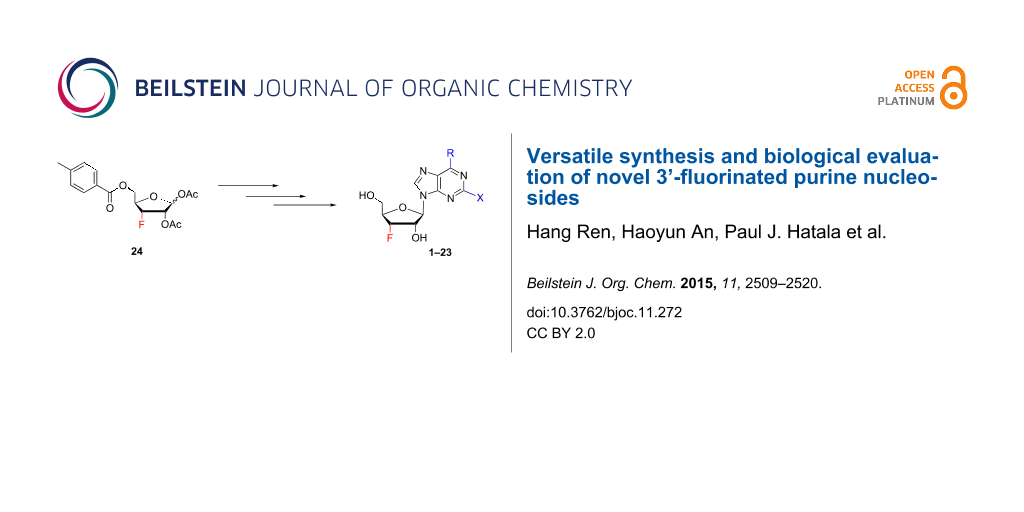
Abstract
A unified synthetic strategy accessing novel 3'-fluorinated purine nucleoside derivatives and their biological evaluation were achieved. Novel 3’-fluorinated analogues were constructed from a common 3’-deoxy-3’-fluororibofuranose intermediate. Employing Suzuki and Stille cross-coupling reactions, fifteen 3’-fluororibose purine nucleosides 1–15 and eight 3’-fluororibose 2-chloro/2-aminopurine nucleosides 16–23 with various substituents at position 6 of the purine ring were efficiently synthesized. Furthermore, 3’-fluorine analogs of natural products nebularine and 6-methylpurine riboside were constructed via our convergent synthetic strategy. Synthesized nucleosides were tested against HT116 (colon cancer) and 143B (osteosarcoma cancer) tumor cell lines. We have demonstrated 3’-fluorine purine nucleoside analogues display potent tumor cell growth inhibition activity at sub- or low micromolar concentration.

Graphical Abstract
Introduction
Antimetabolites are extremely useful for the treatment of cancers and viral infections and are one of the largest classes of drugs. Most antimetabolite drugs are nucleoside derivatives that substitute for endogenous nucleosides and prevent DNA and protein replication [1]. Many of the drugs described by the World Health Organization as "essential medicines" are nucleoside derivatives [2] and nearly 20% of all drugs for the treatment of cancers are nucleoside derivatives [3]. The design of new antimetabolites is an active field of research and several nucleoside derivatives have recently come to market such as gemcitabine, capecitabine, and decitabine [4-6]. Purine nucleoside analogues such as fludarabine, nelarabine, cladribine, and clofarabine are an important subset of nucleoside drugs [7-10]. Purine ribonucleosides substituted at position 6 have exhibited potent antimetabolite activity [11-13] and aryl or heterocyclic substituents have imparted cytostatic activities against various tumor cell-lines [14-16]. Moreover, some 6-heterocyclic substituted purine ribonucleosides also demonstrate strong antiviral activities [17]. Purine derivatives such as, 2’-β-C-methyl-6-substituted purine nucleosides exhibit promising anti-HCV activity by blocking RNA dependent RNA polymerase [18-20]. Design and synthesis of purine-based nucleosides are still needed to enable new therapies for the treatment of drug-resistant tumors and viruses.
One of the first antimetabolite drugs rationally designed from biochemical data was 5-fluorouracil. A hydrogen atom on uracil was replaced with a fluorine atom for specific reasons. The fluorine atom possesses unique characteristics; it enhances the lipophilicity of organic compounds and C–F bonds have low chemical reactivity imparting high enzymatic stability and resistance to metabolic processes. The high electronegativity of fluorine and the lipophilicity it imparts improve the bioavailability of fluorine-containing drugs. Relative to the unfluorinated derivative, fluorinated drugs have demonstrated favorable pharmacological, physicochemical, pharmacokinetic, pharmacodynamic and safety profiles for a number of compounds [21-23]. Several blockbuster drugs such as Lipitor®, Seretide®, Crestor®, Takepron®, Sustiva®, Celebrex®, and the recently described fluorapacin and azvudine [24-29], all contain fluorine atom(s). Not surprisingly, 20% of marketed drugs contain fluorine atom(s). Gemcitabine, the 2’-deoxy-2’,2’-difluorocytidine has been routinely utilized to treat solid tumors [30]. However, 3’-fluorine-modified nucleosides have not been well-studied because of the challenges associated with the synthesis of modified carbohydrate moieties [31-35]. As a result, 3’-fluoro-6-heterocyclic-substituted purine nucleosides are not well represented in the literature. We chose to explore the biological potential of synthetic purine analogues combining substitutions at position 6 and a 3’-fluorine. Novel 6-heterocyclic substituted purine 3’-deoxy-3’-fluororibonucleosides were designed to discover more selective and potent novel antiviral and anticancer therapeutics. Various fluorine-modified ribonucleoside derivatives were designed, synthesized, and tested. The preliminary results are presented herein.
We utilized the structural characteristics of 3’-fluorine and 6-substituted purine nucleosides to expand their biological application. Herein, we report the synthesis of twenty three 3’-deoxy-3’-fluoro-6-modified purine nucleoside derivatives, of which twenty one are novel compounds, including 6-substituted purine 3’-deoxy-3’-fluororibosides 1–15 (Figure 1), 2-chloro-6-substituted purine 3’-deoxy-3’-fluororibosides 16–20 and 2-amino-6-substituted purine 3’-deoxy-3’-fluororiboses 21–23 (Figure 2). In addition, their anticancer activity was evaluated.
![[1860-5397-11-272-1]](/bjoc/content/figures/1860-5397-11-272-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 6-Subsituted purine 3’-deoxy-3’-fluororibosides 1–15.
Figure 1: 6-Subsituted purine 3’-deoxy-3’-fluororibosides 1–15.
![[1860-5397-11-272-2]](/bjoc/content/figures/1860-5397-11-272-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 2-Chloro- and 2-aminopurine 3’-deoxy-3’-fluororibosides 16–23.
Figure 2: 2-Chloro- and 2-aminopurine 3’-deoxy-3’-fluororibosides 16–23.
Results and Discussion
Current strategies to the synthesis of 3’-fluorine and 6-substituted purine require harsh conditions and laborious protecting group manipulation that results in low product yields. It was reported that 3’-deoxy-3’-fluoroadenosine (2) (Figure 1) was synthesized in 3.5% yield starting from adenosine [31,32]. This eight-step synthesis required harsh reaction conditions and HPLC purification of the final product. In addition, the product cannot be utilized for further derivatization to reach our objectives. This 3’-fluorine-modified adenosine 2 has also been synthesized starting from a well-protected xylofuranosyladenine derivative using a complicated strategy [33]. Similarly, 3’-deoxy-3’-fluoroguanosine (23, Figure 2) was isolated in 2% yield after protecting group manipulation from arabinoguanosine [34]. Complicated orthogonally protected adenosine and guanosine derivatives with three or four different protecting groups have also been used for the synthesis of compounds 2 and 23, and the protocols required extensive manipulation of the protecting groups [35]. Compound 2 has also been synthesized starting from xylofuranoside by manipulating the protecting groups on the carbohydrate moiety [36]. De Clercq and co-workers [37] developed a protocol for the synthesis of 3’-fluororibofuranose in 10 steps, and it requires epoxide formation and ring opening as well as reversion of the hydroxy group on the sugar ring. Jeong and co-workers [38-40] synthesized fluorine-substituted ribofuranose but isolation required a challenging separation. Therefore, the synthetic challenges for preparing 3’-fluorine modified sugars and nucleosides have limited the synthesis and biological testing of these promising fluorine modified nucleoside derivatives. To open up research and therapeutic exploration of this class of compounds, we developed efficient synthetic routes for the construction of 3’-deoxy-3’-fluoroadenosine and guanosine derivatives with a number of different modifications on position 6.
