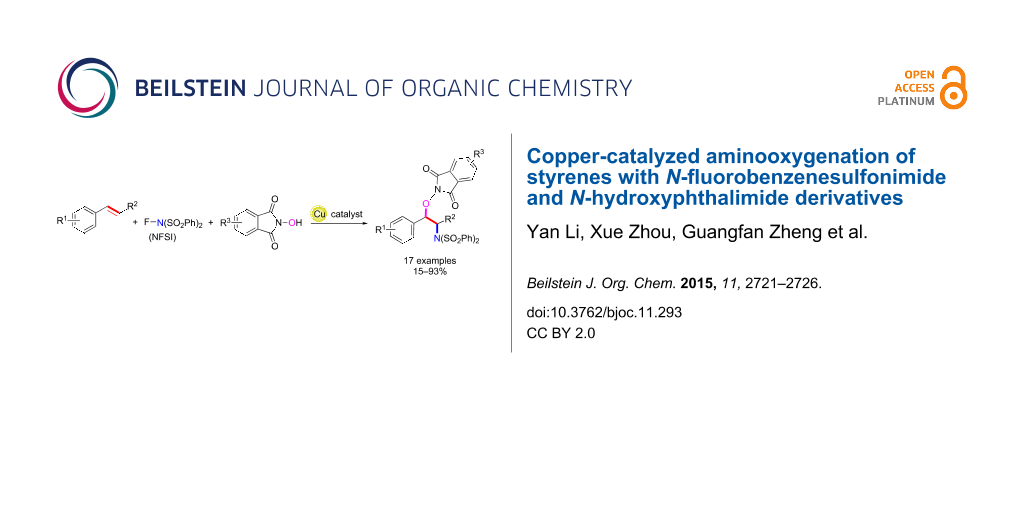
Abstract
A copper-catalyzed aminooxygenation reaction of styrenes with N-fluorobenzenesulfonimide and N-hydroxyphthalimide derivatives has been developed. The aminooxygenation product could be converted into the corresponding alcohol or free amine through the cleavage of the N–O or C–N bond of the N-hydroxyphthalimide moiety.

Graphical Abstract
Findings
Direct aminooxygenation of alkenes provides a straightforward and powerful approach to construct the 1,2-aminoalcohol skeleton [1], which is ubiquitous in bioactive compounds (such as the drugs bestatin (1) and tamiflu (2), the natural products Al-77-B (3) and hapolosin (4); Figure 1) [2] and has also been widely used as chiral ligands and auxiliaries in asymmetric synthesis [3]. Therefore, the development of a new aminooxygenation reaction is still highly attractive [4]. Most of the existing aminooxygenation reactions involve an intramolecular cyclization step [5-33] to provide various valuable cyclic compounds. Comparatively, methods for an intermolecular three-component aminooxygenation reaction are considerably less established. In 2006, Stahl and co-workers reported a Pd-catalyzed aminooxygenation reaction of alkenes with phthalimide and (diacetoxyiodo)benzene through cis-aminopalladation and SN2 C–O bond formation [34]. In 2013, Zhu and co-workers described an n-Bu4NI-catalyzed aminooxygenation of inactive alkenes with benzotriazole and water which underwent a nitrogen-centred radical addition and a nucleophilic oxygen attack [35]. Very recently, Studer and co-workers presented an aminooxygenation of alkenes with N-fluorobenzenesulfonimide (NFSI) and sodium 2,2,6,6-tetramethylpiperidine-1-olate (TEMPONa) via nitrogen-centred radical addition to the alkene followed by trapping of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) [36].
![[1860-5397-11-293-1]](/bjoc/content/figures/1860-5397-11-293-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bioactive compounds containing 1,2-aminoalcohol motif.
Figure 1: Bioactive compounds containing 1,2-aminoalcohol motif.
