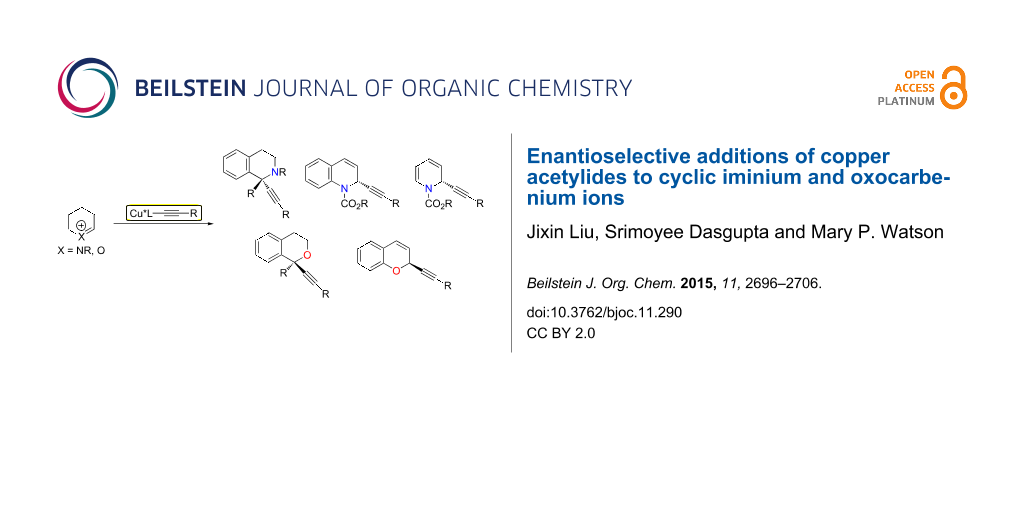
Abstract
The development of enantioselective, copper-catalyzed alkynylations of cyclic iminium and oxocarbenium ions is reviewed. The use of chiral copper-based catalysts has enabled high yields and enantioselectivites in the formation of nitrogen- and oxygen-containing heterocycles with α-stereogenic centers. This review highlights both the accomplishments and the future work needed in this important area.

Graphical Abstract
Introduction
Nitrogen and oxygen heterocycles with α-stereogenic centers represent important classes of biologicially active compounds [1-7]. Enantioselective addition of chiral nucleophiles to imines, iminium ions, carbonyls, or oxocarbenium ions provides efficient access to these scaffolds. In particular, exceptional progress has been made in the addition of chiral metal acetylides. Historically, these reactions required stoichiometric amounts of both metal and chiral ligand [8], but catalytic variants are now available with a variety of metal-based catalysts [9-15]. Among these, chiral copper catalysts have been used with remarkable success in the alkynylation of cyclic iminium ion and oxocarbenium ion intermediates. This review will focus on the development of these enantioselective, copper-catalyzed alkynylations, highlighting both the accomplishments and the future work needed in this important area.
Throughout the discussion below, it is clear that there are privileged ligand architectures of these copper-catalyzed alkynylations. High enantioselectivities have been achieved with pyridine bis(oxazoline) (Pybox), bis(oxazoline) (Box), and Quinap-type ligands (Figure 1).
![[1860-5397-11-290-1]](/bjoc/content/figures/1860-5397-11-290-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chiral ligands utilized in copper-catalyzed alkynylations of cyclic iminium and oxocarbenium ions.
Figure 1: Chiral ligands utilized in copper-catalyzed alkynylations of cyclic iminium and oxocarbenium ions.
Review
Additions to iminium ions
Although this review will focus on enantioselective additions to cyclic electrophiles, it is worth noting that the first enantioselective additions of chiral copper acetylides to imines or iminium ions utilized acyclic imine substrates. In 2002, Li and co-workers reported enantioselective alkynylations of N-aryl aldimines formed in situ from benzaldehydes and anilines (Scheme 1) [16,17]. This reaction employs a CuOTf/Ph-Pybox catalyst system to achieve generally high yields and ee’s of propargylic amines 2. Notably, this reaction can be carried out in H2O, as well as PhMe. Nearly simultaneously, Knochel’s group reported an enantioselective CuBr/Quinap-catalyzed alkynylation to deliver propargylic amines with alkyl substitution at the stereocenter (Scheme 2) [18-20]. In this reaction, proton transfer from the terminal alkyne to the enamine simultaneously generates the copper acetylide and iminium ion, which are proposed to both bind to the chiral copper catalyst (see 4). A broad scope in the acetylene was observed, with arylalkynes resulting in the highest ee’s. To our knowledge, these were the first enantioselective, metal-catalyzed additions of terminal alkynes to imines or iminium ions, and set the stage for subsequent development of enantioselective alkynylations of cyclic iminium ion substrates [21]. As discussed below, the catalyst systems identified in these reactions have largely informed those used for enantioselective alkynylations of cyclic electrophiles.
![[1860-5397-11-290-i1]](/bjoc/content/inline/1860-5397-11-290-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Li’s alkynylation of acyclic N-arylimines.
Scheme 1: Li’s alkynylation of acyclic N-arylimines.
![[1860-5397-11-290-i2]](/bjoc/content/inline/1860-5397-11-290-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Knochel’s alkynylation of acyclic N-alkylenamines.
Scheme 2: Knochel’s alkynylation of acyclic N-alkylenamines.
The first enantioselective, copper-catalyzed alkynylation of a cyclic iminium ion was reported by Li’s research group in 2004 [22]. Building on their development of a cross-dehydrogenative coupling (CDC) reaction between benzylic amines and alkynes to deliver racemic products [23], the Li group developed a CuOTf/Ph-Pybox catalyst system that enables alkynylation of tetrahydroisoquinolines in moderate to good yields and enantioselectivies (Scheme 3). A particularly powerful aspect of this chemistry is that a stable tetrahydroisoquinoline 6 can be utilized as the substrate. Oxidation of the tetrahydroisoquinoline then results in formation of iminium ion 7 in situ. As in Knochel’s reaction above, the highest yields and ee’s were observed with arylacetylenes.
![[1860-5397-11-290-i3]](/bjoc/content/inline/1860-5397-11-290-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Li’s CDC of tetrahydroisoquinolines and alkynes.
Scheme 3: Li’s CDC of tetrahydroisoquinolines and alkynes.
In subsequent studies of this reaction, Li investigated the intermediacy of isoquinolinium ion 9, and found that improved yields and ee’s can be achieved using this substrate and a CuBr/Quinap catalyst, despite the fact that Quinap had proven inferior to Ph-Pybox in the CDC reaction (Scheme 4) [24]. With this new catalyst and electrophile, the catalyst loading, reaction temperature, and reaction time could be reduced. Addition of alkynes with aryl, alkyl and trimethylsilyl substituents were successful, with the highest enantioselectivity observed when (trimethylsilyl)acetylene was used (94% ee).
