
Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. J. Org. Chem. 2014, 79, 230–239. doi:10.1021/jo4024123
Return to citation in text: [1] -
Naka, H.; Uchiyama, M.; Matsumoto, Y.; Wheatley, A. E. H.; McPartlin, M.; Morey, J. V.; Kondo, Y. J. Am. Chem. Soc. 2007, 129, 1921–1930. doi:10.1021/ja064601n
Return to citation in text: [1] -
Getmanenko, Y. A.; Twieg, R. J. J. Org. Chem. 2008, 73, 830–839. doi:10.1021/jo701812t
Return to citation in text: [1] -
Koszinowski, K.; Böhrer, P. Organometallics 2009, 28, 771–779. doi:10.1021/om800947t
Return to citation in text: [1] [2] -
Lipshutz, B. H.; Bülow, G.; Lowe, R. F.; Stevens, K. L. Tetrahedron 1996, 52, 7265–7276. doi:10.1016/0040-4020(96)00250-5
Return to citation in text: [1] -
Blum, J.; Gelman, D.; Baidossi, W.; Shakh, E.; Rosenfeld, A.; Aizenshtat, Z.; Wassermann, B. C.; Frick, M.; Heymer, B.; Schutte, S.; Wernik, S.; Schumann, H. J. Org. Chem. 1997, 62, 8681–8686. doi:10.1021/jo970822n
Return to citation in text: [1] [2] -
Dabrowski, J. A.; Villaume, M. T.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2013, 52, 8156–8159. doi:10.1002/anie.201304035
Return to citation in text: [1] -
Gurung, S. K.; Thapa, S.; Vangala, A. S.; Giri, R. Org. Lett. 2013, 15, 5378–5381. doi:10.1021/ol402701x
Return to citation in text: [1] [2] -
Thapa, S.; Gurung, S. K.; Dickie, D. A.; Giri, R. Angew. Chem., Int. Ed. 2014, 53, 11620–11624. doi:10.1002/anie.201407586
Return to citation in text: [1] [2] -
Gurung, S. K.; Thapa, S.; Kafle, A.; Dickie, D. A.; Giri, R. Org. Lett. 2014, 16, 1264–1267. doi:10.1021/ol500310u
Return to citation in text: [1] [2] -
Thapa, S.; Basnet, P.; Gurung, S. K.; Giri, R. Chem. Commun. 2015, 51, 4009–4012. doi:10.1039/C5CC00116A
Return to citation in text: [1] -
Thapa, S.; Kafle, A.; Gurung, S. K.; Montoya, A.; Riedel, P.; Giri, R. Angew. Chem., Int. Ed. 2015, 54, 8236–8240. doi:10.1002/anie.201502379
Return to citation in text: [1] -
Thathagar, M. B.; Beckers, J.; Rothenberg, G. J. Am. Chem. Soc. 2002, 124, 11858–11859. doi:10.1021/ja027716+
Return to citation in text: [1] -
Li, J.-H.; Li, J.-L.; Wang, D.-P.; Pi, S.-F.; Xie, Y.-X.; Zhang, M.-B.; Hu, X.-C. J. Org. Chem. 2007, 72, 2053–2057. doi:10.1021/jo0623742
Return to citation in text: [1] -
Zhou, Y.; You, W.; Smith, K. B.; Brown, M. K. Angew. Chem., Int. Ed. 2014, 53, 3475–3479. doi:10.1002/anie.201310275
Return to citation in text: [1] -
You, W.; Brown, M. K. J. Am. Chem. Soc. 2014, 136, 14730–14733. doi:10.1021/ja509056j
Return to citation in text: [1] -
Ito, H.; Sensui, H.-o.; Arimoto, K.; Miura, K.; Hosomi, A. Chem. Lett. 1997, 26, 639–640. doi:10.1246/cl.1997.639
Return to citation in text: [1] -
Cornelissen, L.; Lefrancq, M.; Riant, O. Org. Lett. 2014, 16, 3024–3027. doi:10.1021/ol501140p
Return to citation in text: [1] -
Yang, C.-T.; Zhang, Z.-Q.; Liang, J.; Liu, J.-H.; Lu, X.-Y.; Chen, H.-H.; Liu, L. J. Am. Chem. Soc. 2012, 134, 11124–11127. doi:10.1021/ja304848n
Return to citation in text: [1] [2] -
Terao, J.; Todo, H.; Begum, S. A.; Kuniyasu, H.; Kambe, N. Angew. Chem., Int. Ed. 2007, 46, 2086–2089. doi:10.1002/anie.200603451
Return to citation in text: [1] [2] [3] -
Cahiez, G.; Gager, O.; Buendia, J. Angew. Chem., Int. Ed. 2010, 49, 1278–1281. doi:10.1002/anie.200905816
Return to citation in text: [1] -