NFSI is a very interesting reagent. Besides classic electrophilic fluorination reagent [37], it has been used not only as fluoride-atom transfer reagent [38-40] but also as nucleophilic/radical amination reagent [41]. We are highly interested in the multiple reaction modes of NFSI [37-41], especially as a nitrogen-centred radical. In this context, we have realized copper-catalyzed benzylic sp3 C–H amination [42], aminative multiple functionalization of alkynes [43], diamination, aminocyanation [44] and aminofluorination of alkenes [45], as well as amination of allenes [46]. Encouraged by these results, we try to develop copper-catalyzed aminooxygenation of alkenes by using NFSI. Herein, we report a simple and efficient copper-catalyzed three-component aminooxygenation reaction of styrenes with NFSI and N-hydroxyphthalimide (NHPI) derivatives (Scheme 1).
![[1860-5397-11-293-i1]](/bjoc/content/inline/1860-5397-11-293-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Copper-catalyzed radical aminooxygenation reaction of styrenes.
Scheme 1: Copper-catalyzed radical aminooxygenation reaction of styrenes.
Initially, we conducted the three-component amnooxygenation of styrene 1a with NFSI and NHPI (2a). After the reaction of 1a (0.3 mmol), NFSI (0.3 mmol, 1.0 equiv) and 2a (0.45 mmol, 1.5 equiv) was performed in the presence of Cu(OTf)2 (10 mol %) in dichloromethane (DCM, 2 mL) under nitrogen atmosphere at 70 °C for 10.0 h, the desired aminooxygenation product 3a was obtained in 39% yield (Table 1, entry 1). A variety of copper salts such as CuCl, CuBr, CuI, [(CH3CN)4Cu]PF6, CuCN, Cu(acac)2, Cu(OAc)2, CuBr2 and CuCl2 were examined (Table 1, entries 2–10). We found that CuCl2 was the most effective catalyst, affording 3a in 55% yield (Table 1, entry 10). No reaction was observed in the absence of copper salts (Table 1, entry 11). Next, the reaction solvents were scanned. 1,2-Dichloroethane (DCE) and CH3CN were not efficient solvents, providing 3a in 9% and 20% yields, respectively (Table 1, entries 12 and 13). Using CHCl3 as the solvent, only a trace amount of 3a was observed (Table 1, entry 14). No reaction occurred in the solvents DMF, DMSO and THF (Table 1, entries 15–17). A relatively lower temperature (45 °C) only afforded a trace amount of 3a (Table 1, entry 18). Increasing the temperature to 90 °C or 110 °C, 3a was obtained in 45% and 40% yields, respectively (Table 1, entries 19 and 20). The ratio of substrates distinctly influenced the reaction (Table 1, entries 21–23). Changing the ratio from 1:1:1.5 (1a:NFSI:2a) to 1:2:2 or 1:2:3 (2a:NFSI:1a) led to much better yields (Table 1, entries 21 and 22). To our delight, when the ratio was 1:4:3 (2a:NFSI:1a), 3a was obtained in 76% yield (Table 1, entry 23).
Table 1: The optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-293-i4.svg?max-width=637&scale=1.0)
|
||||
| Entrya | Catalyst | Solvent | Temp (°C) | Yieldb (%) |
|---|---|---|---|---|
| 1 | Cu(OTf)2 | DCM | 70 | 39 |
| 2 | CuCl | DCM | 70 | 48 |
| 3 | CuBr | DCM | 70 | 43 |
| 4 | CuI | DCM | 70 | 30 |
| 5 | [(CH3CN)4Cu]PF6 | DCM | 70 | 50 |
| 6 | CuCN | DCM | 70 | 16 |
| 7 | Cu(acac)2 | DCM | 70 | 48 |
| 8 | Cu(OAc)2 | DCM | 70 | 51 |
| 9 | CuBr2 | DCM | 70 | 54 |
| 10 | CuCl2 | DCM | 70 | 55 |
| 11 | none | DCM | 70 | NRc |
| 12 | CuCl2 | DCE | 70 | 9 |
| 13 | CuCl2 | CH3CN | 70 | 20 |
| 14 | CuCl2 | CHCl3 | 70 | trace |
| 15 | CuCl2 | DMF | 70 | NRc |
| 16 | CuCl2 | DMSO | 70 | NRc |
| 17 | CuCl2 | THF | 70 | NRc |
| 18 | CuCl2 | DCM | 45 | trace |
| 19 | CuCl2 | DCM | 90 | 45 |
| 20 | CuCl2 | DCM | 110 | 40 |
| 21d | CuCl2 | DCM | 70 | 70 |
| 22e | CuCl2 | DCM | 70 | 73 |
| 23f | CuCl2 | DCM | 70 | 76 |
aReaction conditions: 1a (0.3 mmol), NFSI (0.3 mmol, 1.0 equiv), 2a (0.45 mmol, 1.5 equiv), catalyst (10 mol %), solvent (2.0 mL), N2, 10.0 h. bIsolated yields. cNR: no reaction. d1a:NFSI:2a = 2.0:2.0:1.0 e1a:NFSI:2a = 3.0:2.0:1.0. f1a:NFSI:2a = 3.0:4.0:1.0.
