Abstract
The synthesis of a new type of Hoveyda–Grubbs 2nd generation catalyst bearing a modified N-heterocyclic carbene ligands is reported. The new catalyst contains an NHC ligand symmetrically substituted with chromanyl moieties. The complex was tested in model CM and RCM reactions. It showed very high activity in CM reactions with electron-deficient α,β-unsaturated compounds even at 0 °C. It was also examined in more demanding systems such as conjugated dienes and polyenes. The catalyst is stable, storable and easy to purify.

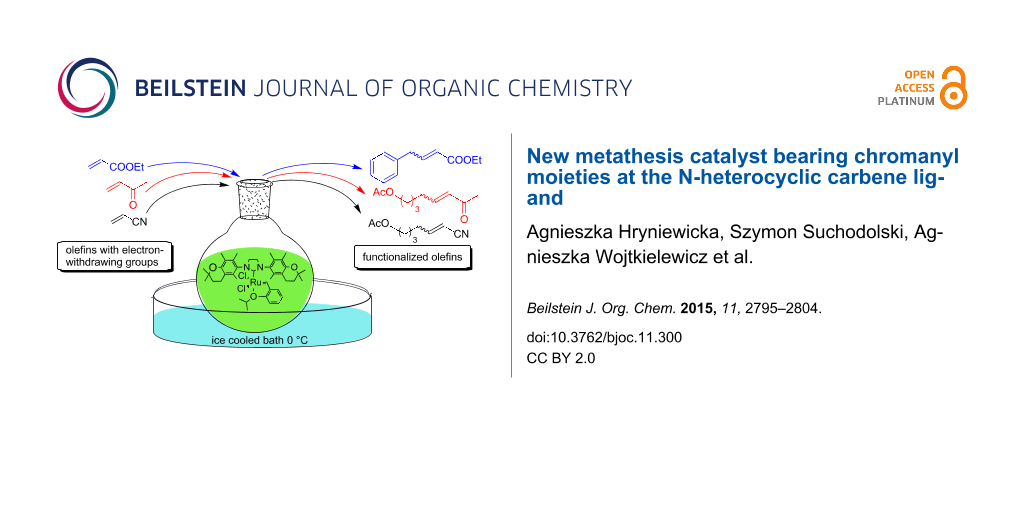
Graphical Abstract
Introduction
Olefin metathesis is still one of the most intensively studied transformations in synthetic organic chemistry. It has been frequently used as a key bond-forming reaction for total syntheses of many natural products [1]. The study on designing new metathesis catalysts and their synthesis has been a very fast developing area of organic chemistry since 1992, when Grubbs discovered the first well-defined ruthenium catalyst [2]. Nearly 400 ruthenium heterocyclic carbene-coordinated olefin metathesis catalysts were prepared until 2010 [3]. Since 2011, when Grubbs reported the synthesis of a Z-selective catalyst [4], several modified stereoselective catalysts were described [5-8]. Over the last few years, considerable attention has also been paid to immobilisation and tagging of catalysts and especially on making them more environmentally friendly [9-14].
Alkene cross-metathesis (CM) is a convenient route to the synthesis of functionalised olefins from simple precursors. Since the discovery of Grubbs 2nd generation catalyst (1, Figure 1) [15], Hoveyda–Grubbs 2nd generation catalyst (2, Figure 1) [16] and some successful modifications, e.g., nitro-Grela catalyst (3, Figure 1) [17] the utility of CM has been continuously expanded. The synthesis of complex structures bearing polar functional groups can be accomplished by CM [18]. Grubbs et al. recognised that CM can be selective when two partners showing different reactivity are used, e.g., reactive terminal olefin (type I) and an electron-deficient olefin (e.g., acrylates or acrylonitriles, type II or III). In these cases, full conversion and high yields can be achieved [18]. Although the problem with the CM reactions of olefins with electron-withdrawing groups, such as α,β-unsaturated ketones and esters, is partly solved [19,20], the conditions of the reactions need some improvement (lower catalyst loading, lower temperature). To the best of our knowledge, there is no such type of reaction performed at 0 °C. A lower temperature of the reaction is important in the synthesis of unstable and thermally-sensitive natural products.
![[1860-5397-11-300-1]](/bjoc/content/figures/1860-5397-11-300-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of olefin metathesis ruthenium catalysts.
Figure 1: Examples of olefin metathesis ruthenium catalysts.