Substituted purine 3’-deoxy-3’-fluoro-β-D-ribofuranosyl nucleosides were successfully constructed from the universal intermediate 1’,2’-di-O-acetyl-5’-O-p-toluyl-3’-fluoro-3’-deoxy-β-D-ribofuranose (25, Figure 3). The final 3’-deoxy-3’-fluoro-β-D-ribofuranose derivatives 1–23 (Figure 1 and Figure 2) were derivatized from the corresponding intermediates 26, 29, 30–40, 42, 48 and 51. All of these compounds are novel derivatives except compounds 2 and 23. The syntheses of these compounds are outlined in Schemes 1–5. 1,2-O-Isopropylidene-5-O-(4-methylbenzoyl)-α-D-xylofuranose (24) was synthesized from D-xylose according to literature procedures [41]. Compound 24 was treated with iodine in methanol, and fluorinated with diethylaminosulfur trifluoride (DAST). The universal intermediate 1’,2’-di-O-acetyl-5’-O-p-toluyl-3’-fluoro-3’-deoxy-β-D-ribofuranose (25) was then obtained in 33.13% overall yield after further treatment with acetic anhydride–acetic acid–sulfuric acid system. More than 200 g scale was achieved for the synthesis, and high purity (98%) product was obtained. This key intermediate 25 can be used to synthesize a variety of 3’-fluoro-modified nucleoside derivatives, and it was utilized for the synthesis of all analogues reported herein. This strategy avoids tedious orthogonal protecting group manipulations previously reported in literature [31-35]. Our strategy provides the desired nucleoside intermediates and also opens up the opportunity for modification on any class of nucleosides with a 3’-fluorine atom to explore their biological and therapeutic potential. While this work is related with purine nucleosides, the strategy can be used for the synthesis of a variety of nucleosides with a wide range of heterocyclic moieties to investigate the impact of a 3’-fluorine atom on the biological activity of nucleosides.
![[1860-5397-11-272-3]](/bjoc/content/figures/1860-5397-11-272-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 3’-Deoxy-3’-fluororibosides constructed from universal intermediate 25.
Figure 3: 3’-Deoxy-3’-fluororibosides constructed from universal intermediate 25.
The 6-chloropurine was silylated with trimethylsilyl triflate and then glycosylated with the universal intermediate 3’-fluoro-3’-deoxy-β-D-ribofuranose 25 using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to provide the desired protected key intermediate 26 in 90% yield (Scheme 1). To construct the first series of fluorinated purine analogues, compound 26 was treated with a saturated solution of ammonia in methanol, which resulted in the amination at the 6-position and deprotection of the protecting groups to furnish 3’-deoxy-3’-fluoroadenosine (2) in 85% yield. Our synthetic strategy provided compound 2 in excellent yield (76%, 2 steps) compared to previously reported literature protocols (3.5%, 8 steps) [31,32]. The 6-chlorine of compound 26 was replaced by hydroxylamine, with concomitant removal of the protecting groups to yield N6-hydroxy-3’-fluoro-3’-deoxyadenosine (3). Hydrogenation of compound 26 under hydrogen pressure (50 psi) over 10% Pd/C resulted in the de-chlorinated compound 27, which was further deprotected in a saturated solution of ammonia in methanol providing the desired 6-deaminated 3’-fluoro-adenosine 1 in 93% yield. We targeted 9-(3-deoxy-3-fluoro-β-D-ribofuranosyl)purine (1) in particular because it is the 3’-fluorine analogue of nebularine, a naturally occurring antibacterial and antineoplastic agent [42,43].
![[1860-5397-11-272-i1]](/bjoc/content/inline/1860-5397-11-272-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3’-deoxy-3’-fluoropurine ribosides 1–3.
Scheme 1: Synthesis of 3’-deoxy-3’-fluoropurine ribosides 1–3.
3’-Fluoro-6-methylpurine riboside 4, a 6-β-D-MPR mimic
6-Methylpurine-β-D-riboside (6-β-D-MPR) is an isolated antibiotic agent that possesses potent antifungal, antiviral, and antitumor activities [44]. In order to explore the effect of fluorine on the biological activity of this pharmacophore, we synthesized 6-methylpurine-3’-deoxy-3’-fluoro-β-D-riboside (4) (Scheme 2). 6-Methylpurine (28) was synthesized from 6-chloropurine according to the reported protocol [44]. Compound 28 was silylated with BSA and glycosylated with the 3’-fluoro-sugar 25 to provide the desired compound 29 in 78% yield. Subsequent deprotection furnished the targeted novel fluorine modified 6-methylpurine riboside 4 in 80% yield.
![[1860-5397-11-272-i2]](/bjoc/content/inline/1860-5397-11-272-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 6-methylpurine 3’-deoxy-3’-fluororiboside 4.
Scheme 2: Synthesis of 6-methylpurine 3’-deoxy-3’-fluororiboside 4.
3’-Fluoro-6-C-substituted purine nucleosides
The 6-C-substituted purine nucleosides have demonstrated anticancer, antiviral and other biological activities [11-20]. In order to explore the effect of fluorine on the biological activity of 6-C-substituted purine nucleosides, we designed and synthesized 3’-deoxy-3’-fluoro-β-D-ribofuranosyl purine derivatives 5–15 with various aromatic and heterocyclic moieties at position 6 of the purine base from the chlorine intermediate 26 (Scheme 3).
![[1860-5397-11-272-i3]](/bjoc/content/inline/1860-5397-11-272-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 6-substituted purine 3’-deoxy-3’-fluororibosides 5–15.
Scheme 3: Synthesis of 6-substituted purine 3’-deoxy-3’-fluororibosides 5–15.
The synthesis of 3’-deoxy-3’-fluoro-β-D-ribofuranosyl purine derivatives began with a palladium-catalyzed cross coupling [45,46] to install the aromatic moieties to the 6-position of the purine ring. In order to accomplish efficient cross-couplings of a wide range of aromatic rings, we utilized three different protocols that were employed for Stille and Suzuki reaction conditions. To this end, 2-(tributylstannyl)furan was coupled with 6-chloropurine nucleoside 26 by Stille cross coupling [47,48] catalyzed by bis(triphenylphosphine)palladium(II) chloride in DMF (Method I) (Scheme 3). The resulting 6-aryl compound 30 was obtained in 91% yield and deprotected with a saturated solution of ammonia in methanol to give the desired analogue 5 with furan-2-yl substituent at position 6 of the purine base. Considering the possible toxicity of organostannyl reagents, we then utilized various organoboronic acids for Suzuki cross couplings [49-51] to synthesize compounds 6–15. The 6-chloropurine intermediate 26 was coupled with 3-thienylboronic acid and phenylboronic acid catalyzed by tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) in toluene (Method II), resulting in the desired protected intermediates 31 and 32 in 56% and 52% yields, respectively. Suzuki coupling efficiency in DMF (entries 2 and 3, Table 1) was not as high as Stille coupling (entry 1, Table 1). Suzuki coupling of intermediate 26 with 1-naphthylboronic acid, pyridine-4-boronic acid, pyridine-3-boronic acid and 5-propynylpyridine-3-boronic acid (Method II) (entries 4, 6, 8, and 10, Table 1) provided no detectable product. After screening various solvent systems, we discovered that dimethoxyethane (DME)–water was the optimal solvent system (Method III), and achieved construction of 6-aryl compounds 33–36 (entries 5, 7, 9 and 11, Table 1), albeit in low to moderate yields. Next, substituted pyridine boronic acids [52-54] were coupled with intermediate 26 using Method III in DME–water to provide the desired 6-aryl products 37–40 (entries 12–15, Table 1). These results indicated that the DME–water solvent system was more favourable for more challenging Suzuki C–C coupling reactions on the 6-chloropurine nucleosides. Intermediate compounds 31–40 were deprotected with a saturated solution of ammonia in methanol to furnish the desired products 6–15 in 70–92% yields (Table 1).