With the optimized reaction conditions in hand (Table 1, entry 23), the scope of this copper-catalyzed aminooxygenation reaction was examined (Figure 2). Styrenes with electron-withdrawing (1a–f) or electron-donating (1h and 1i) groups were viable, providing the corresponding 1,2-aminoalcohol derivatives in good yields. It is worth noting that functionalities such as F, Cl, Br, CN, and NO2 groups, which could easily undergo further transformations, were intact after the reaction (3a–e). The structure of 3e was confirmed by X-ray crystallographic analysis [47]. The substituent at the ortho (3j and 3k) or meta (3l) position of the aromatic ring did not hinder the reaction (41–55% yields). Similarly, for disubstituted (1m) and trisubstituted (1n) substrates, the aminooxygenation underwent smoothly, providing the corresponding products 3m (51%) and 3n (53%). The trans-β-methylstyrene (1o) afforded the desired product 3o in a low yield (15%). In addition, NHPI derivatives 2b and 2c were suitable nitrogen sources and the desired 3p and 3q were obtained in 56% and 64%, respectively. For 4-methoxystyrene (1r), no aminooxygenation reaction occurred.
![[1860-5397-11-293-2]](/bjoc/content/figures/1860-5397-11-293-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The copper-catalyzed three-component aminooxygenation of styrenes with NFSI and NHPI derivatives. Reaction conditions: 1 (0.9 mmol, 3.0 equiv), NFSI (1.2 mmol, 4.0 equiv), 2 (0.3 mmol, 1.0 equiv), CuCl2 (10 mol %), DCM (2.0 mL), N2, 70 °C, 10.0 h. Isolated yields.
Figure 2: The copper-catalyzed three-component aminooxygenation of styrenes with NFSI and NHPI derivatives. R...
Based on these experimental results and our previous investigations [42-46,48], a plausible mechanism for the copper-catalyzed three-component aminooxygenation of styrenes with NFSI an NHPI is shown in Scheme 2. Initially, the oxidation of Cu(I) with NFSI provided F–Cu(III)–N complex I, which could transform into a copper(II)-stabilized benzenesulfonimide radical II through a redox isomerization equilibrium. Next, the intermolecular radical addition of II to styrene 1g took place, producing benzylic radical III and Cu(II)–F species IV. The combination of the intermediates III and IV gave the Cu(III) species V having a C–Cu bond, which reacted with 2a to generate Cu(III)–O species VI, along with the loss of HF. Finally, the reductive elimination of VI afforded aminooxygenation product 3g.
![[1860-5397-11-293-i2]](/bjoc/content/inline/1860-5397-11-293-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Finally, we tried to investigate the synthetic value of our new aminooxygenation method. Then, the selective reduction of 3g was conducted (Scheme 3). The cleavage of the N–O bond in 3g readily occurred with Mo(CO)6/Et3N at 80 °C to give alcohol 4 [36] in 67% yield. Treatment of 3g with NH2NH2·H2O under mild conditions (25 °C) in CHCl3/MeOH gave free amine 5 in 70% yield.