We previously reported a few catalysts bearing the chromanyl moiety, derived from vitamin E [21-23]. In such a system as 2,2,5,7,8-pentamethyl-6-hydroxychromane (α-tocopherol model compound) specific stereoelectronic effects are observed [24,25], which might improve the activity of the catalyst bearing the above-mentioned moieties. The ruthenium complexes 4–6 (Figure 2) that we reported earlier appeared to be the so-called dormant catalysts. Their activity in RCM reactions was low at room temperature and higher at elevated temperature [21]. In catalyst 7 the chelating oxygen atom was provided by the rigid heterocyclic ring of the chromenylmethylidene moiety, whereas in the Hoveyda–Grubbs 2nd generation catalyst, the complexing oxygen atom comes from the freely rotating isopropoxy substituent. This complex proved to be quite efficient and showed activity comparable to that of commercially available catalysts 1 and 2 [23]. The introduction of a nitro group into the 6-position of the chromene moiety in catalyst 8 led to a decrease in the stability of the complex [22].
![[1860-5397-11-300-2]](/bjoc/content/figures/1860-5397-11-300-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected ruthenium metathesis catalyst bearing chromanyl moieties.
Figure 2: Selected ruthenium metathesis catalyst bearing chromanyl moieties.
The aforementioned modifications concerned the benzylidene moiety of the catalysts and affected the initiation rate of the metathesis reaction. Changes in the NHC ligand are also very important because this part of the catalyst participates all along the metathetic process. As a result, the catalyst may gain new properties, increased activity or stereoselectivity. Following this idea, we decided to synthesise a new catalyst bearing two chromanyl moieties symmetrically N,N’-disubstituted in the imidazolinium ring (9, Figure 2). According to Smith et al. [26] α-tocopherol and its amino analogue (α-tocopheramine) have comparable properties, coming from the same stereoelectronic effects mentioned above [24,25]. We expected that these effects may confer new properties of the NHC ligand.
Results and Discussion
Synthesis of the carbene precursor
The synthesis of an imidazolinium salt as a carbene precursor was started from 2,2,5,7,8-pentamethylchromane (10), which was prepared by the reaction between 2,3,5-trimethylphenol and 3-methylbut-2-enol [27]. Chromane 10 was nitrated with fuming nitric acid to give 6-nitrochromane 11 in 58% yield according to Mahdavian [28] (Scheme 1). Nitration using the Smith procedure [29] led to the expected nitrochromane 11, however, formation of an admixture of 5a,6-dinitrochromane was observed. Reduction of the nitro group in 11 was slightly troublesome, probably due to steric hindrance. The tellurium-rongalit system was found to be the most efficient [30], and 6-chromanylamine 12 was obtained in 50% yield. Imidazolinium salt 14 was prepared according to the classical protocol. Chromanylamine 12 was subjected to reaction with 2,3-dihydroxy-1,4-dioxane (glyoxal equivalent) followed by reduction of the intermediate diimines by NaBH3CN to give ethylenediamine 13 in 90% yield. Usage of the more convenient sodium borohydride led to a prolonged reaction time (up to 20 h) and a lower reaction yield. Imidazolinium salt 14 was obtained by treatment of 13 with trimethyl orthoformate in 73% yield. It is worth noting that ethylenediamine 13 and imidazolinium salt 14 were sufficiently pure after precipitation, thus chromatographic purification was not necessary.
![[1860-5397-11-300-i1]](/bjoc/content/inline/1860-5397-11-300-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the new NHC precursor. Reagents and conditions: a) HNO3, CH2Cl2, 0 °C, 58%; b) HOCH2SOONa, Te, NaOH, dioxane, 50 °C, 50%; c) 2,3-dihydroxy-1,4-dioxane, EtOH, HCOOH, rt, then NaBH3CN, rt, 90%; d) NH4Cl, HC(OMe)3, reflux, 73%; e) I) t-AmOK, toluene, rt; II) Hoveyda–Grubbs 1st generation, toluene, 65 °C, 68%.
Scheme 1: Synthesis of the new NHC precursor. Reagents and conditions: a) HNO3, CH2Cl2, 0 °C, 58%; b) HOCH2SO...