Mohapatra, S.; Bandyopadhyay, A.; Barma, D. K.; Capdevila, J. H.; Falck, J. R. Org. Lett. 2003, 5, 4759–4762. doi:10.1021/ol035458v
Return to citation in text: [1] -
Allred, G. D.; Liebeskind, L. S. J. Am. Chem. Soc. 1996, 118, 2748–2749. doi:10.1021/ja9541239
Return to citation in text: [1] -
Li, J.-H.; Tang, B.-X.; Tao, L.-M.; Xie, Y.-X.; Liang, Y.; Zhang, M.-B. J. Org. Chem. 2006, 71, 7488–7490. doi:10.1021/jo061220j
Return to citation in text: [1] -
Thapa, S.; Shrestha, B.; Gurung, S. K.; Giri, R. Org. Biomol. Chem. 2015, 13, 4816–4827. doi:10.1039/C5OB00200A
Return to citation in text: [1] -
Beletskaya, I. P.; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337–2364. doi:10.1016/j.ccr.2004.09.014
Return to citation in text: [1] -
Yang, C.-T.; Zhang, Z.-Q.; Liu, Y.-C.; Liu, L. Angew. Chem., Int. Ed. 2011, 50, 3904–3907. doi:10.1002/anie.201008007
Return to citation in text: [1] -
Cornelissen, L.; Cirriez, V.; Vercruysse, S.; Riant, O. Chem. Commun. 2014, 50, 8018–8020. doi:10.1039/c4cc02923b
Return to citation in text: [1] [2] [3] -
The reaction of Ph3Al with 1-iodoocctane using the Schlenk technique afforded cross-coupled product 3 in 72% yield.
Return to citation in text: [1] -
Cross-coupling of electron-deficient (4-FC6H4)3Al with benzyl bromide afforded the product only in 18% yield.
Return to citation in text: [1] -
The conversion of bromoolefins is generally over 90%. The low to moderate yields of the cross-coupled products could result from protodebromination of the bromoolefins.
Return to citation in text: [1] -
Giri, R.; Hartwig, J. F. J. Am. Chem. Soc. 2010, 132, 15860–15863. doi:10.1021/ja105695s
Return to citation in text: [1] -
Tye, J. W.; Weng, Z.; Giri, R.; Hartwig, J. F. Angew. Chem., Int. Ed. 2010, 49, 2185–2189. doi:10.1002/anie.200902245
Return to citation in text: [1] -
Tye, J. W.; Weng, Z.; Johns, A. M.; Incarvito, C. D.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 9971–9983. doi:10.1021/ja076668w
Return to citation in text: [1] -
Strieter, E. R.; Bhayana, B.; Buchwald, S. L. J. Am. Chem. Soc. 2009, 131, 78–88. doi:10.1021/ja0781893
Return to citation in text: [1] -
Do, H.-Q.; Khan, R. M. K.; Daugulis, O. J. Am. Chem. Soc. 2008, 130, 15185–15192. doi:10.1021/ja805688p
Return to citation in text: [1] -
Schiefer, M.; Hatop, H.; Roesky, H. W.; Schmidt, H.-G.; Noltemeyer, M. Organometallics 2002, 21, 1300–1303. doi:10.1021/om010690v
Return to citation in text: [1] -
Pour, N.; Gofer, Y.; Major, D. T.; Keinan-Adamsky, K.; Gottlieb, H. E.; Aurbach, D. Organometallics 2013, 32, 3165–3173. doi:10.1021/om300865a
Return to citation in text: [1] -
Pour, N.; Gofer, Y.; Major, D. T.; Aurbach, D. J. Am. Chem. Soc. 2011, 133, 6270–6278. doi:10.1021/ja1098512
Return to citation in text: [1] -
Damrauer, R.; Krempp, M.; Damrauer, N. H.; Schmidt, M. W.; Gordon, M. S. J. Am. Chem. Soc. 1993, 115, 5218–5226. doi:10.1021/ja00065a038
Return to citation in text: [1] -
Krieck, S.; Görls, H.; Westerhausen, M. Organometallics 2008, 27, 5052–5057. doi:10.1021/om800509p
Return to citation in text: [1] [2] -
We analyzed a mixture of Ph3Al and LiCl using 27Al NMR. No significant change was observed in the 27Al NMR spectra. However, due to the strong Lewis acidic nature of Ph3Al, formation of a small amount of [Ph3AlCl]−[Li]+ cannot be ruled out without further experiments.