![[1860-5397-11-293-i3]](/bjoc/content/inline/1860-5397-11-293-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Selective reduction of the aminooxygenation product.
Scheme 3: Selective reduction of the aminooxygenation product.
In summary, we have developed a novel copper-catalyzed three-component aminooxygenation reaction of styrenes with NFSI and NHPI derivatives. Furthermore, the aminooxygenation product could be easily converted into the corresponding alcohol or free amine through the cleavage of the N–O or C–N bond of the NHPI moiety. Further studies are underway in our lab.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6
Return to citation in text: [1] -
Shimojima, Y.; Hayashi, H. J. Med. Chem. 1983, 26, 1370–1374. doi:10.1021/jm00364a007
Return to citation in text: [1] -
Ager, D. J.; Prakash, I.; Schaad, D. R. Chem. Rev. 1996, 96, 835–876. doi:10.1021/cr9500038
Return to citation in text: [1] -
Donohoe, T. J.; Callens, C. K. A.; Flores, A.; Lacy, A. R.; Rathi, A. H. Chem. – Eur. J. 2011, 17, 58–76. doi:10.1002/chem.201002323
Return to citation in text: [1] -
Noack, M.; Göttlich, R. Chem. Commun. 2002, 536–537. doi:10.1039/b111656h
Return to citation in text: [1] -
Szolcsányi, P.; Gracza, T. Chem. Commun. 2005, 3948–3950. doi:10.1039/b506731f
Return to citation in text: [1] -
Cochran, B. M.; Michael, F. E. Org. Lett. 2008, 10, 5039–5042. doi:10.1021/ol8022165
Return to citation in text: [1] -
Muñiz, K.; Iglesias, A.; Fang, Y. Chem. Commun. 2009, 5591–5593. doi:10.1039/B912139K
Return to citation in text: [1] -
Borsini, E.; Broggini, G.; Fasana, A.; Galli, S.; Khansaa, M.; Piarulli, U.; Rigamonti, M. Adv. Synth. Catal. 2011, 353, 985–994. doi:10.1002/adsc.201000889
Return to citation in text: [1] -
Broggini, G.; Barbera, V.; Beccalli, E. M.; Chiacchio, U.; Fasana, A.; Galli, S.; Gazzola, S. Adv. Synth. Catal. 2013, 355, 1640–1648. doi:10.1002/adsc.201300104
Return to citation in text: [1] -
Liu, G.-S.; Zhang, Y.-Q.; Yuan, Y.-A.; Xu, H. J. Am. Chem. Soc. 2013, 135, 3343–3346. doi:10.1021/ja311923z
Return to citation in text: [1] -
Zhang, Y.-Q.; Yuan, Y.-A.; Liu, G.-S.; Xu, H. Org. Lett. 2013, 15, 3910–3913. doi:10.1021/ol401666e
Return to citation in text: [1] -
Alexanian, E. J.; Lee, C.; Sorensen, E. J. J. Am. Chem. Soc. 2005, 127, 7690–7691. doi:10.1021/ja051406k
Return to citation in text: [1] -
Sherman, E. S.; Fuller, P. H.; Kasi, D.; Chemler, S. R. J. Org. Chem. 2007, 72, 3896–3905. doi:10.1021/jo070321u
Return to citation in text: [1] -
Chemler, S. R. J. Organomet. Chem. 2011, 696, 150–158. doi:10.1016/j.jorganchem.2010.08.041
Return to citation in text: [1] -
Fuller, P. H.; Kim, J.-W.; Chemler, S. R. J. Am. Chem. Soc. 2008, 130, 17638–17639. doi:10.1021/ja806585m
Return to citation in text: [1] -
Chemler, S. R. Org. Biomol. Chem. 2009, 7, 3009–3019. doi:10.1039/b907743j