![[1860-5397-11-290-i4]](/bjoc/content/inline/1860-5397-11-290-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Li’s alkynylation of N-aryldihydroisoquinolinium ions.
Scheme 4: Li’s alkynylation of N-aryldihydroisoquinolinium ions.
Just before Li’s report of the alkynlation of N-arylisoquinolinium ions, Taylor and Schreiber reported a CuBr/Quinap-catalyzed alkynylation of N-alkylisoquinolinium ions (Scheme 5) [25]. Similar to Li’s alkynylation of N-arylisoquinolinium ions, alkynes with various substituents can be used successfully, and the highest ee’s were observed when (trimethylsilyl)acetylene was used (99% ee). Most of the iminium ion substrates were dihydroisoquinolinium ions, but alkynylation of the aromatic isoquinolinium ion was also achieved in 67% yield and 83% ee at a higher reaction temperature (−20 °C). The authors elegantly showed the potential of this reaction in two examples. First, they reduced alkyne 13 to deliver (S)-homolaudanosine, a natural product from an alkaloid family with neurologic activity, in high yield and enantiopurity. They also demonstrated that this alkynylation is amenable to solid phase synthesis; alkyne 14 was prepared by alkynylation of an isoquinolinium ion linked to a polystyrene bead through the C7 hydroxy group.
![[1860-5397-11-290-i5]](/bjoc/content/inline/1860-5397-11-290-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Schreiber’s alkynylation of N-alkylisoquinolinium ions.
Scheme 5: Schreiber’s alkynylation of N-alkylisoquinolinium ions.
In 2007, Ma and co-workers reported the first enantioselective, copper-catalyzed alkynylation of a pyridinium ion (Scheme 6) [26]. This reaction employs an N-acylpyridinium ion generated in situ and a CuI/Indabox catalyst. Ma found that the identity of base and solvent also affect the enantioselectivity, with iPr2N(n-Pr) and CH2Cl2 proving best. Notably, only 1,2-addition was observed. With respect to the alkyne, activated terminal acetylenes, such as ynones and propriolates, are best for this reaction. Unactivated alkynes give products in reasonable yields (63–77%), but poor enantioselectivities (1–11% ee). However, the use of activated acetylenes provides a functional group handle for elaboration, which the authors demonstrate in the preparation of indolizidine 223AB.
![[1860-5397-11-290-i6]](/bjoc/content/inline/1860-5397-11-290-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Ma’s alkynylation of pyridium ions.
Scheme 6: Ma’s alkynylation of pyridium ions.
Building on their initial discovery of a non-asymmetric, copper-catalyzed, three-component coupling of pyridine, benzoyl chloride and phenylacetylene [27], the Arndtsen group tackled the challenge of developing a catalyst for the alkynylation of cyclic N-acyliminium ions with unactivated alkynes [28]. Using the reaction of N-acylquinoline and phenylacetylene as their model, they observed lower ee’s with Pybox and Box ligands, and only 49% ee with Quinap. However, investigation of novel Pinap ligands led to notably higher ee’s (Scheme 7) [29]. These reaction conditions enable alkynylation of quinoline (20), isoquinoline (21), and pyridine (22) substrates, albeit in lower yields with pyridines.
![[1860-5397-11-290-i7]](/bjoc/content/inline/1860-5397-11-290-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Arndtsen’s alkynylation of cyclic iminium ions.
Scheme 7: Arndtsen’s alkynylation of cyclic iminium ions.
In 2011, Maruoka and co-workers investigated the use of isoquinolinium ions protected as azomethine imines [30]. The use of a CuOAc/Ph-Pybox catalyst enables the addition of a wide variety of alkynes to form isoquinolines with tertiary stereocenters in high yields and ee’s (Scheme 8A). Although lower ee’s were observed with o-tolylacetylene (43% ee) and 1-heptyne (75% ee), all other alkynes resulted in ≥85% ee. Even more impressive, the authors discovered conditions for a highly enantioselective alkynylation to form tetrasubstituted stereocenters. With 1-alkylisoquinolinium ions, high ee’s could not be achieved using only a CuOAc/Ph-Pybox catalyst. Postulating that the acetate of CuOAc may facilitate proton transfer from the alkyne to the azomethine imine, a necessary step to form both the cationic iminium and the copper acetylide, Maruoka investigated the use of a chiral acid co-catalyst. With the addition of chiral Brønsted acid co-catalyst 27, high yields and good to excellent levels of enantioselectivity were achieved in the addition of both aryl and aliphatic alkynes (Scheme 8B). The azomethine amine products 24 and 26 can be deprotected using SmI2. To our knowledge, this report is the first and only example to date of alkynylation of an imine or iminium ion to form a chiral tetrasubstituted center with high ee.
![[1860-5397-11-290-i8]](/bjoc/content/inline/1860-5397-11-290-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Maruoka’s alkynylation of azomethine imines.
Scheme 8: Maruoka’s alkynylation of azomethine imines.
Enantioselective, copper-catalyzed alkynylations have also been accomplished under solvent-free conditions. In 2013, Su and co-workers established that the CDC reaction of N-aryltetrahydroisoquinolines and alkynes can be accomplished under high-speed ball-milling conditions with copper balls (Scheme 9) [31]. Under these solvent-free reaction conditions, isoquinolines 28 were formed more quickly than the in-solvent reactions previously reported, but the ee’s are lower.
![[1860-5397-11-290-i9]](/bjoc/content/inline/1860-5397-11-290-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Su’s CDC of tetrahydroisoquinolines and alkynes under ball milling conditions.
Scheme 9: Su’s CDC of tetrahydroisoquinolines and alkynes under ball milling conditions.
Recently, a new entry to isoquinolinium ions has been established. In their studies of the three-component coupling of aldehydes, amines, and alkynes (A3 reaction), the Ma group serendipitiously discovered that isomerization of exocyclic iminium ion 30 results in the formation of endocyclic iminium ion 31 (Scheme 10) [32]. Subsequent alkynylation was accomplished using a CuI/N-Pinap catalyst to give N-benzylisoquinolines in exceptional yields and enantioselectivities. Notably, catalytic benzoic acid is necessary to achieve high yields. The authors hypothesize that this acid additive facilitates the iminium ion isomerization.