Some specific effects are observed in the NMR spectra of salt 14. In the 1H NMR spectrum, the signals from protons of the imidazolinium ring have atypical multiplicity. Two neighbouring triplets are also present besides the expected singlet from the ethylene bridge between the two nitrogen atoms symmetrically substituted by two identical chromenyl moieties. Similarly, the C-2 protons give two singlets instead of one. We suspected that there is a hindered rotation on the N–C(chromenyl) bond, so that two conformers are observed by NMR. However, the 1H NMR spectrum recorded at elevated temperatures (30 and 50 °C) looked the same. In the 13C NMR spectrum, signals from some of the carbon atoms are doubled. The HSQC correlation confirms that the doubled signals derive from one carbon atom. This fact also may suggest that two conformers are observed in the NMR spectra. This issue will be the subject of future detailed investigations.
Synthesis of the catalyst
The new catalyst, 9, was obtained from imidazolinium salt 14 by deprotonation with potassium tert-amylate followed by the tricyclohexylphosphine ligand exchange in Hoveyda–Grubbs 1st generation catalyst to give the target catalyst 9, which was purified on silica gel. The complex was stable in solid state at 0 °C for a few weeks (NMR test).
Attempts to obtain a monocrystal that would be suitable for X-ray analysis failed. The 1H NMR spectrum confirmed the structure of the catalyst. In the 13C NMR spectrum, some signals were broad and weak, e.g., signals of the quaternary aromatic carbon atoms and the primary carbon atoms of the methyl group attached to the aromatic ring of the chromanyl moieties. The HSQC correlation confirmed the structure of the catalyst and showed good correlation of the attached proton signals with the weak carbon peaks. When the 13C NMR spectrum is recorded at 50 °C, the spectrum becomes simpler but still many signals are almost invisible (at the noise level). The mass spectrum (HRMS) of 9 clearly evidences the molecular weight and the elemental composition of the new catalyst. The most likely unusual NMR spectra of 9 can result from specific stereoelectronic effects observed in the chromanyl system [24,25]. The 2p-type lone pair of electrons of the heterocyclic ring oxygen atom adopts an orientation almost perpendicular to the plane of the aromatic ring allowing for an electronic interaction with the para-substituent of the chromanyl system (–OH in 6-hydroxychroman and –NH2 in 6-aminochroman).
Testing of the new catalyst
The catalyst proved very active in model cross-metathesis reactions, especially with olefins containing electron-withdrawing groups. For example the reaction of allylbenzene and ethyl acrylate at room temperature was almost quantitative. These results prompted us to test this reaction at a lower temperature (0 °C). Commercially available catalysts 1 and 2 proved inactive under these conditions, therefore we compared our catalyst with Grela catalyst 3 [17]. As is shown in Table 1 (entry 1), both catalysts, i.e., 3 and 9 proved very active. After reduction of the catalyst loading to 1 mol %, the yields of the ethyl acrylate reaction with allylbenzene catalysed by both complexes 3 and 9 were still very good (close to 80%, Table 1, entry 2). Encouraged by these results, we decided to test 9 in a CM reaction with other α,β-unsaturated compounds, e.g., methyl vinyl ketone (entry 3, Table 1) and acrylonitrile (entry 4, Table 1). The outcome of these reactions was also very promising for 9. It should be added that in all cases a dimer of the electron-deficient olefin was not observed. The homodimerisation product of allylbenzene (entries 1 and 2, Table 1) was formed in less than 10% (in case of the reaction catalysed by 2 no dimeric products were observed), while the yield of the homodimer of hex-5-enyl acetate (entries 3 and 4, Table 1) was below 3%. Low conversion (especially that obtained with 1 and 2) was related to substantial amounts of unreacted substrates.
Table 1: Comparative investigation of catalysts in CM reactions with electron-deficient olefins.
| Entry | Alkene | Electron-deficient olefin | Product | Conditionsb | Catalyst | Yield (E/Z)c |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-300-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-300-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-300-i7.svg?max-width=637&scale=1.0)
|
0 °C, 3 h, CH2Cl2
2.5 mol % [Ru] |
1
2 3 9 |
11% (E/Z 100:1)
13% (E/Z 32:1) 87% (E/Z 29:1) 91% (E/Z 23:1) |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-300-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-300-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-300-i10.svg?max-width=637&scale=1.0)
|
0 °C, 3 h, CH2Cl2
1 mol % [Ru] |
3
9 |
77%
76% |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-300-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-300-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-300-i13.svg?max-width=637&scale=1.0)
|
0 °C, 3 h, CH2Cl2
1 mol % [Ru] |
1
2 3 9 |
0%
38% (only E) 98% (only E) 99% (only E) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-300-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-300-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-300-i16.svg?max-width=637&scale=1.0)
|
0 °C, 1 h, CH2Cl2
1 mol % [Ru] |
1
2 3 9 |
0%
59% (E/Z 2.5:1) 75% (E/Z 3:1) 97% (E/Z 2.5:1) |
aElectron-deficient olefin was used in excess (2 equiv). bConcentration of alkene amounted 0.1 M. cDetermined by 1H NMR.