Table 1: C–C bond formation by Stille and Suzuki cross-coupling.
| Entry | Methoda | Reagents | Coupling product (yield) | Final product (yield) |
|---|---|---|---|---|
| 1 | I |
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-272-i6.svg?max-width=637&scale=1.0)
|
30 (91%) | 5 (70%) |
| 2 | II |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-272-i7.svg?max-width=637&scale=1.0)
|
31 (56%) | 6 (92%) |
| 3 | II |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-272-i8.svg?max-width=637&scale=1.0)
|
32 (52%) | 7 (84%) |
| 4 | II |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-272-i9.svg?max-width=637&scale=1.0)
|
33 (no product) | – |
| 5 | III | 33 (57%) | 8 (92%) | |
| 6 | II |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-272-i10.svg?max-width=637&scale=1.0)
|
34 (no product) | – |
| 7 | III | 34 (32%) | 9 (89%) | |
| 8 | II |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-272-i11.svg?max-width=637&scale=1.0)
|
35 (no product) | – |
| 9 | III | 35 (29%) | 10 (84%) | |
| 10 | II |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-272-i12.svg?max-width=637&scale=1.0)
|
36 (no product) | – |
| 11 | III | 36 (17%) | 11 (70%) | |
| 12 | III |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-272-i13.svg?max-width=637&scale=1.0)
|
37 (21%) | 12 (77%) |
| 13 | III |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-272-i14.svg?max-width=637&scale=1.0)
|
38 (46%) | 13 (87%) |
| 14 | III |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-272-i15.svg?max-width=637&scale=1.0)
|
39 (49%) | 14 (90%) |
| 15 | III |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-272-i16.svg?max-width=637&scale=1.0)
|
40 (16%) | 15 (72%) |
| 16 | I |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-272-i17.svg?max-width=637&scale=1.0)
|
43 (24%) | 17 (85%) |
| 17 | II |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-272-i18.svg?max-width=637&scale=1.0)
|
44 (16%) | 18 (70%) |
| 18 | II |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-272-i19.svg?max-width=637&scale=1.0)
|
45 (21%) | 19 (72%) |
| 19 | III |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-272-i20.svg?max-width=637&scale=1.0)
|
46 (22%) | 20 (71%) |
aMethod I: (PPh3)2Pd(II)Cl2, DMF; Method II: Pd(PPh3)4, toluene; Method III: Pd(PPh3)4, DME–water.
3’-Fluoro-2-chloropurine nucleosides – cladribine and clofarabine mimics
Anticancer drugs cladribine and clofarabine [55-57] are purine nucleoside derivatives having a chlorine atom at position 2 of the purine moiety. With this in mind, we designed and synthesized 2-chloropurine nucleosides 16–20 (Figure 2 and Scheme 4) with 3’-fluorine and 6-C-aromatic or heterocyclic modifications to explore a broad biological space for this class of nucleoside derivatives. The 2,6-dichloropurine (41) was glycosylated with the 3’-fluororibose intermediate 25 to furnish the 2,6-dichloropurine intermediate 42 in 89% yield (Scheme 4). Chemoselective amination of the 6-position over the 2-position of the purine and deprotection of 2,6-dichloropurine 42 was achieved with a saturated solution of ammonia in methanol to furnish 2-chloro-3’-deoxy-3’-fluoroadenosine (16). The 2,6-dichloro-intermediate 42 was coupled with 2-(tributylstannyl)furan catalyzed by bis(triphenylphosphine)palladium(II) chloride in DMF to provide monochloro-intermediate 43 (Method I, entry 16, Table 1). Monochloro-intermediates 44 and 45 were constructed by Suzuki coupling of dichloro-intermediate 42 with 3-thienylboronic acid and phenylboronic acid in toluene (Method II; entries 17 and 18, Table 1), and monochloro-intermediate 46 was synthesized by coupling of 42 with 1-naphthylboronic acid in DME–water (Method III; entry 19, Table 1). The resulted coupling products 43–46 were deprotected with a saturated solution of ammonia in methanol to provide corresponding final products 17–20 in 70–90% yields.
![[1860-5397-11-272-i4]](/bjoc/content/inline/1860-5397-11-272-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 6-substituted 2-chloropurine 3’-deoxy-3’-fluororibosides 16–20.
Scheme 4: Synthesis of 6-substituted 2-chloropurine 3’-deoxy-3’-fluororibosides 16–20.
Of particular importance, the 2-chlorine atom on the intermediate 42 was stable under the Stille and Suzuki reaction conditions and under ammonia deprotection conditions. However, the palladium-catalyzed cross coupling of dichloro-intermediate 42 with organostannane and organoboronic acid reagents resulted in lower yields when compared to the cross coupling of monochloro-intermediate 26. From the amination studies of 2,6-dichloropurines, the 6-position of the purine possesses higher reactivity towards nucleophiles than the 2-position. In addition, the selectivity for the 6-position is also higher for the Stille than for the Suzuki cross coupling. The lower yields obtained from the cross-coupling of 2,6-dichloropurines is most likely contributed to the 2-chlorine reducing the reactivity of the 6-chlorine, or undesired cross-coupled products at the 2-position of the purine that may have resulted, but were not isolated.
3’-Fluoro-2-aminopurine nucleosides
As mentioned above, chloropurine [55-57] and deaminopurine [42,43] nucleosides have exhibited potent biological activities. We synthesized 2-amino-6-chloropurine 3’-deoxy-3’-fluororiboside 22 and 2-aminopurine-3’-deoxy-3’-fluororiboside 21 to explore the SAR of 3’-fluorine-substituted purine nucleoside derivatives (Scheme 5). The 2-amino-6-chloropurine (47) was glycosylated with the 3’-fluorine riboside 25 under DBU and trimethylsilyl triflate conditions. The corresponding protected intermediate nucleoside 48 was obtained in 78% yield and utilized for further derivatization. Deprotection of compound 48 with a saturated solution of ammonia in methanol at 0 °C resulted in the desired product 22 in 85% yield. In addition, the 6-chlorine atom was left untouched during the deprotection. Compound 48 was hydrogenated over 10% Pd/C under hydrogen pressure (50 psi) giving the de-chlorinated compound 49, which was deprotected under ammonia treatment to complete the desired final product 21. In order to further explore the SAR of this pharmacophore, we constructed 3’-deoxy-3’-fluoroguanosine (23, Scheme 5). N2-Acetyl-6-O-(diphenylcarbamoyl)guanine (50) was prepared according to reported protocol [58,59]. Compound 50 was silylated and glycosylated with 3’-fluorine riboside 25 providing the fully protected intermediate 51 in 72% yield. The final product 23 was obtained in 96% yield after global deprotection of 51 using a saturated solution of ammonia in methanol. This protocol of direct glycosylation for the synthesis of 3’-fluorine modified guanosine derivatives is highly advantageous compared to the previously reported methods utilizing orthogonal protecting groups, selective deprotection, and fluorination of the starting guanosine [34].
![[1860-5397-11-272-i5]](/bjoc/content/inline/1860-5397-11-272-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 2-aminopurine 3’-deoxy-3’-fluororibosides 21–23.
Scheme 5: Synthesis of 2-aminopurine 3’-deoxy-3’-fluororibosides 21–23.