![[1860-5397-11-290-i10]](/bjoc/content/inline/1860-5397-11-290-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
This year, several examples of improved conditions for CDC reactions have been reported. With the emergence of photoredox catalysis as a powerful technique for organic synthesis, Li’s research group has discovered improved conditions for the CDC reaction of N-aryltetrahydroisoquinolines with alkynes (Scheme 11) [33]. By using an iridium-based photoredox catalyst in combination with benzoyl peroxide, iminium ion 7 is formed in situ. This strategy enables reduction of the reaction temperature, ultimately enabling higher enantioselectivities. With respect to the scope of alkynes, high ee’s were observed with both aryl- and alkylacetylenes, but lower yields were seen with alkylacetylenes. In addition, (trimethylsilyl)acetylene can be used but in lower yield and ee (40%, 60% ee).
![[1860-5397-11-290-i11]](/bjoc/content/inline/1860-5397-11-290-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Li’s CDC reaction using photoredox catalysis.
Scheme 11: Li’s CDC reaction using photoredox catalysis.
Also in 2015, Liu and co-workers have discovered conditions that enable the use of N-carbamoyltetrahydroisoquinolines in CDC reactions with alkynes (Scheme 12) [34]. These reactions utilize a CuBr/iPr-Pybox catalyst with 2,2,6,6-tetramethylpiperidine N-oxide as the oxidant. Liu’s mechanistic experiments indicate that the reaction likely proceeds via hemiaminal 35, formed via oxidation to an iminium ion and subsequent trapping by either EtOH or H2O. With respect to the substrate scope, addition of arylalkynes proceeds in high yields and ee’s, including those with some heteroaryl groups (37). Enynes can also be added, but result in lower yields and ee’s, as do octyne and methyl propriolate. A variety of substituents are tolerated on the isoquinoline, including halides, which provides a handle for further manipulation. Furthermore, the Cbz group can be easily removed via hydrogenation, as demonstrated in Liu’s efficient synthesis of homoprotoberberine.
![[1860-5397-11-290-i12]](/bjoc/content/inline/1860-5397-11-290-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Liu’s CDC reaction of N-carbamoyltetrahydroisoquinolines. T+BF4– = 2,2,6,6-tetramethylpiperidine N-oxide tetrafluoroborate.
Scheme 12: Liu’s CDC reaction of N-carbamoyltetrahydroisoquinolines. T+BF4– = 2,2,6,6-tetramethylpiperidine N-...
Very recently, Aponick’s group reported the use of a new P,N ligand, StackPhos, for enantioselective alkynylations of quinolinium ions [35]. They hypothesized that their imidazole-based P,N ligand would provide a different bite angle than Quinap or Pinap ligands and thus enable higher ee’s than previously obtained in the alkynylation of these challenging aromatic iminium ions [28]. Indeed, the CuBr/StackPhos catalyst provides generally high yields and exceptional levels of enantioselectivity in the alkynylation of N-carbamoylquinolinium ions 40 (Scheme 13). A broad scope was observed with the alkyne partner; additions of alkynes with aryl, heteroaryl, alkyl, and trimethylsilyl substituents result in 90–98% ee. The synthetic utility of these products, as well as their absolute configuration, was demonstrated by their reduction to the natural products (+)-galipinine, (+)-cuspareine, and (−)-angustureine.
![[1860-5397-11-290-i13]](/bjoc/content/inline/1860-5397-11-290-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Aponick’s alkynylation of N-carbomoylquinolinium ions using StackPhos as ligand.
Scheme 13: Aponick’s alkynylation of N-carbomoylquinolinium ions using StackPhos as ligand.
Additions to oxocarbenium ions
Similar to the development of enantioselective, metal-catalyzed alkynylations of iminium ions, enantioselective alkynylations of cyclic oxocarbenium ions were preceded by enantioselective alkynylations of acyclic aldehyde and ketone substrates [36-54]. In particular, seminal reports by the Carreira lab demonstrated that the use of stoichiometric metal acetylides was not required to achieve addition to aldehydes; catalytic Zn(OTf)2 and N-methylephedrine in combination with Et3N enabled in situ formation of chiral zinc acetylides, which underwent addition to aldehydes in good yields and high enantioselectivities (Scheme 14) [38]. Since this report, a variety of metal catalysts, including copper-based catalysts [36,37,50-52], have been employed in enantioselective alkynylations of aldehydes and ketones [36-54]. However, in contrast to the efforts made in enantioselective, metal-catalyzed additions to cyclic iminium ions, much less attention has been focused on analogous reactions of cyclic oxocarbenium ions. Given the precedent in alkynylations of aldehydes and ketones, as well as the alkynylations of iminium ions discussed above, we have pursued the development of enantioselective, metal-catalyzed alkynylation of cyclic oxocarbenium ion intermediates. In the course of these studies, we have found that copper-based catalysts are uniquely effective in promoting these alkynylations in good yields and ee’s.
![[1860-5397-11-290-i14]](/bjoc/content/inline/1860-5397-11-290-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Carreira’s enantioselective, catalytic alkynylation of aldehydes.
Scheme 14: Carreira’s enantioselective, catalytic alkynylation of aldehydes.
In 2011, we reported the first example of enantioselective alkynylation of a cyclic oxocarbenium ion intermediate (Scheme 15) [55]. Isochroman oxocarbenium ion 41 was formed in situ via Lewis acid-mediated ionization of a racemic acetal precursor. By using a Cu(MeCN)4PF6/BnBox catalyst, moderate to high yields and enantioselectivities were achieved in the addition of arylalkynes. Both yields and ee’s drop with vinyl- or alkylalkynes.
![[1860-5397-11-290-i15]](/bjoc/content/inline/1860-5397-11-290-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Watson’s alkynylation of isochroman oxocarbenium ions.
Scheme 15: Watson’s alkynylation of isochroman oxocarbenium ions.
Recently, we have demonstrated that enantioselective, copper-catalyzed alkynylations of oxocarbenium ions derived from chromene acetals 43 can also be achieved in high yields and ee’s (Scheme 16) [56]. In this case, the reaction concentration has a significant impact on the enantioselectivity. We also observed that the alkynylation of chromene acetals with electron-donating substituents proceeds in higher ee’s than less electron-rich substrates, suggesting that more stable oxocarbenium ions result in more selective reactions, potentially because they lead to later enantiodetermining transition states.
![[1860-5397-11-290-i16]](/bjoc/content/inline/1860-5397-11-290-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Watson’s alkynylation of chromene oxocarbenium ions.
Scheme 16: Watson’s alkynylation of chromene oxocarbenium ions.