The high activity of Grela catalyst 3 was a result of faster initiation of the catalytic cycle arising from the electron-withdrawing effect of the nitro group. Consequently, lowered electron density at the oxygen atom in the isopropoxybenzylidene fragment weakens the coordination to the ruthenium atom, and finally, facilitates the initiating process. In the new catalyst 9, which is modified in the NHC ligand, different effects are responsible for its activity. According to Grubbs [31], catalyst 1 is able to react with α,β-unsaturated carbonyl compounds to form an enoic carbene [Ru]=CHCOX, which is kinetically favourable. As a result, a stronger electron-donating ligand should stabilise the electron-deficient enoic carbene [31]. One can speculate that specific stereoelectronic effects occurring in the chromanyl system, known from the vitamin E chemistry, contribute to high activity of 9. The N-(2,2,5,7,8-pentamethyl-6-chromanyl) substituents, bearing electron releasing methyl groups as well as interplaying dihydropyranyl oxygen and nitrogen atoms in the imidazolidine cycle, can stabilise the enoic carbene (Scheme 2).
![[1860-5397-11-300-i2]](/bjoc/content/inline/1860-5397-11-300-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: CM with electron-deficient olefin.
Scheme 2: CM with electron-deficient olefin.
The CM products of terminal olefins (entries 1–3, Table 2) were obtained in high and very high yields. Alkenes were converted almost quantitatively and the excess of (Z)-but-2-ene-1,4-diol diacetate was recovered. Some side products of self-metathesis (SM) of terminal alkenes were isolated. It should be added that two terminal olefins (entry 4, Table 2) gave also SM products besides the desired CM products. Furthermore, more dimeric products gave allyloxybenzene than hex-5-enyl acetate. It is worth noting that the CM reaction between styrene and (Z)-but-2-ene-1,4-diol diacetate (entry 1, Table 2) was highly efficient (97% yield) using catalyst 9. Moreover, in the CM of allyloxybenzene and hex-5-enyl acetate, the yield with 9 was almost twice higher than that obtained for 1 or 2 (entry 4, Table 2). In the model RCM, the activity of catalyst 9 was slightly lower than that of commercial complexes, supposedly due to steric reasons (Table 3).
Table 2: Comparative investigation of the catalysts’ performance in CM reactions.
| Entry | Substrates | Product | Catalyst | Yield (E/Z)a | |
|---|---|---|---|---|---|
| 1b |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-300-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-300-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-300-i19.svg?max-width=637&scale=1.0)
|
1
2 9 |
81% (E/Z 58:1)
80% (E/Z 43:1) 97% (E/Z 42:1) |
| 2b |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-300-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-300-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-300-i22.svg?max-width=637&scale=1.0)
|
1
2 9 |
76% (E/Z 12:1)
75% (E/Z 12:1) 73% (E/Z 18:1) |
| 3b |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-300-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-300-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-300-i25.svg?max-width=637&scale=1.0)
|
1
2 9 |
82% (E/Z 5:1)
86% (E/Z 5:1) 75% (E/Z 6:1) |
| 4c |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-300-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-300-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-300-i28.svg?max-width=637&scale=1.0)
|
1
2 9 |
47% (E/Z 7:1)
53% (E/Z 5:1) 83% (E/Z 9:1) |
aE/Z ratio determined by 1H NMR, isolated yield. bReaction conditions: 20 °C, 3 h, CH2Cl2, 0.1 M (terminal alkene), 2.5 mol % [Ru], (Z)-but-2-ene-1,4-diol diacetate (2 equiv). cReaction conditions: 20 °C, 3 h, CH2Cl2, 0.1 M (both alkenes), 2.5 mol % [Ru].