Biological evaluation
Newly synthesized compounds were tested against the human colon cancer cell line HCT-116 and the human osteosarcoma cancer cell line 143B (Table 2). The fluorinated nebularine analog 1 and its protected analog 27 demonstrated potent tumor cell growth inhibition at sub-micromolar concentration. The protected 6-methylpurine riboside 29 exhibited better antitumor activity than its deprotected analog 4, the 3’-fluoro-6-MPR analog. The 6-chlorine compound 22, as well as protected purine nucleosides 32, 39 and 40, also showed potent inhibitory activity. 3’-Fluorine purine nucleosides 2–4, 12, 30, and 49 showed moderate levels of inhibitory activity, but other derivatives did not show detectable activity against the tested tumor cell lines. Antiviral and other biological evaluation of these 3’-fluorine modified nucleosides is in progress and will be reported in due course.
Table 2: Antitumor activity of newly synthesized compounds.
| Compound | Cytotoxicity IC50 (μM) | Compound | Cytotoxicity IC50 (μM) | ||
|---|---|---|---|---|---|
| HCT116 | 143B | HCT116 | 143B | ||
| 1 | 0.5–1.0 | 0.5–1.0 | 22 | 0.5–1.0 | 0.5–1.0 |
| 2 | 1.0–5.0 | 1.0–5.0 | 27 | 0.5–1.0 | 0.5–1.0 |
| 3 | 5.0–10 | 5.0–10 | 29 | 0.5–1.0 | 0.5–1.0 |
| 4 | 5.0–10 | 5.0–10 | 30 | 1.0–5.0 | 1.0–5.0 |
| 5 | >10 | >10 | 31 | >10 | >10 |
| 6 | >10 | >10 | 32 | 0.5–1.0 | 0.5–1.0 |
| 7 | >20 | >20 | 33 | >20 | >30 |
| 8 | >20 | >20 | 34 | >10 | >10 |
| 9 | >10 | >10 | 35 | >10 | >10 |
| 10 | >10 | >10 | 36 | >20 | >20 |
| 11 | >20 | >20 | 37 | >20 | >20 |
| 12 | 1.0–5.0 | 1.0–5.0 | 38 | >10 | >10 |
| 13 | >10 | >10 | 39 | 0.5–1.0 | 0.5–1.0 |
| 14 | >10 | >10 | 40 | 0.5–5.0 | 0.5–5.0 |
| 15 | >20 | >20 | 43 | >10 | >10 |
| 16 | >10 | >10 | 44 | >10 | >10 |
| 17 | >10 | >10 | 45 | >20 | >20 |
| 18 | >10 | >10 | 46 | >20 | >20 |
| 19 | >20 | >20 | 49 | 1.0–5.0 | 1.0–5.0 |
| 20 | >20 | >20 | Camptothecin | 0.30 | 1.20 |
Conclusion
The 6-chloropurine, 2,6-dichloropurine, and 2-amino-6-chloropurine were glycosylated with the protected 3’-deoxy-3’-fluororibose 25 to provide the corresponding key intermediates 26, 42, and 48. These intermediates were then further derivatized to furnish final products 1–3 and 5–23. The glycosylation of 6-methylpurine 28 with 25 furnished 3’-deoxy-3’-fluoro-6-methylpurine riboside 4, the analogue of biologically active natural product, 6-methylpurine-β-D-riboside (6-β-D-MPR). We constructed 3’-deoxy-3’-fluororibofuranosylpurine nucleosides 5–11 with various aromatic and heterocyclic moieties at position 6 from the key intermediate 26 by Stille and Suzuki cross-coupling reactions. 3’-Deoxy-3’-fluororibofuranosyl 2-chloropurine nucleosides 17–20 were synthesized by the similar strategy from the key intermediate 42. DME–water was found to be an efficient solvent system for Suzuki reaction on challenging substrates. De-chlorination of the 6-chloro-intermediates 26 and 48, followed by deprotection, resulted in 6-deaminopurine nucleosides 1 and 21, new analogues of the naturally isolated antibacterial and antineoplastic agent 6-deaminoadenosine, nebularine. Newly synthesized compounds were evaluated for their antitumor activity. Eleven compounds exhibited potent tumour cell growth inhibition activity against the human colon tumor cell line HCT116 and the human osteosarcoma cell line 143B at sub- or low micromolar concentration.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and 1H NMR and mass spectral data for new compounds. | ||
| Format: PDF | Size: 3.5 MB | Download |
References
-
Henry, J. R.; Mader, M. M. Recent Advances in Antimetabolite Cancer Chemotherapies. In Annual Reports in Medicinal Chemistry; Doherty, A. M., Ed.; Elsevier: San Diego, CA, U.S.A., 2004; Vol. 39, pp 159–172. doi:10.1016/s0065-7743(04)39013-5
Return to citation in text: [1] -
19th WHO Model List of Essential Medicines. WHO, 2015; http://www.who.int/medicines/publications/essentialmedicines/EML2015_8-May-15.pdf (accessed July 8, 2015).
Return to citation in text: [1] -
Parker, W. B. Chem. Rev. 2009, 109, 2880–2893. doi:10.1021/cr900028p
Return to citation in text: [1] -
Johnson, S. A.; Thomas, W. Hematol. Oncol. 2000, 18, 141–153. doi:10.1002/1099-1069(200012)18:4<141::AID-HON666>3.0.CO;2-#
Return to citation in text: [1] -
Johnson, S. A. Expert Opin. Pharmacother. 2001, 2, 929–943. doi:10.1517/14656566.2.6.929
Return to citation in text: [1] -
Parker, W. B.; Secrist, J. A., III; Waud, W. R. Curr. Opin. Invest. Drugs 2004, 5, 592–596.
Return to citation in text: [1] -
Keating, M. J.; Kantarjian, H.; Talpaz, M.; Redman, J.; Koller, C.; Barlogie, B.; Velasquez, W.; Plunkett, W.; Freireich, E. J.; McCredie, K. B. Blood 1989, 74, 19–25.
Return to citation in text: [1] -
Rai, K. R.; Peterson, B. L.; Appelbaum, F. R.; Kolitz, J.; Elias, L.; Shepherd, L.; Hines, J.; Threatte, G. A.; Larson, R. A.; Cheson, B. D.; Schiffer, C. A. N. Engl. J. Med. 2000, 343, 1750–1757. doi:10.1056/NEJM200012143432402
Return to citation in text: [1] -
Bonate, P. L.; Arthaud, L.; Cantrell, W. R., Jr.; Stephenson, K.; Secrist, J. A., III; Weitman, S. Nat. Rev. Drug Discovery 2006, 5, 855–863. doi:10.1038/nrd2055
Return to citation in text: [1] -
Jeha, S.; Kantarjian, H. Expert Rev. Anticancer Ther. 2007, 7, 113–118. doi:10.1586/14737140.7.2.113
Return to citation in text: [1] -
Gundersen, L.-L.; Nissen-Meyer, J.; Spilsberg, B. J. Med. Chem. 2002, 45, 1383–1386. doi:10.1021/jm0110284
Return to citation in text: [1] [2] -
Nauš, P.; Perliková, P.; Pohl, R.; Hocek, M. Collect. Czech. Chem. Commun. 2011, 76, 957–988. doi:10.1135/cccc2011082
Return to citation in text: [1] [2] -
Montgomery, J. A.; Hewson, K. J. Med. Chem. 1968, 11, 48–52. doi:10.1021/jm00307a010
Return to citation in text: [1] [2] -
Hocek, M.; Holý, A.; Votruba, I.; Dvořáková, H. J. Med. Chem. 2000, 43, 1817–1825. doi:10.1021/jm991167+
Return to citation in text: [1] [2] -
Hocek, M.; Holý, A.; Votruba, I.; Dvořáková, H. Collect. Czech. Chem. Commun. 2001, 66, 483–499. doi:10.1135/cccc20010483
Return to citation in text: [1] [2] -
Nauš, P.; Pohl, R.; Votruba, I.; Džubák, P.; Hajdúch, M.; Ameral, R.; Birkuš, G.; Wang, T.; Ray, A. S.; Mackman, R.; Cihlar, T.; Hocek, M. J. Med. Chem. 2010, 53, 460–470. doi:10.1021/jm901428k
Return to citation in text: [1] [2] -
Hocek, M.; Nauš, P.; Pohl, R.; Votruba, I.; Furman, P. A.; Tharnish, P. M.; Otto, M. J. J. Med. Chem. 2005, 48, 5869–5873. doi:10.1021/jm050335x
Return to citation in text: [1] [2] -
Smith, K. L.; Lai, V. C. H.; Prigaro, B. J.; Ding, Y.; Gunic, E.; Girardet, J.-L.; Zhong, W.; Hong, Z.; Lang, S.; An, H. Bioorg. Med. Chem. Lett. 2004, 14, 3517–3520. doi:10.1016/j.bmcl.2004.04.067
Return to citation in text: [1] [2] -
Ding, Y.; Girardet, J.-L.; Hong, Z.; Lai, V. C. H.; An, H.; Koh, Y.-h.; Shaw, S. Z.; Zhong, W. Bioorg. Med. Chem. Lett. 2005, 15, 709–713. doi:10.1016/j.bmcl.2004.11.020
Return to citation in text: [1] [2] -
Olsen, D. B.; Eldrup, A. B.; Bartholomew, L.; Bhat, B.; Bosserman, M. R.; Ceccacci, A.; Colwell, L. F.; Fay, J. F.; Flores, O. A.; Getty, K. L.; Grobler, J. A.; LaFemina, R. L.; Markel, E. J.; Migliaccio, G.; Prhavc, M.; Stahlhut, M. W.; Tomassini, J. E.; MacCoss, M.; Hazuda, D. J.; Carroll, S. S. Antimicrob. Agents Chemother. 2004, 48, 3944–3953. doi:10.1128/AAC.48.10.3944-3953.2004
Return to citation in text: [1] [2] -
Fustero, S. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Wiley-Blackwell: Chichester, U.K., 2009; pp 527–583.