Return to citation in text: [1] -
Dyer, P. W.; Fawcett, J.; Hanton, M. J. Organometallics 2008, 27, 5082–5087. doi:10.1021/om8005933
Return to citation in text: [1] -
Brown, H. C.; Grayson, M. J. Am. Chem. Soc. 1953, 75, 20–24. doi:10.1021/ja01097a006
Return to citation in text: [1] -
Periasamy, M.; Seenivasaperumal, M.; Padmaja, M.; Rao, V. D. ARKIVOC 2004, No. viii, 4–11.
Return to citation in text: [1] -
Lindner, R.; van den Bosch, B.; Lutz, M.; Reek, J. N. H.; van der Vlugt, J. I. Organometallics 2011, 30, 499–510. doi:10.1021/om100804k
Return to citation in text: [1] -
Breitenfeld, J.; Wodrich, M. D.; Hu, X. Organometallics 2014, 33, 5708–5715. doi:10.1021/om500506y
Return to citation in text: [1] -
Uemura, M.; Yorimitsu, H.; Oshima, K. Chem. Commun. 2006, 4726–4728. doi:10.1039/b612173j
Return to citation in text: [1] -
Di Franco, T.; Boutin, N.; Hu, X. Synthesis 2013, 45, 2949–2958. doi:10.1055/s-0033-1338544
Return to citation in text: [1] -
Maruyama, K.; Noguchi-Yachide, T.; Sugita, K.; Hashimoto, Y.; Ishikawa, M. Bioorg. Med. Chem. Lett. 2010, 20, 6661–6666. doi:10.1016/j.bmcl.2010.09.011
Return to citation in text: [1] -
Lipshutz, B. H.; Blomgren, P. A.; Kim, S.-K. Tetrahedron Lett. 1999, 40, 197–200. doi:10.1016/S0040-4039(98)02271-0
Return to citation in text: [1] -
Liu, J.-H.; Yang, C.-T.; Lu, X.-Y.; Zhang, Z.-Q.; Xu, L.; Cui, M.; Lu, X.; Xiao, B.; Fu, Y.; Liu, L. Chem. – Eur. J. 2014, 20, 15334–15338. doi:10.1002/chem.201405223
Return to citation in text: [1] -
Kori, M.; Hamamura, K.; Fuse, H.; Yamamoto, T. JP Patent JP2002/000532, 2002; p 748.