Table 3: Comparative investigation of catalysts in RCM.
| Entry | Substrate | Product | Conditions | Catalyst | Conversiona |
|---|---|---|---|---|---|
| 1 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-300-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-300-i30.svg?max-width=637&scale=1.0)
|
20 °C, CH2Cl2, 0.1 M,
1 h, 1 mol % [Ru] |
1
2 9 |
95%
99% 70% |
| 2 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-300-i31.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-300-i32.svg?max-width=637&scale=1.0)
|
20 °C, CH2Cl2, 0.1 M,
1 h, 1 mol % [Ru] |
1
2 9 |
83%
85% 65% |
| 3 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-300-i33.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-300-i34.svg?max-width=637&scale=1.0)
|
80 °C, toluene, 0.06 M,
16 h, 5 mol % [Ru] |
1
2 9 |
38%
15% 22% |
aDetermined by 1H NMR.
The potency of the new catalyst 9 was tested not only in standard, model metathesis reactions but was also examined in more demanding systems, such as conjugated dienes and polyenes (Table 4). The CM reaction between alkene and diene (or polyene) often suffers from low regio- and stereoselectivity control. The CM reaction may be accompanied by various self-metathesis processes. Additionally, due to the competitive cleavage of both double bonds of the diene substrate, two different products may be formed in the CM reaction between alkene and diene (Scheme 3). A further complication is a Z/E isomer mixture formation.
Table 4: CM reactions between dienes and alkenes in the presence of various 2nd-generation catalystsa.
| Entry | Diene | Alkene | Products | [Ru] |
Yieldb (E/Z)
product A |
Yield (E/Z)
product B |
|---|---|---|---|---|---|---|
| 1c |
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-300-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-300-i36.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-300-i37.svg?max-width=637&scale=1.0)
|
1
2 9 |
89% (only E)
72% (only E) 70% (only E) |
–
– – |
| 2d |
![[Graphic 34]](/bjoc/content/inline/1860-5397-11-300-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 35]](/bjoc/content/inline/1860-5397-11-300-i39.svg?max-width=637&scale=1.0)
|
![[Graphic 36]](/bjoc/content/inline/1860-5397-11-300-i40.svg?max-width=637&scale=1.0)
|
1
2 9 |
95% (only E)
92% (only E) 94% (only E) |
–
– – |
| 3d |
![[Graphic 37]](/bjoc/content/inline/1860-5397-11-300-i41.svg?max-width=637&scale=1.0)
|
![[Graphic 38]](/bjoc/content/inline/1860-5397-11-300-i42.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-11-300-i43.svg?max-width=637&scale=1.0)
|
1
2 9 |
42% (only E)
40% (only E) 51% (only E) |
–
– – |
| 4c,e |
![[Graphic 40]](/bjoc/content/inline/1860-5397-11-300-i44.svg?max-width=637&scale=1.0)
|
![[Graphic 41]](/bjoc/content/inline/1860-5397-11-300-i45.svg?max-width=637&scale=1.0)
|
![[Graphic 42]](/bjoc/content/inline/1860-5397-11-300-i46.svg?max-width=637&scale=1.0)
|
1
2 9 |
40% (E/Z 12:1)
20% (E/Z 11:1) 17% (E/Z 8:1) |
10% (only E)
22% (E/Z 8:1) 49% (E/Z 18:1) |
aReaction conditions: diene (3 equiv, 0.36 M), alkene (1 equiv, 0.12 M), and 10 mol % of catalyst in DCM or toluene at 45 °C for 16 h. bIsolated yield. Yields were calculated in relation to alkene. cE/Z ratio determined by GC/MS. dE/Z ratio determined by 1H NMR. eIsolated as inseparable mixture of A and B. Yield calculated from 1H NMR.
![[1860-5397-11-300-i3]](/bjoc/content/inline/1860-5397-11-300-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible products of metathesis reaction between diene and alkene.
Scheme 3: Possible products of metathesis reaction between diene and alkene.
The reactions of ethyl sorbate and its 3-methyl substituted analogue (ethyl (2E,4Z/E)-3-methylhexa-2,4-dienoate) with various alkenes were chosen to examine the activity and selectivity profile of catalyst 9 (Table 4). The results clearly indicate that carbene 9 can promote the CM reactions of dienes with different olefins as efficiently as commercial Grubbs 2nd generation and Hoveyda–Grubbs 2nd generation complexes 1 and 2. Complex 9 catalysed the reactions of ethyl 3-methylhexa-2,4-dienoate in a completely selective manner taking into account product regio- and stereoselectivity. In all reactions the E-isomer of compound A was formed as a main product (Table 4, entries 1–3). Homodimerisation products of diene and alkene were obtained in very small amounts (<2% and <4% for entry 1 and 2, respectively, Table 4). The SM product of methyl vinyl ketone was not observed (entry 3, Table 4). Additionally, isomerised diene (ethyl (2E,4E)-3-methylhexa-2,4-dienoate, used in excess) was isolated from the reaction mixture. When ethyl sorbate was used, the formation of two CM products, A and B, was observed due to unselective metathetic scission of the C2–C3 and C4–C5 double bonds (Table 4, entry 4) [32]. The low product yield was caused not only by the unselective attack of catalyst on diene substrate but also by competitive SM reactions of both substrates.