Return to citation in text: [1] -
Sherman, E. S.; Chemler, S. R. Adv. Synth. Catal. 2009, 351, 467–471. doi:10.1002/adsc.200800705
Return to citation in text: [1] -
Paderes, M. C.; Chemler, S. R. Org. Lett. 2009, 11, 1915–1918. doi:10.1021/ol9003492
Return to citation in text: [1] -
Karyakarte, S. D.; Smith, T. P.; Chemler, S. R. J. Org. Chem. 2012, 77, 7755–7760. doi:10.1021/jo3013226
Return to citation in text: [1] -
Paderes, M. C.; Chemler, S. R. Eur. J. Org. Chem. 2011, 3679–3684. doi:10.1002/ejoc.201100444
Return to citation in text: [1] -
de Haro, T.; Nevado, C. Angew. Chem., Int. Ed. 2011, 123, 936–940. doi:10.1002/ange.201005763
Return to citation in text: [1] -
Mancheno, D. E.; Thornton, A. R.; Stoll, A. H.; Kong, A.; Blakey, S. B. Org. Lett. 2010, 12, 4110–4113. doi:10.1021/ol101702w
Return to citation in text: [1] -
Wang, H.; Wang, Y.; Liang, D.; Liu, L.; Zhang, J.; Zhu, Q. Angew. Chem., Int. Ed. 2011, 50, 5678–5681. doi:10.1002/anie.201100362
Return to citation in text: [1] -
Sanjaya, S.; Chiba, S. Org. Lett. 2012, 14, 5342–5345. doi:10.1021/ol302525m
Return to citation in text: [1] -
Desai, L. V.; Sanford, M. S. Angew. Chem., Int. Ed. 2007, 46, 5737–5740. doi:10.1002/anie.200701454
Return to citation in text: [1] -
Schmidt, V. A.; Alexanian, E. J. J. Am. Chem. Soc. 2011, 133, 11402–11405. doi:10.1021/ja204255e
Return to citation in text: [1] -
Han, B.; Yang, X.-L.; Fang, R.; Yu, W.; Wang, C.; Duan, X.-Y.; Liu, S. Angew. Chem., Int. Ed. 2012, 51, 8816–8820. doi:10.1002/anie.201203799
Return to citation in text: [1] -
Sequeira, F. C.; Chemler, S. R. Org. Lett. 2012, 14, 4482–4485. doi:10.1021/ol301984b
Return to citation in text: [1] -
Michaelis, D. J.; Shaffer, C. J.; Yoon, T. P. J. Am. Chem. Soc. 2007, 129, 1866–1867. doi:10.1021/ja067894t
Return to citation in text: [1] -
Michaelis, D. J.; Ischay, M. A.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 6610–6615. doi:10.1021/ja800495r
Return to citation in text: [1] -
Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 4570–4571. doi:10.1021/ja1013536
Return to citation in text: [1] -
Benkovics, T.; Guzei, I. A.; Yoon, T. P. Angew. Chem., Int. Ed. 2010, 49, 9153–9157. doi:10.1002/anie.201004635
Return to citation in text: [1] -
Liu, G.; Stahl, S. S. J. Am. Chem. Soc. 2006, 128, 7179–7181. doi:10.1021/ja061706h
Return to citation in text: [1] -
Xue, Q.; Xie, J.; Xu, P.; Hu, K.; Cheng, Y.; Zhu, C. ACS Catal. 2013, 3, 1365–1368. doi:10.1021/cs400250m
Return to citation in text: [1] -
Li, Y.; Hartmann, M.; Daniliuc, C. G.; Studer, A. Chem. Commun. 2015, 51, 5706–5709. doi:10.1039/C5CC00591D
Return to citation in text: [1] [2] -
Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v
Return to citation in text: [1] [2] -