Return to citation in text: [1] -
Dunsford, J. J.; Clark, E. R.; Ingleson, M. J. Angew. Chem., Int. Ed. 2015, 54, 5688–5692. doi:10.1002/anie.201411403
Return to citation in text: [1] -
Tobisu, M.; Takahira, T.; Chatani, N. Org. Lett. 2015, 17, 4352–4355. doi:10.1021/acs.orglett.5b02200
Return to citation in text: [1] -
Ellis-Davies, G. C. R.; Gilbert, A.; Heath, P.; Lane, J. C.; Warrington, J. V.; Westover, D. L. J. Chem. Soc., Perkin Trans. 2 1984, 1833–1841. doi:10.1039/p29840001833
Return to citation in text: [1]
| 64. | Di Franco, T.; Boutin, N.; Hu, X. Synthesis 2013, 45, 2949–2958. doi:10.1055/s-0033-1338544 |
| 65. | Maruyama, K.; Noguchi-Yachide, T.; Sugita, K.; Hashimoto, Y.; Ishikawa, M. Bioorg. Med. Chem. Lett. 2010, 20, 6661–6666. doi:10.1016/j.bmcl.2010.09.011 |
| 66. | Lipshutz, B. H.; Blomgren, P. A.; Kim, S.-K. Tetrahedron Lett. 1999, 40, 197–200. doi:10.1016/S0040-4039(98)02271-0 |
| 1. | Heck, R. F. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 833 ff. |
| 2. | Diederich, F.; Stang, P. J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: New York, 1998. |
| 3. | Negishi, E.-i.; Hu, Q.; Huang, Z.; Qian, M.; Wang, G. Aldrichimica Acta 2005, 38, 71. |
| 4. | Fu, G. C. Acc. Chem. Res. 2008, 41, 1555–1564. doi:10.1021/ar800148f |
| 5. | Knochel, P.; Jones, P., Eds. Organozinc Reagents: A Practical Approach; Oxford University Press: New York, 1999. |
| 6. | Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417–1492. doi:10.1021/cr100327p |
| 7. | Chang, W.-T. T.; Smith, R. C.; Regens, C. S.; Bailey, A. D.; Werner, N. S.; Denmark, S. E. Org. React. 2011, 75, 213–746. |
| 8. | Nakao, Y.; Hiyama, T. Chem. Soc. Rev. 2011, 40, 4893–4901. doi:10.1039/c1cs15122c |
| 9. | Sore, H. F.; Galloway, W. R. J. D.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 1845–1866. doi:10.1039/C1CS15181A |
| 19. | Koszinowski, K.; Böhrer, P. Organometallics 2009, 28, 771–779. doi:10.1021/om800947t |
| 43. | Cornelissen, L.; Cirriez, V.; Vercruysse, S.; Riant, O. Chem. Commun. 2014, 50, 8018–8020. doi:10.1039/c4cc02923b |
| 13. | Huo, S. Org. Lett. 2003, 5, 423–425. doi:10.1021/ol0272693 |
| 14. | Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442–4489. doi:10.1002/anie.200500368 |
| 16. | Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. J. Org. Chem. 2014, 79, 230–239. doi:10.1021/jo4024123 |
| 17. | Naka, H.; Uchiyama, M.; Matsumoto, Y.; Wheatley, A. E. H.; McPartlin, M.; Morey, J. V.; Kondo, Y. J. Am. Chem. Soc. 2007, 129, 1921–1930. doi:10.1021/ja064601n |
| 18. | Getmanenko, Y. A.; Twieg, R. J. J. Org. Chem. 2008, 73, 830–839. doi:10.1021/jo701812t |
| 19. | Koszinowski, K.; Böhrer, P. Organometallics 2009, 28, 771–779. doi:10.1021/om800947t |
| 20. | Lipshutz, B. H.; Bülow, G.; Lowe, R. F.; Stevens, K. L. Tetrahedron 1996, 52, 7265–7276. doi:10.1016/0040-4020(96)00250-5 |