Interestingly, the new ruthenium complex showed a different regioselectivity compared to catalysts 1 and 2. The metathetic scission of the less reactive C2–C3 double bond prevailed with complex 9 (Table 4, entry 4). This result could be explained by the higher affinity of the new catalyst toward electron-deficient olefins. It may also be assumed that the presence of the chromane system changes the electronic properties of the complex compared to other catalysts. It may favour chelation of the ruthenium centre of the catalyst by the carbonyl ester oxygen from the diene substrate. It seems that the oxygen–ruthenium bond is rather labile. The oxygen coordination does not decrease the activity of the catalyst but rather stabilises the π-complex and rutenacyclobutane intermediate (Figure 3), which promotes the metathesis reaction on the C2–C3 double bond. The formation of four- and five-membered chelates has been postulated previously [33,34].
![[1860-5397-11-300-3]](/bjoc/content/figures/1860-5397-11-300-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: π-Complex and rutenacyclobutane intermediate with a five-membered ring chelate.
Figure 3: π-Complex and rutenacyclobutane intermediate with a five-membered ring chelate.
The CM reactions of polyenes, β-carotene and retinyl acetate with ethyl 3-methylhexa-2,4-dienoate in the presence of catalyst 9 were also studied (Scheme 4). A previous investigation of the CM reaction of β-carotene promoted by commercial Grubbs and Hoveyda–Grubbs 2nd generation catalysts (1 and 2) proved that the two double bonds, C11–C12 and C15–C15’ of the β-carotene molecule were reactive in CM [35]. In the case of metathetic fragmentation of β-carotene, the use of 9 instead of 1 or 2 improved the regioselectivity. The product of the central C15–C15’ double bond scission was formed preferably. The activity of 9 was comparable to that of 1, albeit lower than 2 (Scheme 4). Regio- and stereoselectivity (>95% E-isomer) of retinyl acetate CM catalysed by 9 appeared to be the same as in analogous reactions that were carried out in the presence of catalysts 1 and 2 [36] (Scheme 4). However, the product was obtained in a slightly lower yield.
![[1860-5397-11-300-i4]](/bjoc/content/inline/1860-5397-11-300-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: CM reaction of β-carotene and retinyl acetate with ethyl (2E,4Z/E)-3-methylhexa-2,4-dienoate. Reaction conditions: diene (4 equiv, 0.325 M), alkene (1 eqiuv, 0.08 M). Yield was obtained by quantative HPLC analysis. Yields were calculated in relation to polyene (retinyl acetate or β-carotene). In case of the β-carotene reaction, yields were divided by two due to the symmetry of the β-carotene molecule.
Scheme 4: CM reaction of β-carotene and retinyl acetate with ethyl (2E,4Z/E)-3-methylhexa-2,4-dienoate. React...
Conclusion
In summary, an efficient synthesis of new olefin metathesis catalyst 9 is reported. The catalyst contains a NHC ligand generated from the imidazolinium salt 14 bearing two symmetrically substituted 6-chromanyl moieties. The catalyst 9 was tested in model CM and RCM reactions and showed an activity comparable or superior to that of the commercial Grubbs and Hoveyda–Grubbs 2nd generation complexes. The new complex was also active in metathesis of more demanding systems such as conjugated dienes and polyenes. It proved reactive toward electron-deficient olefins even at lowered temperature (0 °C).