Return to citation in text: [1] -
Filler, R.; Saha, R. Future Med. Chem. 2009, 1, 777–791. doi:10.4155/fmc.09.65
Return to citation in text: [1] -
Guo, X.; Li, Y.; Tao, L.; Wang, Q.; Wang, S.; Hu, W.; Pan, Z.; Yang, Q.; Cui, Y.; Ge, Z.; Dong, L.; Yu, X.; An, H.; Song, C.; Chang, J. Bioorg. Med. Chem. Lett. 2011, 21, 6770–6772. doi:10.1016/j.bmcl.2011.09.040
Return to citation in text: [1] -
An, H.; Zhu, J.; Wang, X.; Xu, X. Bioorg. Med. Chem. Lett. 2006, 16, 4826–4829. doi:10.1016/j.bmcl.2006.06.070
Return to citation in text: [1] -
Xu, W.; Xi, B.; Wu, J.; An, H.; Zhu, J.; Abassi, Y.; Feinstein, S. C.; Gaylord, M.; Geng, B.; Yan, H.; Fan, W.; Sui, M.; Wang, X.; Xu, X. Mol. Cancer Ther. 2009, 8, 3318–3330. doi:10.1158/1535-7163.MCT-09-0548
Return to citation in text: [1] -
Bao, Y.; Mo, X.; Xu, X.; He, Y.; Xu, X.; An, H. J. Pharm. Biomed. Anal. 2008, 48, 664–671. doi:10.1016/j.jpba.2008.06.013
Return to citation in text: [1] -
An, H.; Hu, X.; Gu, J.; Chen, L.; Xu, W.; Mo, X.; Xu, W.; Wang, X.; Xu, X. AAPS PharmSciTech 2008, 9, 551–556. doi:10.1208/s12249-008-9081-5
Return to citation in text: [1] -
Zheng, L.; Wang, Q.; Yang, X.; Guo, X.; Chen, L.; Tao, L.; Dong, L.; Li, Y.; An, H.; Yu, X.; Wang, Q.; Chang, J. Antiviral Ther. 2012, 17, 679–687. doi:10.3851/IMP2094
Return to citation in text: [1] -
Wang, Q.; Hu, W.; Wang, S.; Pan, Z.; Tao, L.; Guo, X.; Qian, K.; Chen, C.-H.; Lee, K.-H.; Chang, J. Eur. J. Med. Chem. 2011, 46, 4178–4183. doi:10.1016/j.ejmech.2011.06.020
Return to citation in text: [1] -
Yokoyama, A.; Nakai, Y.; Yoneda, S.; Kurita, Y.; Niitani, H. Anti-Cancer Drugs 1997, 8, 574–580. doi:10.1097/00001813-199707000-00004
Return to citation in text: [1] -
Pankiewicz, K. W.; Krzeminski, J.; Watanabe, K. A. J. Org. Chem. 1992, 57, 7315–7321. doi:10.1021/jo00052a055
Return to citation in text: [1] [2] [3] [4] -
Giordani, A.; Ermoli, A.; Battisthi, C. Nucleosides, Nucleotides Nucleic Acids 1991, 10, 719–721. doi:10.1080/07328319108046581
Return to citation in text: [1] [2] [3] [4] -
van Aerschot, A.; Herdewijn, P.; Janssen, G.; Cools, M.; De Clercq, E. Antiviral Res. 1989, 12, 133–150. doi:10.1016/0166-3542(89)90047-8
Return to citation in text: [1] [2] [3] -
Puech, F.; Gosselin, G.; Imbach, J.-L. J. Chem. Soc., Chem. Commun. 1989, 955–957. doi:10.1039/c39890000955
Return to citation in text: [1] [2] [3] [4] -
Battistini, C.; Giordani, A.; Ermoli, A.; Franceschi, G. Synthesis 1990, 900–905. doi:10.1055/s-1990-27048
Return to citation in text: [1] [2] [3] -
Morizawa, Y.; Nakayama, T.; Yasuda, A.; Uchida, K. Bull. Chem. Soc. Jpn. 1989, 62, 2119–2120. doi:10.1246/bcsj.62.2119
Return to citation in text: [1] -
Mikhailopulo, I. A.; Poopeiko, N. E.; Pricota, T. I.; Sivets, G. G.; Kvasyuk, E. I.; Balzarini, J.; De Clercq, E. J. Med. Chem. 1991, 34, 2195–2202. doi:10.1021/jm00111a040
Return to citation in text: [1] -
Kim, H. O.; Lim, M. H.; Park, J. G.; Moon, H. R.; Jacobson, K. A.; Kim, H.-D.; Chun, M. W.; Jeong, L. S. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 923–925. doi:10.1081/NCN-120022701
Return to citation in text: [1] -
Lim, M. H.; Kim, H. O.; Moon, H. R.; Lee, S. J.; Chun, M. W.; Gao, Z.-G.; Melman, N.; Jacobson, K. A.; Kim, J. H.; Jeong, L. S. Bioorg. Med. Chem. Lett. 2003, 13, 817–820. doi:10.1016/S0960-894X(03)00027-1
Return to citation in text: [1] -
Mikhailopulo, I. A.; Sivets, G. G. Helv. Chim. Acta 1999, 82, 2052–2065. doi:10.1002/(SICI)1522-2675(19991110)82:11<2052::AID-HLCA2052>3.0.CO;2-7
Return to citation in text: [1] -
Ozols, A. M.; Azhayev, A. V.; Dyatkina, N. B.; Krayevsky, A. A. Synthesis 1980, 557–559. doi:10.1055/s-1980-29120
Return to citation in text: [1] -
Bennett, L. L., Jr.; Schnebli, H. P.; Vail, M. H.; Allan, P. W.; Montgomery, J. A. Mol. Pharmacol. 1966, 2, 432–443.
Return to citation in text: [1] [2] -
Lynch, T. P.; Paran, J. H.; Paterson, A. R. P. Cancer Res. 1981, 41, 560–565.