| 21. | Blum, J.; Gelman, D.; Baidossi, W.; Shakh, E.; Rosenfeld, A.; Aizenshtat, Z.; Wassermann, B. C.; Frick, M.; Heymer, B.; Schutte, S.; Wernik, S.; Schumann, H. J. Org. Chem. 1997, 62, 8681–8686. doi:10.1021/jo970822n |
| 23. | Gurung, S. K.; Thapa, S.; Vangala, A. S.; Giri, R. Org. Lett. 2013, 15, 5378–5381. doi:10.1021/ol402701x |
| 24. | Thapa, S.; Gurung, S. K.; Dickie, D. A.; Giri, R. Angew. Chem., Int. Ed. 2014, 53, 11620–11624. doi:10.1002/anie.201407586 |
| 25. | Gurung, S. K.; Thapa, S.; Kafle, A.; Dickie, D. A.; Giri, R. Org. Lett. 2014, 16, 1264–1267. doi:10.1021/ol500310u |
| 13. | Huo, S. Org. Lett. 2003, 5, 423–425. doi:10.1021/ol0272693 |
| 14. | Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442–4489. doi:10.1002/anie.200500368 |
| 15. | Blümke, T. D.; Groll, K.; Karaghiosoff, K.; Knochel, P. Org. Lett. 2011, 13, 6440–6443. doi:10.1021/ol202733v |
| 45. | Cross-coupling of electron-deficient (4-FC6H4)3Al with benzyl bromide afforded the product only in 18% yield. |
| 71. | Ellis-Davies, G. C. R.; Gilbert, A.; Heath, P.; Lane, J. C.; Warrington, J. V.; Westover, D. L. J. Chem. Soc., Perkin Trans. 2 1984, 1833–1841. doi:10.1039/p29840001833 |
| 10. | Tsubouchi, A.; Muramatsu, D.; Takeda, T. Angew. Chem., Int. Ed. 2013, 52, 12719–12722. doi:10.1002/anie.201306882 |
| 11. | Thaler, T.; Knochel, P. Angew. Chem., Int. Ed. 2009, 48, 645–648. doi:10.1002/anie.200804446 |
| 12. | Krasovskiy, A.; Malakhov, V.; Gavryushin, A.; Knochel, P. Angew. Chem., Int. Ed. 2006, 45, 6040–6044. doi:10.1002/anie.200601450 |
| 46. | The conversion of bromoolefins is generally over 90%. The low to moderate yields of the cross-coupled products could result from protodebromination of the bromoolefins. |
| 23. | Gurung, S. K.; Thapa, S.; Vangala, A. S.; Giri, R. Org. Lett. 2013, 15, 5378–5381. doi:10.1021/ol402701x |
| 24. | Thapa, S.; Gurung, S. K.; Dickie, D. A.; Giri, R. Angew. Chem., Int. Ed. 2014, 53, 11620–11624. doi:10.1002/anie.201407586 |
| 25. | Gurung, S. K.; Thapa, S.; Kafle, A.; Dickie, D. A.; Giri, R. Org. Lett. 2014, 16, 1264–1267. doi:10.1021/ol500310u |
| 26. | Thapa, S.; Basnet, P.; Gurung, S. K.; Giri, R. Chem. Commun. 2015, 51, 4009–4012. doi:10.1039/C5CC00116A |
| 27. | Thapa, S.; Kafle, A.; Gurung, S. K.; Montoya, A.; Riedel, P.; Giri, R. Angew. Chem., Int. Ed. 2015, 54, 8236–8240. doi:10.1002/anie.201502379 |
| 40. | Thapa, S.; Shrestha, B.; Gurung, S. K.; Giri, R. Org. Biomol. Chem. 2015, 13, 4816–4827. doi:10.1039/C5OB00200A |
| 41. | Beletskaya, I. P.; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337–2364. doi:10.1016/j.ccr.2004.09.014 |
| 69. | Dunsford, J. J.; Clark, E. R.; Ingleson, M. J. Angew. Chem., Int. Ed. 2015, 54, 5688–5692. doi:10.1002/anie.201411403 |
| 22. | Dabrowski, J. A.; Villaume, M. T.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2013, 52, 8156–8159. doi:10.1002/anie.201304035 |
| 10. | Tsubouchi, A.; Muramatsu, D.; Takeda, T. Angew. Chem., Int. Ed. 2013, 52, 12719–12722. doi:10.1002/anie.201306882 |