Experimental
General
All manipulations of organometallic compounds were performed using standard Schlenk techniques under an atmosphere of dry argon. CH2Cl2 was dried by distillation over CaH2, toluene over Na. 1,4-Dioxane, ethanol (96%) and trimethyl orthoformate were used as received. Melting points were determined on a Kofler apparatus of the Boetius type and were uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance II spectrometer (400 and 100 MHz, respectively). Spectra are referenced relative to the chemical shift (δ) of TMS. Mass spectra were obtained with Micromass LCT TOF and AutoSpec Premier (Waters) spectrometers. IR spectra were recorded on a Nicolet series II Magna-IR 550 FTIR spectrometer. Flash chromatography (FC) was performed on silica gel 230–400 mesh. Catalysts: 1, 2, 3 and the Hoveyda–Grubbs 1st generation complex were purchased from Apeiron Synthesis. 2,2,5,7,8-Pentamethylchromane (10) was prepared from 2,3,5-trimethylphenol analogously to the procedure described for 2,2,5,8-tetramethyl-6-chromanol by Dean [27]. 2,3-Dihydroxy-1,4-dioxane was obtain according to Venuti [37]. Substrates for testing catalysts in RCM reactions were prepared by allylation of commercial diethyl malonate with allyl bromide and/or 3-chloro-2-methylpropene according to Hensle [38]. Their purity was estimated by 1H NMR spectroscopy and found to be at least 95%. Other chemicals are commercially available and used as received. The numbering of carbon atoms in the chromanyl moiety was used as shown in Figure 4.
![[1860-5397-11-300-4]](/bjoc/content/figures/1860-5397-11-300-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Numbering of carbon atoms in the chromanyl moiety.
Figure 4: Numbering of carbon atoms in the chromanyl moiety.
6-Nitro-2,2,5,7,8-pentamethylchromane (11)
To the solution of 2,2,5,7,8-pentamethylchromane (10) (2.5 g, 0.012 mol) in dry CH2Cl2 (150 mL) cooled to 0 °C fuming nitric acid (1.5 mL, 3 equiv, 0.036 mol) was added in one portion. The reaction was carried out at 0 °C for 1.5 h. Next the reaction mixture was washed with saturated NaHCO3, dried over Na2SO4 and evaporated. The crude product was purified by column chromatography (hexane/ethyl acetate 20:1) to give a yellow solid (1.75 g, 58%). Mp 120–123 °C; IR (KBr) ν: 2977, 2934, 1522, 1368, 1112 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.64 (t, J = 6.8 Hz, 2H, H-4), 2.15 and 2.12 (2s, 9H, H-5a, 7a, 8b), 1.83 (t, J = 6.8 Hz, 2H, H-3), 1.33 (s, 6H H-2a, 2b) ppm; 13C NMR (100 MHz, CDCl3) δ 152.6 (C-8a), 146.2 (C-6), 126.1 (C-8), 125.0 (C-5), 123.7 (C-7), 117.4 (C-4a), 74.0 (C-2), 32.4 (C-3), 26.7 (C-2a and 2b), 20.8 (C-4), 14.5, 13.7, 11.8 (C-5a, 7a and 8b) ppm. HRMS (ESI): [M + Na]+ calcd for C14H19NNaO3: 272.1257, found: 272.1252. Literature mp 125–126 °C; IR, 1H NMR and 13C NMR data are compatible with the data in [39].
2,2,5,7,8-Pentamethylchromanyl-6-amine (12)
To the solution of rongalite (sodium hydroxymethylsulfinate dihydrate) (4.62 g, 5 equiv, 30 mmol) in a solution of 1 M NaOH (150 mL) tellurium powder (154 mg, 0.2 equiv, 1.2 mmol) was added. Then the solution of 6-nitro-2,2,5,7,8-pentamethylchromane (11, 1.5 g, 6 mmol) in dioxane (20 mL) was added to the reaction mixture. The reaction was carried out at 50 °C for 8 h. The reaction mixture was filtered through a pad of Celite and the filtrate was extracted with methylene chloride (3 × 20 mL). The organic layers were combined and washed with brine and water, dried over Na2SO4 and evaporated. The crude product was purified by column chromatography (hexane/ethyl acetate 8:1) to give a white solid (660 mg, 50%). Mp 38–40 °C; IR (KBr) ν: 3455, 3364, 2975, 2921, 1625, 1447, 1420, 1269 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.31 (s, 2H NH2), 2.67 (t, J = 6.9 Hz, 2H, H-4), 2.15, 2.12 and 2.08 (3s, 9H, H-5a, 7a, 8b), 1.80 (t, J = 6.9 Hz, 2H, H-3), 1.30 (s, 6H, H-2a, 2b) ppm; 13C NMR (100 MHz, CDCl3) δ 144.9 (C-8a), 134.9 (C-6), 122.2 (C-8), 120.4 (C-7), 117.6 (C-5), 116.9 (C-4a), 72.2 (C-2), 33.3 (C-3), 26.7 (C-2a and 2b), 21.4 (C-4), 13.5, 12.5, 11.9 (C-5a, 7a, 8b) ppm. HRMS (ESI): [M + H]+ calcd for C14H22NO: 220.1696, found: 220.1691. Literature mp 51.5–53.5 °C; IR, 1H NMR and 13C NMR data are compatible with the data in [39].