Return to citation in text: [1] [2] -
Marasco, C., Jr.; Pera, P.; Spiess, A. J.; Bernacki, R.; Sufrin, J. Molecules 2005, 10, 1015–1020. doi:10.3390/10081015
Return to citation in text: [1] [2] -
Söderberg, B. C. G. Coord. Chem. Rev. 2006, 250, 300–387. doi:10.1016/j.ccr.2005.03.033
Return to citation in text: [1] -
Agrofoglio, L. A.; Gillaizeau, I.; Saito, Y. Chem. Rev. 2003, 103, 1875–1916. doi:10.1021/cr010374q
Return to citation in text: [1] -
Greco, N. J.; Tor, Y. Tetrahedron 2007, 63, 3515–3527. doi:10.1016/j.tet.2007.01.073
Return to citation in text: [1] -
Srivatsan, S. G.; Tor, Y. J. Am. Chem. Soc. 2007, 129, 2044–2053. doi:10.1021/ja066455r
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Strouse, J. J.; Jeselnik, M.; Arterburn, J. B. Acta Chim. Slov. 2005, 52, 187–199.
Return to citation in text: [1] -
Bellina, F.; Carpita, A.; Rossi, R. Synthesis 2004, 2419–2440. doi:10.1055/s-2004-831223
Return to citation in text: [1] -
Parry, P. R.; Bryce, M. R.; Tarbit, B. Synthesis 2003, 1035–1038. doi:10.1055/s-2003-39158
Return to citation in text: [1] -
Li, W.; Nelson, D. P.; Jensen, M. S.; Hoerrner, R. S.; Cai, D.; Larsen, R. D.; Reider, P. J. J. Org. Chem. 2002, 67, 5394–5397. doi:10.1021/jo025792p
Return to citation in text: [1] -
Cioffi, C. L.; Spencer, W. T.; Richards, J. J.; Herr, R. J. J. Org. Chem. 2004, 69, 2210–2212. doi:10.1021/jo034664d
Return to citation in text: [1] -
Bauta, W. E.; Schulmeier, B. E.; Burke, B.; Puente, J. F.; Cantrell, W. R., Jr.; Lovett, D.; Goebel, J.; Anderson, B.; Ionescu, D.; Guo, R. Org. Process Res. Dev. 2004, 8, 889–896. doi:10.1021/op049884n
Return to citation in text: [1] [2] -
Kluin-Nelemans, H. C.; Oldhoff, J. M.; van Doormaal, J. J.; van’t Wout, J. W.; Verhoef, G.; Gerrits, W. B. J.; van Dobbenburgh, O. A.; Pasmans, S. G.; Fijnheer, R. Blood 2003, 102, 4270–4276. doi:10.1182/blood-2003-05-1699
Return to citation in text: [1] [2] -
Aleem, A.; Anjum, F.; Algahtani, F.; Iqbal, Z.; Alsaleh, K.; AlMomen, A. Asian Pac. J. Cancer Prev. 2013, 14, 1089–1092. doi:10.7314/APJCP.2013.14.2.1089
Return to citation in text: [1] [2] -
Dalpozzo, R.; De Nino, A.; Maiuolo, L.; Procopio, A.; De Munno, G.; Sindona, G. Tetrahedron 2001, 57, 4035–4038. doi:10.1016/S0040-4020(01)00284-8
Return to citation in text: [1] -
Robins, M. J.; Zou, R.; Guo, Z.; Wnuk, S. F. J. Org. Chem. 1996, 61, 9207–9212. doi:10.1021/jo9617023
Return to citation in text: [1]
| 45. | Söderberg, B. C. G. Coord. Chem. Rev. 2006, 250, 300–387. doi:10.1016/j.ccr.2005.03.033 |
| 46. | Agrofoglio, L. A.; Gillaizeau, I.; Saito, Y. Chem. Rev. 2003, 103, 1875–1916. doi:10.1021/cr010374q |
| 47. | Greco, N. J.; Tor, Y. Tetrahedron 2007, 63, 3515–3527. doi:10.1016/j.tet.2007.01.073 |
| 48. | Srivatsan, S. G.; Tor, Y. J. Am. Chem. Soc. 2007, 129, 2044–2053. doi:10.1021/ja066455r |
| 49. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 50. | Strouse, J. J.; Jeselnik, M.; Arterburn, J. B. Acta Chim. Slov. 2005, 52, 187–199. |
| 51. | Bellina, F.; Carpita, A.; Rossi, R. Synthesis 2004, 2419–2440. doi:10.1055/s-2004-831223 |
| 1. | Henry, J. R.; Mader, M. M. Recent Advances in Antimetabolite Cancer Chemotherapies. In Annual Reports in Medicinal Chemistry; Doherty, A. M., Ed.; Elsevier: San Diego, CA, U.S.A., 2004; Vol. 39, pp 159–172. doi:10.1016/s0065-7743(04)39013-5 |
| 7. | Keating, M. J.; Kantarjian, H.; Talpaz, M.; Redman, J.; Koller, C.; Barlogie, B.; Velasquez, W.; Plunkett, W.; Freireich, E. J.; McCredie, K. B. Blood 1989, 74, 19–25. |
| 8. | Rai, K. R.; Peterson, B. L.; Appelbaum, F. R.; Kolitz, J.; Elias, L.; Shepherd, L.; Hines, J.; Threatte, G. A.; Larson, R. A.; Cheson, B. D.; Schiffer, C. A. N. Engl. J. Med. 2000, 343, 1750–1757. doi:10.1056/NEJM200012143432402 |
| 9. | Bonate, P. L.; Arthaud, L.; Cantrell, W. R., Jr.; Stephenson, K.; Secrist, J. A., III; Weitman, S. Nat. Rev. Drug Discovery 2006, 5, 855–863. doi:10.1038/nrd2055 |
| 10. | Jeha, S.; Kantarjian, H. Expert Rev. Anticancer Ther. 2007, 7, 113–118. doi:10.1586/14737140.7.2.113 |
| 33. | van Aerschot, A.; Herdewijn, P.; Janssen, G.; Cools, M.; De Clercq, E. Antiviral Res. 1989, 12, 133–150. doi:10.1016/0166-3542(89)90047-8 |
| 4. | Johnson, S. A.; Thomas, W. Hematol. Oncol. 2000, 18, 141–153. doi:10.1002/1099-1069(200012)18:4<141::AID-HON666>3.0.CO;2-# |
| 5. | Johnson, S. A. Expert Opin. Pharmacother. 2001, 2, 929–943. doi:10.1517/14656566.2.6.929 |
| 6. | Parker, W. B.; Secrist, J. A., III; Waud, W. R. Curr. Opin. Invest. Drugs 2004, 5, 592–596. |
| 34. | Puech, F.; Gosselin, G.; Imbach, J.-L. J. Chem. Soc., Chem. Commun. 1989, 955–957. doi:10.1039/c39890000955 |
| 31. | Pankiewicz, K. W.; Krzeminski, J.; Watanabe, K. A. J. Org. Chem. 1992, 57, 7315–7321. doi:10.1021/jo00052a055 |
| 32. | Giordani, A.; Ermoli, A.; Battisthi, C. Nucleosides, Nucleotides Nucleic Acids 1991, 10, 719–721. doi:10.1080/07328319108046581 |
| 33. | van Aerschot, A.; Herdewijn, P.; Janssen, G.; Cools, M.; De Clercq, E. Antiviral Res. 1989, 12, 133–150. doi:10.1016/0166-3542(89)90047-8 |
| 34. | Puech, F.; Gosselin, G.; Imbach, J.-L. J. Chem. Soc., Chem. Commun. 1989, 955–957. doi:10.1039/c39890000955 |