| 35. | Terao, J.; Todo, H.; Begum, S. A.; Kuniyasu, H.; Kambe, N. Angew. Chem., Int. Ed. 2007, 46, 2086–2089. doi:10.1002/anie.200603451 |
| 42. | Yang, C.-T.; Zhang, Z.-Q.; Liu, Y.-C.; Liu, L. Angew. Chem., Int. Ed. 2011, 50, 3904–3907. doi:10.1002/anie.201008007 |
| 43. | Cornelissen, L.; Cirriez, V.; Vercruysse, S.; Riant, O. Chem. Commun. 2014, 50, 8018–8020. doi:10.1039/c4cc02923b |
| 44. | The reaction of Ph3Al with 1-iodoocctane using the Schlenk technique afforded cross-coupled product 3 in 72% yield. |
| 70. | Tobisu, M.; Takahira, T.; Chatani, N. Org. Lett. 2015, 17, 4352–4355. doi:10.1021/acs.orglett.5b02200 |
| 15. | Blümke, T. D.; Groll, K.; Karaghiosoff, K.; Knochel, P. Org. Lett. 2011, 13, 6440–6443. doi:10.1021/ol202733v |
| 67. | Liu, J.-H.; Yang, C.-T.; Lu, X.-Y.; Zhang, Z.-Q.; Xu, L.; Cui, M.; Lu, X.; Xiao, B.; Fu, Y.; Liu, L. Chem. – Eur. J. 2014, 20, 15334–15338. doi:10.1002/chem.201405223 |
| 21. | Blum, J.; Gelman, D.; Baidossi, W.; Shakh, E.; Rosenfeld, A.; Aizenshtat, Z.; Wassermann, B. C.; Frick, M.; Heymer, B.; Schutte, S.; Wernik, S.; Schumann, H. J. Org. Chem. 1997, 62, 8681–8686. doi:10.1021/jo970822n |
| 10. | Tsubouchi, A.; Muramatsu, D.; Takeda, T. Angew. Chem., Int. Ed. 2013, 52, 12719–12722. doi:10.1002/anie.201306882 |
| 28. | Thathagar, M. B.; Beckers, J.; Rothenberg, G. J. Am. Chem. Soc. 2002, 124, 11858–11859. doi:10.1021/ja027716+ |
| 29. | Li, J.-H.; Li, J.-L.; Wang, D.-P.; Pi, S.-F.; Xie, Y.-X.; Zhang, M.-B.; Hu, X.-C. J. Org. Chem. 2007, 72, 2053–2057. doi:10.1021/jo0623742 |
| 30. | Zhou, Y.; You, W.; Smith, K. B.; Brown, M. K. Angew. Chem., Int. Ed. 2014, 53, 3475–3479. doi:10.1002/anie.201310275 |
| 31. | You, W.; Brown, M. K. J. Am. Chem. Soc. 2014, 136, 14730–14733. doi:10.1021/ja509056j |
| 32. | Ito, H.; Sensui, H.-o.; Arimoto, K.; Miura, K.; Hosomi, A. Chem. Lett. 1997, 26, 639–640. doi:10.1246/cl.1997.639 |
| 33. | Cornelissen, L.; Lefrancq, M.; Riant, O. Org. Lett. 2014, 16, 3024–3027. doi:10.1021/ol501140p |
| 34. | Yang, C.-T.; Zhang, Z.-Q.; Liang, J.; Liu, J.-H.; Lu, X.-Y.; Chen, H.-H.; Liu, L. J. Am. Chem. Soc. 2012, 134, 11124–11127. doi:10.1021/ja304848n |
| 35. | Terao, J.; Todo, H.; Begum, S. A.; Kuniyasu, H.; Kambe, N. Angew. Chem., Int. Ed. 2007, 46, 2086–2089. doi:10.1002/anie.200603451 |
| 36. | Cahiez, G.; Gager, O.; Buendia, J. Angew. Chem., Int. Ed. 2010, 49, 1278–1281. doi:10.1002/anie.200905816 |
| 37. | Mohapatra, S.; Bandyopadhyay, A.; Barma, D. K.; Capdevila, J. H.; Falck, J. R. Org. Lett. 2003, 5, 4759–4762. doi:10.1021/ol035458v |
| 38. | Allred, G. D.; Liebeskind, L. S. J. Am. Chem. Soc. 1996, 118, 2748–2749. doi:10.1021/ja9541239 |
| 39. | Li, J.-H.; Tang, B.-X.; Tao, L.-M.; Xie, Y.-X.; Liang, Y.; Zhang, M.-B. J. Org. Chem. 2006, 71, 7488–7490. doi:10.1021/jo061220j |
| 68. | Kori, M.; Hamamura, K.; Fuse, H.; Yamamoto, T. JP Patent JP2002/000532, 2002; p 748. |
| 11. | Thaler, T.; Knochel, P. Angew. Chem., Int. Ed. 2009, 48, 645–648. doi:10.1002/anie.200804446 |