We have also discovered a CuSPh/Ph-Pybox catalyst that enables the formation of diaryl, tetrasubstituted stereocenters via an enantioselective alkynylation reaction (Scheme 17) [57]. Remarkably, by tethering one aryl group to the oxygen atom, the two faces of oxocarbenium ion 46 can be distinguished by the catalyst. In general, excellent yields and enantioselectivities were observed, although lower yields and ee’s are seen with certain aliphatic alkynes. The addition of (dimethylphenylsilyl)acetylene proceeds in 53% yield and 81% ee, providing a silylalkyne that can be readily deprotected in quantitative yield for further elaboration. Notably, comparison of the ee of ligand vs ee of product revealed a significant positive nonlinear effect, indicting that catalyst aggregation occurs under these reaction conditions [58]. It is unclear at this point, whether these catalyst aggregates are on or off the catalytic cycle.
![[1860-5397-11-290-i17]](/bjoc/content/inline/1860-5397-11-290-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Watson’s alkynylation to set diaryl tetrasubstituted stereocenters.
Scheme 17: Watson’s alkynylation to set diaryl tetrasubstituted stereocenters.
Conclusion
As described above, highly enantioselective, copper-catalyzed alkynylations of cyclic iminium and oxocarbenium ion intermediates have been achieved. α-Chiral piperidines, quinolines, isoquinolines, and benzopyrans are accessible via these reactions. The utility of these alkyne-substituted products has been demonstrated via elaboration to biologically active natural products. As evidenced by the most recent reports, particularly for iminium ions, progress is on-going to determine stable precursors to the requisite iminium ion intermediates and to identify readily removed protecting groups.
Given the potential of enantioselective, copper-catalyzed alkynylations to deliver important scaffolds, significant effort is still required to develop this class of reactions. In particular, all cyclic iminium and oxocarbenium ions utilized to date have been limited to those that form stabilized cationic intermediates (benzylic or aromatic). With few exceptions, the vast majority lack β-hydrogens, so competitive elimination reactions are not possible. Conditions to enable the use of non-stabilized iminium and oxocarbenium ions with β-hydrogens would represent an exceptional advance in this field and allow access to a wide variety of useful compounds.
In addition, little is understood about how these chiral copper catalysts provide high levels of enantioselectivity. This lack is likely due to the multiple possibilities for copper acetylide structures (monomer, dimer, dicopper acetylide, etc.) [18,59-65]. Careful mechanistic studies to elucidate the structures of chiral copper acetylides and to provide stereochemical rationale for the enantioselectivities of these reactions is needed to enable further development of catalysts in this important area.
References
-
Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 3341–3370. doi:10.1021/cr030692k
Return to citation in text: [1] -
Bentley, K. W. Nat. Prod. Rep. 2004, 21, 395–424. doi:10.1039/b212259f
Return to citation in text: [1] -
Daly, J. W. J. Med. Chem. 2003, 46, 445–452. doi:10.1021/jm0204845
Return to citation in text: [1] -
Albrecht, U.; Lalk, M.; Langer, P. Bioorg. Med. Chem. 2005, 13, 1531–1536. doi:10.1016/j.bmc.2004.12.031
Return to citation in text: [1] -
TenBrink, R. E.; Bergh, C. L.; Duncan, J. N.; Harris, D. W.; Huff, R. M.; Lahti, R. A.; Lawson, C. F.; Lutzke, B. S.; Martin, I. J.; Rees, S. A.; Schlachter, S. K.; Sih, J. C.; Smith, M. W. J. Med. Chem. 1996, 39, 2435–2437. doi:10.1021/jm960084f
Return to citation in text: [1] -
Xu, J.; Kjer, J.; Sendker, J.; Wray, V.; Guan, H.; Edrada, R.; Müller, W. E. G.; Bayer, M.; Lin, W.; Wu, J.; Proksch, P. Bioorg. Med. Chem. 2009, 17, 7362–7367. doi:10.1016/j.bmc.2009.08.031
Return to citation in text: [1] -
Pierce, M. E.; Parsons, R. L., Jr.; Radesca, L. A.; Lo, Y. S.; Silverman, S.; Moore, J. R.; Islam, Q.; Choudhury, A.; Fortunak, J. M. D.; Nguyen, D.; Luo, C.; Morgan, S. J.; Davis, W. P.; Confalone, P. N.; Chen, C.-y.; Tillyer, R. D.; Frey, L.; Tan, L.; Xu, F.; Zhao, D.; Thompson, A. S.; Corley, E. G.; Grabowski, E. J. J.; Reamer, R.; Reider, P. J. J. Org. Chem. 1998, 63, 8536–8543. doi:10.1021/jo981170l
Return to citation in text: [1] -
Tan, L.; Chen, C.-y.; Tillyer, R. D.; Grabowski, E. J. J.; Reider, P. J. Angew. Chem., Int. Ed. 1999, 38, 711–713. doi:10.1002/(SICI)1521-3773(19990301)38:5<711::AID-ANIE711>3.0.CO;2-W
See for example.
Return to citation in text: [1] -
Ahamed, M.; Todd, M. H. Eur. J. Org. Chem. 2010, 5935–5942. doi:10.1002/ejoc.201000877
Return to citation in text: [1] -
Cozzi, P. G.; Hilgraf, R.; Zimmermann, N. Eur. J. Org. Chem. 2004, 4095–4105. doi:10.1002/ejoc.200400246
Return to citation in text: [1] -
Kumagai, N.; Shibasaki, M. Bull. Chem. Soc. Jpn. 2015, 88, 503–517. doi:10.1246/bcsj.20140302
Return to citation in text: [1] -
Ohshima, T. To Catalytic Asymmetric 1,2-Alkynylation. In Comprehensive Chirality; Carreira, E. M.; Yamamoto, H., Eds.; Elsevier: Amsterdam, 2012; pp 355–377. doi:10.1016/B978-0-08-095167-6.00418-3
Return to citation in text: [1] -
Pu, L. Tetrahedron 2003, 59, 9873–9886. doi:10.1016/j.tet.2003.10.042
Return to citation in text: [1] -
Riant, O.; Hannedouche, J. Org. Biomol. Chem. 2007, 5, 873–888. doi:10.1039/b617746h
Return to citation in text: [1] -
Trost, B. M.; Bartlett, M. J. Catalytic Enantioselective Addition of Terminal Alkynes to Carbonyls. In Modern Alkyne Chemistry; Trost, B. M.; Li, C.-J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2014; pp 201–238. doi:10.1002/9783527677894.ch8
Return to citation in text: [1] -
Wei, C.; Li, C.-J. J. Am. Chem. Soc. 2002, 124, 5638–5639. doi:10.1021/ja026007t
Return to citation in text: [1] -
Wei, C.; Mague, J. T.; Li, C.-J. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5749–5754. doi:10.1073/pnas.0307150101
Return to citation in text: [1] -
Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M
Return to citation in text: [1] [2] -
Koradin, C.; Gommermann, N.; Polborn, K.; Knochel, P. Chem. – Eur. J. 2003, 9, 2797–2811. doi:10.1002/chem.200204691
Return to citation in text: [1] -
Gommermann, N.; Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2003, 42, 5763–5766. doi:10.1002/anie.200352578
Return to citation in text: [1] -
Yin, L.; Otsuka, Y.; Takada, H.; Mouri, S.; Yazaki, R.; Kumagai, N.; Shibasaki, M. Org. Lett. 2013, 15, 698–701. doi:10.1021/ol3035609
See for a recent example of enantioselective, copper-catalyzed alkynlation of acyclic ketoimines.