| 35. | Battistini, C.; Giordani, A.; Ermoli, A.; Franceschi, G. Synthesis 1990, 900–905. doi:10.1055/s-1990-27048 |
| 58. | Dalpozzo, R.; De Nino, A.; Maiuolo, L.; Procopio, A.; De Munno, G.; Sindona, G. Tetrahedron 2001, 57, 4035–4038. doi:10.1016/S0040-4020(01)00284-8 |
| 59. | Robins, M. J.; Zou, R.; Guo, Z.; Wnuk, S. F. J. Org. Chem. 1996, 61, 9207–9212. doi:10.1021/jo9617023 |
| 2. | 19th WHO Model List of Essential Medicines. WHO, 2015; http://www.who.int/medicines/publications/essentialmedicines/EML2015_8-May-15.pdf (accessed July 8, 2015). |
| 31. | Pankiewicz, K. W.; Krzeminski, J.; Watanabe, K. A. J. Org. Chem. 1992, 57, 7315–7321. doi:10.1021/jo00052a055 |
| 32. | Giordani, A.; Ermoli, A.; Battisthi, C. Nucleosides, Nucleotides Nucleic Acids 1991, 10, 719–721. doi:10.1080/07328319108046581 |
| 34. | Puech, F.; Gosselin, G.; Imbach, J.-L. J. Chem. Soc., Chem. Commun. 1989, 955–957. doi:10.1039/c39890000955 |
| 18. | Smith, K. L.; Lai, V. C. H.; Prigaro, B. J.; Ding, Y.; Gunic, E.; Girardet, J.-L.; Zhong, W.; Hong, Z.; Lang, S.; An, H. Bioorg. Med. Chem. Lett. 2004, 14, 3517–3520. doi:10.1016/j.bmcl.2004.04.067 |
| 19. | Ding, Y.; Girardet, J.-L.; Hong, Z.; Lai, V. C. H.; An, H.; Koh, Y.-h.; Shaw, S. Z.; Zhong, W. Bioorg. Med. Chem. Lett. 2005, 15, 709–713. doi:10.1016/j.bmcl.2004.11.020 |
| 20. | Olsen, D. B.; Eldrup, A. B.; Bartholomew, L.; Bhat, B.; Bosserman, M. R.; Ceccacci, A.; Colwell, L. F.; Fay, J. F.; Flores, O. A.; Getty, K. L.; Grobler, J. A.; LaFemina, R. L.; Markel, E. J.; Migliaccio, G.; Prhavc, M.; Stahlhut, M. W.; Tomassini, J. E.; MacCoss, M.; Hazuda, D. J.; Carroll, S. S. Antimicrob. Agents Chemother. 2004, 48, 3944–3953. doi:10.1128/AAC.48.10.3944-3953.2004 |
| 24. | An, H.; Zhu, J.; Wang, X.; Xu, X. Bioorg. Med. Chem. Lett. 2006, 16, 4826–4829. doi:10.1016/j.bmcl.2006.06.070 |
| 25. | Xu, W.; Xi, B.; Wu, J.; An, H.; Zhu, J.; Abassi, Y.; Feinstein, S. C.; Gaylord, M.; Geng, B.; Yan, H.; Fan, W.; Sui, M.; Wang, X.; Xu, X. Mol. Cancer Ther. 2009, 8, 3318–3330. doi:10.1158/1535-7163.MCT-09-0548 |
| 26. | Bao, Y.; Mo, X.; Xu, X.; He, Y.; Xu, X.; An, H. J. Pharm. Biomed. Anal. 2008, 48, 664–671. doi:10.1016/j.jpba.2008.06.013 |
| 27. | An, H.; Hu, X.; Gu, J.; Chen, L.; Xu, W.; Mo, X.; Xu, W.; Wang, X.; Xu, X. AAPS PharmSciTech 2008, 9, 551–556. doi:10.1208/s12249-008-9081-5 |
| 28. | Zheng, L.; Wang, Q.; Yang, X.; Guo, X.; Chen, L.; Tao, L.; Dong, L.; Li, Y.; An, H.; Yu, X.; Wang, Q.; Chang, J. Antiviral Ther. 2012, 17, 679–687. doi:10.3851/IMP2094 |
| 29. | Wang, Q.; Hu, W.; Wang, S.; Pan, Z.; Tao, L.; Guo, X.; Qian, K.; Chen, C.-H.; Lee, K.-H.; Chang, J. Eur. J. Med. Chem. 2011, 46, 4178–4183. doi:10.1016/j.ejmech.2011.06.020 |
| 55. | Bauta, W. E.; Schulmeier, B. E.; Burke, B.; Puente, J. F.; Cantrell, W. R., Jr.; Lovett, D.; Goebel, J.; Anderson, B.; Ionescu, D.; Guo, R. Org. Process Res. Dev. 2004, 8, 889–896. doi:10.1021/op049884n |
| 56. | Kluin-Nelemans, H. C.; Oldhoff, J. M.; van Doormaal, J. J.; van’t Wout, J. W.; Verhoef, G.; Gerrits, W. B. J.; van Dobbenburgh, O. A.; Pasmans, S. G.; Fijnheer, R. Blood 2003, 102, 4270–4276. doi:10.1182/blood-2003-05-1699 |
| 57. | Aleem, A.; Anjum, F.; Algahtani, F.; Iqbal, Z.; Alsaleh, K.; AlMomen, A. Asian Pac. J. Cancer Prev. 2013, 14, 1089–1092. doi:10.7314/APJCP.2013.14.2.1089 |
| 17. | Hocek, M.; Nauš, P.; Pohl, R.; Votruba, I.; Furman, P. A.; Tharnish, P. M.; Otto, M. J. J. Med. Chem. 2005, 48, 5869–5873. doi:10.1021/jm050335x |
| 30. | Yokoyama, A.; Nakai, Y.; Yoneda, S.; Kurita, Y.; Niitani, H. Anti-Cancer Drugs 1997, 8, 574–580. doi:10.1097/00001813-199707000-00004 |
| 42. | Bennett, L. L., Jr.; Schnebli, H. P.; Vail, M. H.; Allan, P. W.; Montgomery, J. A. Mol. Pharmacol. 1966, 2, 432–443. |
| 43. | Lynch, T. P.; Paran, J. H.; Paterson, A. R. P. Cancer Res. 1981, 41, 560–565. |
| 14. | Hocek, M.; Holý, A.; Votruba, I.; Dvořáková, H. J. Med. Chem. 2000, 43, 1817–1825. doi:10.1021/jm991167+ |
| 15. | Hocek, M.; Holý, A.; Votruba, I.; Dvořáková, H. Collect. Czech. Chem. Commun. 2001, 66, 483–499. doi:10.1135/cccc20010483 |
| 16. | Nauš, P.; Pohl, R.; Votruba, I.; Džubák, P.; Hajdúch, M.; Ameral, R.; Birkuš, G.; Wang, T.; Ray, A. S.; Mackman, R.; Cihlar, T.; Hocek, M. J. Med. Chem. 2010, 53, 460–470. doi:10.1021/jm901428k |
| 52. | Parry, P. R.; Bryce, M. R.; Tarbit, B. Synthesis 2003, 1035–1038. doi:10.1055/s-2003-39158 |
| 53. | Li, W.; Nelson, D. P.; Jensen, M. S.; Hoerrner, R. S.; Cai, D.; Larsen, R. D.; Reider, P. J. J. Org. Chem. 2002, 67, 5394–5397. doi:10.1021/jo025792p |
| 54. | Cioffi, C. L.; Spencer, W. T.; Richards, J. J.; Herr, R. J. J. Org. Chem. 2004, 69, 2210–2212. doi:10.1021/jo034664d |
| 11. | Gundersen, L.-L.; Nissen-Meyer, J.; Spilsberg, B. J. Med. Chem. 2002, 45, 1383–1386. doi:10.1021/jm0110284 |
| 12. | Nauš, P.; Perliková, P.; Pohl, R.; Hocek, M. Collect. Czech. Chem. Commun. 2011, 76, 957–988. doi:10.1135/cccc2011082 |
| 13. | Montgomery, J. A.; Hewson, K. J. Med. Chem. 1968, 11, 48–52. doi:10.1021/jm00307a010 |
| 21. | Fustero, S. In Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I., Ed.; Wiley-Blackwell: Chichester, U.K., 2009; pp 527–583. |
| 22. | Filler, R.; Saha, R. Future Med. Chem. 2009, 1, 777–791. doi:10.4155/fmc.09.65 |