| 15. | Blümke, T. D.; Groll, K.; Karaghiosoff, K.; Knochel, P. Org. Lett. 2011, 13, 6440–6443. doi:10.1021/ol202733v |
| 43. | Cornelissen, L.; Cirriez, V.; Vercruysse, S.; Riant, O. Chem. Commun. 2014, 50, 8018–8020. doi:10.1039/c4cc02923b |
| 47. | Giri, R.; Hartwig, J. F. J. Am. Chem. Soc. 2010, 132, 15860–15863. doi:10.1021/ja105695s |
| 48. | Tye, J. W.; Weng, Z.; Giri, R.; Hartwig, J. F. Angew. Chem., Int. Ed. 2010, 49, 2185–2189. doi:10.1002/anie.200902245 |
| 49. | Tye, J. W.; Weng, Z.; Johns, A. M.; Incarvito, C. D.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 9971–9983. doi:10.1021/ja076668w |
| 50. | Strieter, E. R.; Bhayana, B.; Buchwald, S. L. J. Am. Chem. Soc. 2009, 131, 78–88. doi:10.1021/ja0781893 |
| 51. | Do, H.-Q.; Khan, R. M. K.; Daugulis, O. J. Am. Chem. Soc. 2008, 130, 15185–15192. doi:10.1021/ja805688p |
| 52. | Schiefer, M.; Hatop, H.; Roesky, H. W.; Schmidt, H.-G.; Noltemeyer, M. Organometallics 2002, 21, 1300–1303. doi:10.1021/om010690v |
| 53. | Pour, N.; Gofer, Y.; Major, D. T.; Keinan-Adamsky, K.; Gottlieb, H. E.; Aurbach, D. Organometallics 2013, 32, 3165–3173. doi:10.1021/om300865a |
| 54. | Pour, N.; Gofer, Y.; Major, D. T.; Aurbach, D. J. Am. Chem. Soc. 2011, 133, 6270–6278. doi:10.1021/ja1098512 |
| 55. | Damrauer, R.; Krempp, M.; Damrauer, N. H.; Schmidt, M. W.; Gordon, M. S. J. Am. Chem. Soc. 1993, 115, 5218–5226. doi:10.1021/ja00065a038 |
| 56. | Krieck, S.; Görls, H.; Westerhausen, M. Organometallics 2008, 27, 5052–5057. doi:10.1021/om800509p |
| 57. | We analyzed a mixture of Ph3Al and LiCl using 27Al NMR. No significant change was observed in the 27Al NMR spectra. However, due to the strong Lewis acidic nature of Ph3Al, formation of a small amount of [Ph3AlCl]−[Li]+ cannot be ruled out without further experiments. |
| 62. | Breitenfeld, J.; Wodrich, M. D.; Hu, X. Organometallics 2014, 33, 5708–5715. doi:10.1021/om500506y |
| 63. | Uemura, M.; Yorimitsu, H.; Oshima, K. Chem. Commun. 2006, 4726–4728. doi:10.1039/b612173j |
| 60. | Periasamy, M.; Seenivasaperumal, M.; Padmaja, M.; Rao, V. D. ARKIVOC 2004, No. viii, 4–11. |
| 61. | Lindner, R.; van den Bosch, B.; Lutz, M.; Reek, J. N. H.; van der Vlugt, J. I. Organometallics 2011, 30, 499–510. doi:10.1021/om100804k |
| 58. | Dyer, P. W.; Fawcett, J.; Hanton, M. J. Organometallics 2008, 27, 5082–5087. doi:10.1021/om8005933 |
| 59. | Brown, H. C.; Grayson, M. J. Am. Chem. Soc. 1953, 75, 20–24. doi:10.1021/ja01097a006 |
| 34. | Yang, C.-T.; Zhang, Z.-Q.; Liang, J.; Liu, J.-H.; Lu, X.-Y.; Chen, H.-H.; Liu, L. J. Am. Chem. Soc. 2012, 134, 11124–11127. doi:10.1021/ja304848n |
| 35. | Terao, J.; Todo, H.; Begum, S. A.; Kuniyasu, H.; Kambe, N. Angew. Chem., Int. Ed. 2007, 46, 2086–2089. doi:10.1002/anie.200603451 |
| 56. | Krieck, S.; Görls, H.; Westerhausen, M. Organometallics 2008, 27, 5052–5057. doi:10.1021/om800509p |
© 2015 Shrestha and Giri; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)