Return to citation in text: [1] -
Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997–4999. doi:10.1021/ol047814v
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763
Return to citation in text: [1] -
Li, Z.; MacLeod, P. D.; Li, C.-J. Tetrahedron: Asymmetry 2006, 17, 590–597. doi:10.1016/j.tetasy.2006.02.007
Return to citation in text: [1] -
Taylor, A. M.; Schreiber, S. L. Org. Lett. 2006, 8, 143–146. doi:10.1021/ol0526165
Return to citation in text: [1] -
Sun, Z.; Yu, S.; Ding, Z.; Ma, D. J. Am. Chem. Soc. 2007, 129, 9300–9301. doi:10.1021/ja0734849
Return to citation in text: [1] -
Black, D. A.; Arndtsen, B. A. Org. Lett. 2004, 6, 1107–1110. doi:10.1021/ol036462+
Return to citation in text: [1] -
Black, D. A.; Beveridge, R. E.; Arndtsen, B. A. J. Org. Chem. 2008, 73, 1906–1910. doi:10.1021/jo702293h
Return to citation in text: [1] [2] -
Knöpfel, T. F.; Aschwanden, P.; Ichikawa, T.; Watanabe, T.; Carreira, E. M. Angew. Chem., Int. Ed. 2004, 43, 5971–5973. doi:10.1002/anie.200461286
See for the initial report of Pinap ligands.
Return to citation in text: [1] -
Hashimoto, T.; Omote, M.; Maruoka, K. Angew. Chem., Int. Ed. 2011, 50, 8952–8955. doi:10.1002/anie.201104017
Return to citation in text: [1] -
Yu, J.; Li, Z.; Jia, K.; Jiang, Z.; Liu, M.; Su, W. Tetrahedron Lett. 2013, 54, 2006–2009. doi:10.1016/j.tetlet.2013.02.007
Return to citation in text: [1] -
Lin, W.; Cao, T.; Fan, W.; Han, Y.; Kuang, J.; Luo, H.; Miao, B.; Tang, X.; Yu, Q.; Yuan, W.; Zhang, J.; Zhu, C.; Ma, S. Angew. Chem., Int. Ed. 2014, 53, 277–281. doi:10.1002/anie.201308699
Return to citation in text: [1] -
Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/C4OB02138J
Return to citation in text: [1] -
Sun, S.; Li, C.; Floreancig, P. E.; Lou, H.; Liu, L. Org. Lett. 2015, 17, 1684–1687. doi:10.1021/acs.orglett.5b00447
Return to citation in text: [1] -
Pappoppula, M.; Cardoso, F. S. P.; Garrett, B. O.; Aponick, A. Angew. Chem., Int. Ed. 2015, 54, 15202–15206. doi:10.1002/anie.201507848
Return to citation in text: [1] -
Asano, Y.; Hara, K.; Ito, H.; Sawamura, M. Org. Lett. 2007, 9, 3901–3904. doi:10.1021/ol701534n
Return to citation in text: [1] [2] [3] -
Asano, Y.; Ito, H.; Hara, K.; Sawamura, M. Organometallics 2008, 27, 5984–5996. doi:10.1021/om800667c
Return to citation in text: [1] [2] [3] -
Anand, N. K.; Carreira, E. M. J. Am. Chem. Soc. 2001, 123, 9687–9688. doi:10.1021/ja016378u
Return to citation in text: [1] [2] [3] -
Boyall, D.; Frantz, D. E.; Carreira, E. M. Org. Lett. 2002, 4, 2605–2606. doi:10.1021/ol026282k
Return to citation in text: [1] [2] -
Frantz, D. E.; Fässler, R.; Carreira, E. M. J. Am. Chem. Soc. 2000, 122, 1806–1807. doi:10.1021/ja993838z
Return to citation in text: [1] [2] -
Ito, J.-i.; Asai, R.; Nishiyama, H. Org. Lett. 2010, 12, 3860–3862. doi:10.1021/ol1015338
Return to citation in text: [1] [2] -
Koyuncu, H.; Dogan, Ö. Org. Lett. 2007, 9, 3477–3479. doi:10.1021/ol701535y
Return to citation in text: [1] [2] -
Takita, R.; Yakura, K.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 13760–13761. doi:10.1021/ja053946n
Return to citation in text: [1] [2] -
Trost, B. M.; Chan, V. S.; Yamamoto, D. J. Am. Chem. Soc. 2010, 132, 5186–5192. doi:10.1021/ja910656b
Return to citation in text: [1] [2] -
Yang, F.; Xi, P.; Yang, L.; Lan, J.; Xie, R.; You, J. J. Org. Chem. 2007, 72, 5457–5460. doi:10.1021/jo0707535
Return to citation in text: [1] [2] -
Li, X.; Lu, G.; Kwok, W. H.; Chan, A. S. C. J. Am. Chem. Soc. 2002, 124, 12636–12637. doi:10.1021/ja025541y
Return to citation in text: [1] [2] -
Lu, G.; Li, X.; Chan, W. L.; Chan, A. S. C. Chem. Commun. 2002, 172–173. doi:10.1039/b107817h
Return to citation in text: [1] [2] -
Moore, D.; Pu, L. Org. Lett. 2002, 4, 1855–1857. doi:10.1021/ol025825n
Return to citation in text: [1] [2] -
Gao, G.; Moore, D.; Xie, R.-G.; Pu, L. Org. Lett. 2002, 4, 4143–4146. doi:10.1021/ol026921r
Return to citation in text: [1] [2] -
Lu, G.; Li, X.; Jia, X.; Chan, W. L.; Chan, A. S. C. Angew. Chem., Int. Ed. 2003, 42, 5057–5058. doi:10.1002/anie.200352013
Return to citation in text: [1] [2] [3] -
Lu, G.; Li, X.; Li, Y.-M.; Kwong, F. Y.; Chan, A. S. C. Adv. Synth. Catal. 2006, 348, 1926–1933. doi:10.1002/adsc.200606078
Return to citation in text: [1] [2] [3] -
Motoki, R.; Kanai, M.; Shibasaki, M. Org. Lett. 2007, 9, 2997–3000. doi:10.1021/ol071003k