Rueda-Becerril, M.; Sazepin, C. C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.; Paquin, J.-F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026–4029. doi:10.1021/ja211679v
Return to citation in text: [1] [2] -
Halperin, S. D.; Fan, H.; Chang, S.; Martin, R. E.; Britton, R. Angew. Chem., Int. Ed. 2014, 53, 4690–4693. doi:10.1002/anie.201400420
Return to citation in text: [1] [2] -
Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583
Return to citation in text: [1] [2] -
Li, Y.; Zhang, Q. Synthesis 2015, 47, 159–174. doi:10.1055/s-0034-1379396
Return to citation in text: [1] [2] -
Ni, Z.; Zhang, Q.; Xiong, T.; Zheng, Y.; Li, Y.; Zhang, H.; Zhang, J.; Liu, Q. Angew. Chem., Int. Ed. 2012, 51, 1244–1247. doi:10.1002/anie.201107427
Return to citation in text: [1] [2] -
Zheng, G.; Li, Y.; Han, J.; Xiong, T.; Zhang, Q. Nat. Commun. 2015, 6, No. 7011. doi:10.1038/ncomms8011
Return to citation in text: [1] [2] -
Zhang, H.; Pu, W.; Xiong, T.; Li, Y.; Zhou, X.; Sun, K.; Liu, Q.; Zhang, Q. Angew. Chem., Int. Ed. 2013, 52, 2529–2533. doi:10.1002/anie.201209142
Return to citation in text: [1] [2] -
Zhang, H.; Song, Y.; Zhao, J.; Zhang, J.; Zhang, Q. Angew. Chem., Int. Ed. 2014, 53, 11079–11083. doi:10.1002/anie.201406797
Return to citation in text: [1] [2] -
Zhang, G.; Xiong, T.; Wang, Z.; Xu, G.; Wang, X.; Zhang, Q. Angew. Chem., Int. Ed. 2015, 54, 12649–12653. doi:10.1002/anie.201506066
Return to citation in text: [1] [2] -
The X-ray crystallographic coordinates for the structure of compound 3e reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 1422654.
Return to citation in text: [1] -
Sun, K.; Li, Y.; Zhang, Q. Sci. China: Chem. 2015, 58, 1354–1358. doi:10.1007/s11426-015-5385-y
Return to citation in text: [1]
| 42. | Ni, Z.; Zhang, Q.; Xiong, T.; Zheng, Y.; Li, Y.; Zhang, H.; Zhang, J.; Liu, Q. Angew. Chem., Int. Ed. 2012, 51, 1244–1247. doi:10.1002/anie.201107427 |
| 43. | Zheng, G.; Li, Y.; Han, J.; Xiong, T.; Zhang, Q. Nat. Commun. 2015, 6, No. 7011. doi:10.1038/ncomms8011 |
| 44. | Zhang, H.; Pu, W.; Xiong, T.; Li, Y.; Zhou, X.; Sun, K.; Liu, Q.; Zhang, Q. Angew. Chem., Int. Ed. 2013, 52, 2529–2533. doi:10.1002/anie.201209142 |
| 45. | Zhang, H.; Song, Y.; Zhao, J.; Zhang, J.; Zhang, Q. Angew. Chem., Int. Ed. 2014, 53, 11079–11083. doi:10.1002/anie.201406797 |
| 46. | Zhang, G.; Xiong, T.; Wang, Z.; Xu, G.; Wang, X.; Zhang, Q. Angew. Chem., Int. Ed. 2015, 54, 12649–12653. doi:10.1002/anie.201506066 |
| 48. | Sun, K.; Li, Y.; Zhang, Q. Sci. China: Chem. 2015, 58, 1354–1358. doi:10.1007/s11426-015-5385-y |
| 46. | Zhang, G.; Xiong, T.; Wang, Z.; Xu, G.; Wang, X.; Zhang, Q. Angew. Chem., Int. Ed. 2015, 54, 12649–12653. doi:10.1002/anie.201506066 |
| 47. | The X-ray crystallographic coordinates for the structure of compound 3e reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 1422654. |
| 1. | Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6 |