| 23. | Guo, X.; Li, Y.; Tao, L.; Wang, Q.; Wang, S.; Hu, W.; Pan, Z.; Yang, Q.; Cui, Y.; Ge, Z.; Dong, L.; Yu, X.; An, H.; Song, C.; Chang, J. Bioorg. Med. Chem. Lett. 2011, 21, 6770–6772. doi:10.1016/j.bmcl.2011.09.040 |
| 55. | Bauta, W. E.; Schulmeier, B. E.; Burke, B.; Puente, J. F.; Cantrell, W. R., Jr.; Lovett, D.; Goebel, J.; Anderson, B.; Ionescu, D.; Guo, R. Org. Process Res. Dev. 2004, 8, 889–896. doi:10.1021/op049884n |
| 56. | Kluin-Nelemans, H. C.; Oldhoff, J. M.; van Doormaal, J. J.; van’t Wout, J. W.; Verhoef, G.; Gerrits, W. B. J.; van Dobbenburgh, O. A.; Pasmans, S. G.; Fijnheer, R. Blood 2003, 102, 4270–4276. doi:10.1182/blood-2003-05-1699 |
| 57. | Aleem, A.; Anjum, F.; Algahtani, F.; Iqbal, Z.; Alsaleh, K.; AlMomen, A. Asian Pac. J. Cancer Prev. 2013, 14, 1089–1092. doi:10.7314/APJCP.2013.14.2.1089 |
| 37. | Mikhailopulo, I. A.; Poopeiko, N. E.; Pricota, T. I.; Sivets, G. G.; Kvasyuk, E. I.; Balzarini, J.; De Clercq, E. J. Med. Chem. 1991, 34, 2195–2202. doi:10.1021/jm00111a040 |
| 35. | Battistini, C.; Giordani, A.; Ermoli, A.; Franceschi, G. Synthesis 1990, 900–905. doi:10.1055/s-1990-27048 |
| 36. | Morizawa, Y.; Nakayama, T.; Yasuda, A.; Uchida, K. Bull. Chem. Soc. Jpn. 1989, 62, 2119–2120. doi:10.1246/bcsj.62.2119 |
| 44. | Marasco, C., Jr.; Pera, P.; Spiess, A. J.; Bernacki, R.; Sufrin, J. Molecules 2005, 10, 1015–1020. doi:10.3390/10081015 |
| 11. | Gundersen, L.-L.; Nissen-Meyer, J.; Spilsberg, B. J. Med. Chem. 2002, 45, 1383–1386. doi:10.1021/jm0110284 |
| 12. | Nauš, P.; Perliková, P.; Pohl, R.; Hocek, M. Collect. Czech. Chem. Commun. 2011, 76, 957–988. doi:10.1135/cccc2011082 |
| 13. | Montgomery, J. A.; Hewson, K. J. Med. Chem. 1968, 11, 48–52. doi:10.1021/jm00307a010 |
| 14. | Hocek, M.; Holý, A.; Votruba, I.; Dvořáková, H. J. Med. Chem. 2000, 43, 1817–1825. doi:10.1021/jm991167+ |
| 15. | Hocek, M.; Holý, A.; Votruba, I.; Dvořáková, H. Collect. Czech. Chem. Commun. 2001, 66, 483–499. doi:10.1135/cccc20010483 |
| 16. | Nauš, P.; Pohl, R.; Votruba, I.; Džubák, P.; Hajdúch, M.; Ameral, R.; Birkuš, G.; Wang, T.; Ray, A. S.; Mackman, R.; Cihlar, T.; Hocek, M. J. Med. Chem. 2010, 53, 460–470. doi:10.1021/jm901428k |
| 17. | Hocek, M.; Nauš, P.; Pohl, R.; Votruba, I.; Furman, P. A.; Tharnish, P. M.; Otto, M. J. J. Med. Chem. 2005, 48, 5869–5873. doi:10.1021/jm050335x |
| 18. | Smith, K. L.; Lai, V. C. H.; Prigaro, B. J.; Ding, Y.; Gunic, E.; Girardet, J.-L.; Zhong, W.; Hong, Z.; Lang, S.; An, H. Bioorg. Med. Chem. Lett. 2004, 14, 3517–3520. doi:10.1016/j.bmcl.2004.04.067 |
| 19. | Ding, Y.; Girardet, J.-L.; Hong, Z.; Lai, V. C. H.; An, H.; Koh, Y.-h.; Shaw, S. Z.; Zhong, W. Bioorg. Med. Chem. Lett. 2005, 15, 709–713. doi:10.1016/j.bmcl.2004.11.020 |
| 20. | Olsen, D. B.; Eldrup, A. B.; Bartholomew, L.; Bhat, B.; Bosserman, M. R.; Ceccacci, A.; Colwell, L. F.; Fay, J. F.; Flores, O. A.; Getty, K. L.; Grobler, J. A.; LaFemina, R. L.; Markel, E. J.; Migliaccio, G.; Prhavc, M.; Stahlhut, M. W.; Tomassini, J. E.; MacCoss, M.; Hazuda, D. J.; Carroll, S. S. Antimicrob. Agents Chemother. 2004, 48, 3944–3953. doi:10.1128/AAC.48.10.3944-3953.2004 |
| 42. | Bennett, L. L., Jr.; Schnebli, H. P.; Vail, M. H.; Allan, P. W.; Montgomery, J. A. Mol. Pharmacol. 1966, 2, 432–443. |
| 43. | Lynch, T. P.; Paran, J. H.; Paterson, A. R. P. Cancer Res. 1981, 41, 560–565. |
| 44. | Marasco, C., Jr.; Pera, P.; Spiess, A. J.; Bernacki, R.; Sufrin, J. Molecules 2005, 10, 1015–1020. doi:10.3390/10081015 |
| 31. | Pankiewicz, K. W.; Krzeminski, J.; Watanabe, K. A. J. Org. Chem. 1992, 57, 7315–7321. doi:10.1021/jo00052a055 |
| 32. | Giordani, A.; Ermoli, A.; Battisthi, C. Nucleosides, Nucleotides Nucleic Acids 1991, 10, 719–721. doi:10.1080/07328319108046581 |
| 33. | van Aerschot, A.; Herdewijn, P.; Janssen, G.; Cools, M.; De Clercq, E. Antiviral Res. 1989, 12, 133–150. doi:10.1016/0166-3542(89)90047-8 |
| 34. | Puech, F.; Gosselin, G.; Imbach, J.-L. J. Chem. Soc., Chem. Commun. 1989, 955–957. doi:10.1039/c39890000955 |
| 35. | Battistini, C.; Giordani, A.; Ermoli, A.; Franceschi, G. Synthesis 1990, 900–905. doi:10.1055/s-1990-27048 |
| 31. | Pankiewicz, K. W.; Krzeminski, J.; Watanabe, K. A. J. Org. Chem. 1992, 57, 7315–7321. doi:10.1021/jo00052a055 |
| 32. | Giordani, A.; Ermoli, A.; Battisthi, C. Nucleosides, Nucleotides Nucleic Acids 1991, 10, 719–721. doi:10.1080/07328319108046581 |
| 38. | Kim, H. O.; Lim, M. H.; Park, J. G.; Moon, H. R.; Jacobson, K. A.; Kim, H.-D.; Chun, M. W.; Jeong, L. S. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 923–925. doi:10.1081/NCN-120022701 |
| 39. | Lim, M. H.; Kim, H. O.; Moon, H. R.; Lee, S. J.; Chun, M. W.; Gao, Z.-G.; Melman, N.; Jacobson, K. A.; Kim, J. H.; Jeong, L. S. Bioorg. Med. Chem. Lett. 2003, 13, 817–820. doi:10.1016/S0960-894X(03)00027-1 |
| 40. | Mikhailopulo, I. A.; Sivets, G. G. Helv. Chim. Acta 1999, 82, 2052–2065. doi:10.1002/(SICI)1522-2675(19991110)82:11<2052::AID-HLCA2052>3.0.CO;2-7 |
| 41. | Ozols, A. M.; Azhayev, A. V.; Dyatkina, N. B.; Krayevsky, A. A. Synthesis 1980, 557–559. doi:10.1055/s-1980-29120 |
© 2015 Ren et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)