Return to citation in text: [1] [2] [3] -
Chen, C.; Hong, L.; Xu, Z.-Q.; Liu, L.; Wang, R. Org. Lett. 2006, 8, 2277–2280. doi:10.1021/ol060526+
Return to citation in text: [1] [2] -
Zhou, S.; Chen, C.-R.; Gau, H.-M. Org. Lett. 2010, 12, 48–51. doi:10.1021/ol902454n
Return to citation in text: [1] [2] -
Maity, P.; Srinivas, H. D.; Watson, M. P. J. Am. Chem. Soc. 2011, 133, 17142–17145. doi:10.1021/ja207585p
Return to citation in text: [1] -
Srinivas, H. D.; Maity, P.; Yap, G. P. A.; Watson, M. P. J. Org. Chem. 2015, 80, 4003–4016. doi:10.1021/acs.joc.5b00364
Return to citation in text: [1] -
Dasgupta, S.; Rivas, T.; Watson, M. P. Angew. Chem., Int. Ed. 2015, 54, 14154–14158. doi:10.1002/anie.201507373
Return to citation in text: [1] -
Girard, C.; Kagan, H. B. Angew. Chem., Int. Ed. 1998, 37, 2922–2959. doi:10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1
Return to citation in text: [1] -
Díez, J.; Gamasa, M. P.; Panera, M. Inorg. Chem. 2006, 45, 10043–10045. doi:10.1021/ic061453t
Return to citation in text: [1] -
Panera, M. a.; Díez, J.; Merino, I.; Rubio, E.; Gamasa, M. P. Inorg. Chem. 2009, 48, 11147–11160. doi:10.1021/ic901527x
Return to citation in text: [1] -
Nakajima, K.; Shibata, M.; Nishibayashi, Y. J. Am. Chem. Soc. 2015, 137, 2472–2475. doi:10.1021/jacs.5b00004
Return to citation in text: [1] -
Ahlquist, M.; Fokin, V. V. Organometallics 2007, 26, 4389–4391. doi:10.1021/om700669v
Return to citation in text: [1] -
Díez, J.; Gamasa, M. P.; Gimeno, J.; Aguirre, A.; García-Granda, S.; Holubova, J.; Falvello, L. R. Organometallics 1999, 18, 662–669. doi:10.1021/om980592i
Return to citation in text: [1] -
Mealli, C.; Godinho, S. S. M. C.; Calhorda, M. J. Organometallics 2001, 20, 1734–1742. doi:10.1021/om0009132
Return to citation in text: [1] -
Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed. 2005, 44, 2210–2215. doi:10.1002/anie.200461496
Return to citation in text: [1]
| 57. | Dasgupta, S.; Rivas, T.; Watson, M. P. Angew. Chem., Int. Ed. 2015, 54, 14154–14158. doi:10.1002/anie.201507373 |
| 58. | Girard, C.; Kagan, H. B. Angew. Chem., Int. Ed. 1998, 37, 2922–2959. doi:10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1 |
| 18. | Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M |
| 59. | Díez, J.; Gamasa, M. P.; Panera, M. Inorg. Chem. 2006, 45, 10043–10045. doi:10.1021/ic061453t |
| 60. | Panera, M. a.; Díez, J.; Merino, I.; Rubio, E.; Gamasa, M. P. Inorg. Chem. 2009, 48, 11147–11160. doi:10.1021/ic901527x |
| 61. | Nakajima, K.; Shibata, M.; Nishibayashi, Y. J. Am. Chem. Soc. 2015, 137, 2472–2475. doi:10.1021/jacs.5b00004 |
| 62. | Ahlquist, M.; Fokin, V. V. Organometallics 2007, 26, 4389–4391. doi:10.1021/om700669v |
| 63. | Díez, J.; Gamasa, M. P.; Gimeno, J.; Aguirre, A.; García-Granda, S.; Holubova, J.; Falvello, L. R. Organometallics 1999, 18, 662–669. doi:10.1021/om980592i |
| 64. | Mealli, C.; Godinho, S. S. M. C.; Calhorda, M. J. Organometallics 2001, 20, 1734–1742. doi:10.1021/om0009132 |
| 65. | Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed. 2005, 44, 2210–2215. doi:10.1002/anie.200461496 |
| 1. | Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 3341–3370. doi:10.1021/cr030692k |
| 2. | Bentley, K. W. Nat. Prod. Rep. 2004, 21, 395–424. doi:10.1039/b212259f |
| 3. | Daly, J. W. J. Med. Chem. 2003, 46, 445–452. doi:10.1021/jm0204845 |
| 4. | Albrecht, U.; Lalk, M.; Langer, P. Bioorg. Med. Chem. 2005, 13, 1531–1536. doi:10.1016/j.bmc.2004.12.031 |
| 5. | TenBrink, R. E.; Bergh, C. L.; Duncan, J. N.; Harris, D. W.; Huff, R. M.; Lahti, R. A.; Lawson, C. F.; Lutzke, B. S.; Martin, I. J.; Rees, S. A.; Schlachter, S. K.; Sih, J. C.; Smith, M. W. J. Med. Chem. 1996, 39, 2435–2437. doi:10.1021/jm960084f |
| 6. | Xu, J.; Kjer, J.; Sendker, J.; Wray, V.; Guan, H.; Edrada, R.; Müller, W. E. G.; Bayer, M.; Lin, W.; Wu, J.; Proksch, P. Bioorg. Med. Chem. 2009, 17, 7362–7367. doi:10.1016/j.bmc.2009.08.031 |
| 7. | Pierce, M. E.; Parsons, R. L., Jr.; Radesca, L. A.; Lo, Y. S.; Silverman, S.; Moore, J. R.; Islam, Q.; Choudhury, A.; Fortunak, J. M. D.; Nguyen, D.; Luo, C.; Morgan, S. J.; Davis, W. P.; Confalone, P. N.; Chen, C.-y.; Tillyer, R. D.; Frey, L.; Tan, L.; Xu, F.; Zhao, D.; Thompson, A. S.; Corley, E. G.; Grabowski, E. J. J.; Reamer, R.; Reider, P. J. J. Org. Chem. 1998, 63, 8536–8543. doi:10.1021/jo981170l |
| 18. | Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M |