| 5. | Noack, M.; Göttlich, R. Chem. Commun. 2002, 536–537. doi:10.1039/b111656h |
| 6. | Szolcsányi, P.; Gracza, T. Chem. Commun. 2005, 3948–3950. doi:10.1039/b506731f |
| 7. | Cochran, B. M.; Michael, F. E. Org. Lett. 2008, 10, 5039–5042. doi:10.1021/ol8022165 |
| 8. | Muñiz, K.; Iglesias, A.; Fang, Y. Chem. Commun. 2009, 5591–5593. doi:10.1039/B912139K |
| 9. | Borsini, E.; Broggini, G.; Fasana, A.; Galli, S.; Khansaa, M.; Piarulli, U.; Rigamonti, M. Adv. Synth. Catal. 2011, 353, 985–994. doi:10.1002/adsc.201000889 |
| 10. | Broggini, G.; Barbera, V.; Beccalli, E. M.; Chiacchio, U.; Fasana, A.; Galli, S.; Gazzola, S. Adv. Synth. Catal. 2013, 355, 1640–1648. doi:10.1002/adsc.201300104 |
| 11. | Liu, G.-S.; Zhang, Y.-Q.; Yuan, Y.-A.; Xu, H. J. Am. Chem. Soc. 2013, 135, 3343–3346. doi:10.1021/ja311923z |
| 12. | Zhang, Y.-Q.; Yuan, Y.-A.; Liu, G.-S.; Xu, H. Org. Lett. 2013, 15, 3910–3913. doi:10.1021/ol401666e |
| 13. | Alexanian, E. J.; Lee, C.; Sorensen, E. J. J. Am. Chem. Soc. 2005, 127, 7690–7691. doi:10.1021/ja051406k |
| 14. | Sherman, E. S.; Fuller, P. H.; Kasi, D.; Chemler, S. R. J. Org. Chem. 2007, 72, 3896–3905. doi:10.1021/jo070321u |
| 15. | Chemler, S. R. J. Organomet. Chem. 2011, 696, 150–158. doi:10.1016/j.jorganchem.2010.08.041 |
| 16. | Fuller, P. H.; Kim, J.-W.; Chemler, S. R. J. Am. Chem. Soc. 2008, 130, 17638–17639. doi:10.1021/ja806585m |
| 17. | Chemler, S. R. Org. Biomol. Chem. 2009, 7, 3009–3019. doi:10.1039/b907743j |
| 18. | Sherman, E. S.; Chemler, S. R. Adv. Synth. Catal. 2009, 351, 467–471. doi:10.1002/adsc.200800705 |
| 19. | Paderes, M. C.; Chemler, S. R. Org. Lett. 2009, 11, 1915–1918. doi:10.1021/ol9003492 |
| 20. | Karyakarte, S. D.; Smith, T. P.; Chemler, S. R. J. Org. Chem. 2012, 77, 7755–7760. doi:10.1021/jo3013226 |
| 21. | Paderes, M. C.; Chemler, S. R. Eur. J. Org. Chem. 2011, 3679–3684. doi:10.1002/ejoc.201100444 |
| 22. | de Haro, T.; Nevado, C. Angew. Chem., Int. Ed. 2011, 123, 936–940. doi:10.1002/ange.201005763 |
| 23. | Mancheno, D. E.; Thornton, A. R.; Stoll, A. H.; Kong, A.; Blakey, S. B. Org. Lett. 2010, 12, 4110–4113. doi:10.1021/ol101702w |
| 24. | Wang, H.; Wang, Y.; Liang, D.; Liu, L.; Zhang, J.; Zhu, Q. Angew. Chem., Int. Ed. 2011, 50, 5678–5681. doi:10.1002/anie.201100362 |
| 25. | Sanjaya, S.; Chiba, S. Org. Lett. 2012, 14, 5342–5345. doi:10.1021/ol302525m |
| 26. | Desai, L. V.; Sanford, M. S. Angew. Chem., Int. Ed. 2007, 46, 5737–5740. doi:10.1002/anie.200701454 |
| 27. | Schmidt, V. A.; Alexanian, E. J. J. Am. Chem. Soc. 2011, 133, 11402–11405. doi:10.1021/ja204255e |
| 28. | Han, B.; Yang, X.-L.; Fang, R.; Yu, W.; Wang, C.; Duan, X.-Y.; Liu, S. Angew. Chem., Int. Ed. 2012, 51, 8816–8820. doi:10.1002/anie.201203799 |
| 29. | Sequeira, F. C.; Chemler, S. R. Org. Lett. 2012, 14, 4482–4485. doi:10.1021/ol301984b |