| 19. | Koradin, C.; Gommermann, N.; Polborn, K.; Knochel, P. Chem. – Eur. J. 2003, 9, 2797–2811. doi:10.1002/chem.200204691 |
| 20. | Gommermann, N.; Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2003, 42, 5763–5766. doi:10.1002/anie.200352578 |
| 30. | Hashimoto, T.; Omote, M.; Maruoka, K. Angew. Chem., Int. Ed. 2011, 50, 8952–8955. doi:10.1002/anie.201104017 |
| 16. | Wei, C.; Li, C.-J. J. Am. Chem. Soc. 2002, 124, 5638–5639. doi:10.1021/ja026007t |
| 17. | Wei, C.; Mague, J. T.; Li, C.-J. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5749–5754. doi:10.1073/pnas.0307150101 |
| 31. | Yu, J.; Li, Z.; Jia, K.; Jiang, Z.; Liu, M.; Su, W. Tetrahedron Lett. 2013, 54, 2006–2009. doi:10.1016/j.tetlet.2013.02.007 |
| 9. | Ahamed, M.; Todd, M. H. Eur. J. Org. Chem. 2010, 5935–5942. doi:10.1002/ejoc.201000877 |
| 10. | Cozzi, P. G.; Hilgraf, R.; Zimmermann, N. Eur. J. Org. Chem. 2004, 4095–4105. doi:10.1002/ejoc.200400246 |
| 11. | Kumagai, N.; Shibasaki, M. Bull. Chem. Soc. Jpn. 2015, 88, 503–517. doi:10.1246/bcsj.20140302 |
| 12. | Ohshima, T. To Catalytic Asymmetric 1,2-Alkynylation. In Comprehensive Chirality; Carreira, E. M.; Yamamoto, H., Eds.; Elsevier: Amsterdam, 2012; pp 355–377. doi:10.1016/B978-0-08-095167-6.00418-3 |
| 13. | Pu, L. Tetrahedron 2003, 59, 9873–9886. doi:10.1016/j.tet.2003.10.042 |
| 14. | Riant, O.; Hannedouche, J. Org. Biomol. Chem. 2007, 5, 873–888. doi:10.1039/b617746h |
| 15. | Trost, B. M.; Bartlett, M. J. Catalytic Enantioselective Addition of Terminal Alkynes to Carbonyls. In Modern Alkyne Chemistry; Trost, B. M.; Li, C.-J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2014; pp 201–238. doi:10.1002/9783527677894.ch8 |
| 28. | Black, D. A.; Beveridge, R. E.; Arndtsen, B. A. J. Org. Chem. 2008, 73, 1906–1910. doi:10.1021/jo702293h |
| 8. |
Tan, L.; Chen, C.-y.; Tillyer, R. D.; Grabowski, E. J. J.; Reider, P. J. Angew. Chem., Int. Ed. 1999, 38, 711–713. doi:10.1002/(SICI)1521-3773(19990301)38:5<711::AID-ANIE711>3.0.CO;2-W
See for example. |
| 29. |
Knöpfel, T. F.; Aschwanden, P.; Ichikawa, T.; Watanabe, T.; Carreira, E. M. Angew. Chem., Int. Ed. 2004, 43, 5971–5973. doi:10.1002/anie.200461286
See for the initial report of Pinap ligands. |
| 24. | Li, Z.; MacLeod, P. D.; Li, C.-J. Tetrahedron: Asymmetry 2006, 17, 590–597. doi:10.1016/j.tetasy.2006.02.007 |
| 26. | Sun, Z.; Yu, S.; Ding, Z.; Ma, D. J. Am. Chem. Soc. 2007, 129, 9300–9301. doi:10.1021/ja0734849 |
| 23. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763 |
| 27. | Black, D. A.; Arndtsen, B. A. Org. Lett. 2004, 6, 1107–1110. doi:10.1021/ol036462+ |
| 21. |
Yin, L.; Otsuka, Y.; Takada, H.; Mouri, S.; Yazaki, R.; Kumagai, N.; Shibasaki, M. Org. Lett. 2013, 15, 698–701. doi:10.1021/ol3035609
See for a recent example of enantioselective, copper-catalyzed alkynlation of acyclic ketoimines. |
| 25. | Taylor, A. M.; Schreiber, S. L. Org. Lett. 2006, 8, 143–146. doi:10.1021/ol0526165 |
| 34. | Sun, S.; Li, C.; Floreancig, P. E.; Lou, H.; Liu, L. Org. Lett. 2015, 17, 1684–1687. doi:10.1021/acs.orglett.5b00447 |
| 32. | Lin, W.; Cao, T.; Fan, W.; Han, Y.; Kuang, J.; Luo, H.; Miao, B.; Tang, X.; Yu, Q.; Yuan, W.; Zhang, J.; Zhu, C.; Ma, S. Angew. Chem., Int. Ed. 2014, 53, 277–281. doi:10.1002/anie.201308699 |
| 33. | Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/C4OB02138J |
| 55. | Maity, P.; Srinivas, H. D.; Watson, M. P. J. Am. Chem. Soc. 2011, 133, 17142–17145. doi:10.1021/ja207585p |
| 56. | Srinivas, H. D.; Maity, P.; Yap, G. P. A.; Watson, M. P. J. Org. Chem. 2015, 80, 4003–4016. doi:10.1021/acs.joc.5b00364 |
| 36. | Asano, Y.; Hara, K.; Ito, H.; Sawamura, M. Org. Lett. 2007, 9, 3901–3904. doi:10.1021/ol701534n |
| 37. | Asano, Y.; Ito, H.; Hara, K.; Sawamura, M. Organometallics 2008, 27, 5984–5996. doi:10.1021/om800667c |
| 50. | Lu, G.; Li, X.; Jia, X.; Chan, W. L.; Chan, A. S. C. Angew. Chem., Int. Ed. 2003, 42, 5057–5058. doi:10.1002/anie.200352013 |
| 51. | Lu, G.; Li, X.; Li, Y.-M.; Kwong, F. Y.; Chan, A. S. C. Adv. Synth. Catal. 2006, 348, 1926–1933. doi:10.1002/adsc.200606078 |
| 52. | Motoki, R.; Kanai, M.; Shibasaki, M. Org. Lett. 2007, 9, 2997–3000. doi:10.1021/ol071003k |
| 36. | Asano, Y.; Hara, K.; Ito, H.; Sawamura, M. Org. Lett. 2007, 9, 3901–3904. doi:10.1021/ol701534n |
| 37. | Asano, Y.; Ito, H.; Hara, K.; Sawamura, M. Organometallics 2008, 27, 5984–5996. doi:10.1021/om800667c |
| 38. | Anand, N. K.; Carreira, E. M. J. Am. Chem. Soc. 2001, 123, 9687–9688. doi:10.1021/ja016378u |
| 39. | Boyall, D.; Frantz, D. E.; Carreira, E. M. Org. Lett. 2002, 4, 2605–2606. doi:10.1021/ol026282k |
| 40. | Frantz, D. E.; Fässler, R.; Carreira, E. M. J. Am. Chem. Soc. 2000, 122, 1806–1807. doi:10.1021/ja993838z |
| 41. | Ito, J.-i.; Asai, R.; Nishiyama, H. Org. Lett. 2010, 12, 3860–3862. doi:10.1021/ol1015338 |
| 42. | Koyuncu, H.; Dogan, Ö. Org. Lett. 2007, 9, 3477–3479. doi:10.1021/ol701535y |
| 43. | Takita, R.; Yakura, K.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 13760–13761. doi:10.1021/ja053946n |
| 44. | Trost, B. M.; Chan, V. S.; Yamamoto, D. J. Am. Chem. Soc. 2010, 132, 5186–5192. doi:10.1021/ja910656b |
| 45. | Yang, F.; Xi, P.; Yang, L.; Lan, J.; Xie, R.; You, J. J. Org. Chem. 2007, 72, 5457–5460. doi:10.1021/jo0707535 |
| 46. | Li, X.; Lu, G.; Kwok, W. H.; Chan, A. S. C. J. Am. Chem. Soc. 2002, 124, 12636–12637. doi:10.1021/ja025541y |
| 47. | Lu, G.; Li, X.; Chan, W. L.; Chan, A. S. C. Chem. Commun. 2002, 172–173. doi:10.1039/b107817h |
| 48. | Moore, D.; Pu, L. Org. Lett. 2002, 4, 1855–1857. doi:10.1021/ol025825n |
| 49. | Gao, G.; Moore, D.; Xie, R.-G.; Pu, L. Org. Lett. 2002, 4, 4143–4146. doi:10.1021/ol026921r |
| 50. | Lu, G.; Li, X.; Jia, X.; Chan, W. L.; Chan, A. S. C. Angew. Chem., Int. Ed. 2003, 42, 5057–5058. doi:10.1002/anie.200352013 |
| 51. | Lu, G.; Li, X.; Li, Y.-M.; Kwong, F. Y.; Chan, A. S. C. Adv. Synth. Catal. 2006, 348, 1926–1933. doi:10.1002/adsc.200606078 |
| 52. | Motoki, R.; Kanai, M.; Shibasaki, M. Org. Lett. 2007, 9, 2997–3000. doi:10.1021/ol071003k |
| 53. | Chen, C.; Hong, L.; Xu, Z.-Q.; Liu, L.; Wang, R. Org. Lett. 2006, 8, 2277–2280. doi:10.1021/ol060526+ |
| 54. | Zhou, S.; Chen, C.-R.; Gau, H.-M. Org. Lett. 2010, 12, 48–51. doi:10.1021/ol902454n |
| 36. | Asano, Y.; Hara, K.; Ito, H.; Sawamura, M. Org. Lett. 2007, 9, 3901–3904. doi:10.1021/ol701534n |
| 37. | Asano, Y.; Ito, H.; Hara, K.; Sawamura, M. Organometallics 2008, 27, 5984–5996. doi:10.1021/om800667c |
| 38. | Anand, N. K.; Carreira, E. M. J. Am. Chem. Soc. 2001, 123, 9687–9688. doi:10.1021/ja016378u |
| 39. | Boyall, D.; Frantz, D. E.; Carreira, E. M. Org. Lett. 2002, 4, 2605–2606. doi:10.1021/ol026282k |
| 40. | Frantz, D. E.; Fässler, R.; Carreira, E. M. J. Am. Chem. Soc. 2000, 122, 1806–1807. doi:10.1021/ja993838z |
| 41. | Ito, J.-i.; Asai, R.; Nishiyama, H. Org. Lett. 2010, 12, 3860–3862. doi:10.1021/ol1015338 |
| 42. | Koyuncu, H.; Dogan, Ö. Org. Lett. 2007, 9, 3477–3479. doi:10.1021/ol701535y |
| 43. | Takita, R.; Yakura, K.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 13760–13761. doi:10.1021/ja053946n |
| 44. | Trost, B. M.; Chan, V. S.; Yamamoto, D. J. Am. Chem. Soc. 2010, 132, 5186–5192. doi:10.1021/ja910656b |
| 45. | Yang, F.; Xi, P.; Yang, L.; Lan, J.; Xie, R.; You, J. J. Org. Chem. 2007, 72, 5457–5460. doi:10.1021/jo0707535 |
| 46. | Li, X.; Lu, G.; Kwok, W. H.; Chan, A. S. C. J. Am. Chem. Soc. 2002, 124, 12636–12637. doi:10.1021/ja025541y |
| 47. | Lu, G.; Li, X.; Chan, W. L.; Chan, A. S. C. Chem. Commun. 2002, 172–173. doi:10.1039/b107817h |
| 48. | Moore, D.; Pu, L. Org. Lett. 2002, 4, 1855–1857. doi:10.1021/ol025825n |
| 49. | Gao, G.; Moore, D.; Xie, R.-G.; Pu, L. Org. Lett. 2002, 4, 4143–4146. doi:10.1021/ol026921r |
| 50. | Lu, G.; Li, X.; Jia, X.; Chan, W. L.; Chan, A. S. C. Angew. Chem., Int. Ed. 2003, 42, 5057–5058. doi:10.1002/anie.200352013 |
| 51. | Lu, G.; Li, X.; Li, Y.-M.; Kwong, F. Y.; Chan, A. S. C. Adv. Synth. Catal. 2006, 348, 1926–1933. doi:10.1002/adsc.200606078 |
| 52. | Motoki, R.; Kanai, M.; Shibasaki, M. Org. Lett. 2007, 9, 2997–3000. doi:10.1021/ol071003k |
| 53. | Chen, C.; Hong, L.; Xu, Z.-Q.; Liu, L.; Wang, R. Org. Lett. 2006, 8, 2277–2280. doi:10.1021/ol060526+ |
| 54. | Zhou, S.; Chen, C.-R.; Gau, H.-M. Org. Lett. 2010, 12, 48–51. doi:10.1021/ol902454n |
| 38. | Anand, N. K.; Carreira, E. M. J. Am. Chem. Soc. 2001, 123, 9687–9688. doi:10.1021/ja016378u |
| 35. | Pappoppula, M.; Cardoso, F. S. P.; Garrett, B. O.; Aponick, A. Angew. Chem., Int. Ed. 2015, 54, 15202–15206. doi:10.1002/anie.201507848 |
| 28. | Black, D. A.; Beveridge, R. E.; Arndtsen, B. A. J. Org. Chem. 2008, 73, 1906–1910. doi:10.1021/jo702293h |
© 2015 Liu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)