| 30. | Michaelis, D. J.; Shaffer, C. J.; Yoon, T. P. J. Am. Chem. Soc. 2007, 129, 1866–1867. doi:10.1021/ja067894t |
| 31. | Michaelis, D. J.; Ischay, M. A.; Yoon, T. P. J. Am. Chem. Soc. 2008, 130, 6610–6615. doi:10.1021/ja800495r |
| 32. | Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 4570–4571. doi:10.1021/ja1013536 |
| 33. | Benkovics, T.; Guzei, I. A.; Yoon, T. P. Angew. Chem., Int. Ed. 2010, 49, 9153–9157. doi:10.1002/anie.201004635 |
| 44. | Zhang, H.; Pu, W.; Xiong, T.; Li, Y.; Zhou, X.; Sun, K.; Liu, Q.; Zhang, Q. Angew. Chem., Int. Ed. 2013, 52, 2529–2533. doi:10.1002/anie.201209142 |
| 4. | Donohoe, T. J.; Callens, C. K. A.; Flores, A.; Lacy, A. R.; Rathi, A. H. Chem. – Eur. J. 2011, 17, 58–76. doi:10.1002/chem.201002323 |
| 45. | Zhang, H.; Song, Y.; Zhao, J.; Zhang, J.; Zhang, Q. Angew. Chem., Int. Ed. 2014, 53, 11079–11083. doi:10.1002/anie.201406797 |
| 3. | Ager, D. J.; Prakash, I.; Schaad, D. R. Chem. Rev. 1996, 96, 835–876. doi:10.1021/cr9500038 |
| 42. | Ni, Z.; Zhang, Q.; Xiong, T.; Zheng, Y.; Li, Y.; Zhang, H.; Zhang, J.; Liu, Q. Angew. Chem., Int. Ed. 2012, 51, 1244–1247. doi:10.1002/anie.201107427 |
| 2. | Shimojima, Y.; Hayashi, H. J. Med. Chem. 1983, 26, 1370–1374. doi:10.1021/jm00364a007 |
| 43. | Zheng, G.; Li, Y.; Han, J.; Xiong, T.; Zhang, Q. Nat. Commun. 2015, 6, No. 7011. doi:10.1038/ncomms8011 |
| 36. | Li, Y.; Hartmann, M.; Daniliuc, C. G.; Studer, A. Chem. Commun. 2015, 51, 5706–5709. doi:10.1039/C5CC00591D |
| 37. | Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v |
| 38. | Rueda-Becerril, M.; Sazepin, C. C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.; Paquin, J.-F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026–4029. doi:10.1021/ja211679v |
| 39. | Halperin, S. D.; Fan, H.; Chang, S.; Martin, R. E.; Britton, R. Angew. Chem., Int. Ed. 2014, 53, 4690–4693. doi:10.1002/anie.201400420 |
| 40. | Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583 |
| 41. | Li, Y.; Zhang, Q. Synthesis 2015, 47, 159–174. doi:10.1055/s-0034-1379396 |
| 35. | Xue, Q.; Xie, J.; Xu, P.; Hu, K.; Cheng, Y.; Zhu, C. ACS Catal. 2013, 3, 1365–1368. doi:10.1021/cs400250m |
| 36. | Li, Y.; Hartmann, M.; Daniliuc, C. G.; Studer, A. Chem. Commun. 2015, 51, 5706–5709. doi:10.1039/C5CC00591D |
| 34. | Liu, G.; Stahl, S. S. J. Am. Chem. Soc. 2006, 128, 7179–7181. doi:10.1021/ja061706h |
| 38. | Rueda-Becerril, M.; Sazepin, C. C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.; Paquin, J.-F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026–4029. doi:10.1021/ja211679v |
| 39. | Halperin, S. D.; Fan, H.; Chang, S.; Martin, R. E.; Britton, R. Angew. Chem., Int. Ed. 2014, 53, 4690–4693. doi:10.1002/anie.201400420 |
| 40. | Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583 |
© 2015 Li et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)