N,N-Bis(2,2,5,7,8-pentamethylchroman-6-yl)ethane-1,2-diamine (13)
To a solution of 2,2,5,7,8-pentamethylchromanyl-6-amine (12, 500 mg, 2.3 mmol) in ethanol (96%, 20 mL), 2,3-dihydroxy-1,4-dioxane (138 mg, 1.15 mmol) and two drops of formic acid were added in a manner similar to a that described in [40]. The reaction mixture was stirred for 24 h. A yellow precipitate appeared. Then sodium cyanoborohydride (217 mg, 3 equiv, 3.45 mmol) was added and the reaction mixture was stirred for 1.5 h. The reaction mixture was quenched with water (20 mL) and extracted with dichloromethane (3 × 20 mL). The organic layers were washed with brine and water, dried over Na2SO4 and evaporated to dryness. After crystallization from ethanol a white solid was obtained (480 mg, 90%). Mp 182–183 °C; IR (KBr) ν: 3369, 2968, 2925, 2779, 1450, 1420, 1265 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.04 (s, 4H, -NCH2CH2N-), 2.64 (t, J = 6.8 Hz, 4H, H-4), 2.26, 2.22 and 2.13 (3s, 18H, H-5a, 7a, 8b), 1.81 (t, J = 6.8 Hz, 4H, H-3), 1.31 (s, 12H, H-2a, 2b) ppm; 13C NMR (100 MHz, CDCl3) δ 147.9 (C-8a), 137.6 (C-6), 129.1 (C-8), 127.1 (C-7), 122.6 (C-5), 117.2 (C-4a), 72.6 (C-2), 50.4 (-NH-CH2-CH2-NH-), 33.1 (C-3), 26.8 (C-2a and 2b), 21.4 (C-4), 14.4, 13.5, 12.1 (C-5a, 7a, 8b) ppm. HRMS (ESI): [M + H]+ calcd for C30H45N2O2: 465.3476, found: 465.3465.
1,3-Bis(2,2,5,7,8-pentamethylchroman-6-yl)-4,5-dihydro-1H-imidazol-3-ium chloride (14)
To a solution of ethylenediamine 13 (200 mg, 0.43 mmol) in trimethyl orthoformate (5 mL), ammonium chloride (35 mg, 0.65 mmol) was added in a similar manner as described in [40]. The reaction mixture was refluxed for 2 h. Solids were filtered off and the filtrate was evaporated to dryness. The resulting oil was stirred overnight with diethyl ether causing precipitation of a white solid (160 mg, 73% yield). Mp 246–247 °C; IR (KBr) ν: 3397, 2974, 2929, 1626, 1456, 1411, 1269 cm−1; 1H NMR (400 MHz, CDCl3) δ 8.56 and 8.52 (2s, 1H, N-CH=N), 4.75–4.50 (m, 4H, -NCH2CH2N-), 2.64 (t, J = 6.8 Hz, 4H, H-4), 2.30, 2.28 and 2.12 (18H, 3s, H-5a, 7a, 8b), 1.82 (t, J = 6.8 Hz, 4H, H-3), 1.32 (s, 12H, H-2a, 2b) ppm; 13C NMR (100 MHz, CDCl3) δ 159.3, 159.2 (-N=CH-N-), 153.2 (C-8a), 131.8, 131.6 (C-6), 131.3, 131.0 (C-8), 124.4, 124.3 (C-7), 124.2 (C-5), 118.3, 118.2 (C-4a), 74.0 (C-2), 53.0 (-NH-CH2-CH2-NH-), 32.2 (C-3), 26.9, 26.8, 26.5, 26.47 (C-2a and 2b), 21.0 (C-4), 15.1, 15.0, 14.4, 14.3, 12.0 (C-5a, 7a, 8b) ppm; HRMS (ESI): [M − Cl]+ calcd for C31H43N2O2: 475.3319, found: 475.3308. Some signals in 13C NMR spectrum are doubled (see text “Synthesis of the carbene precursor”).
1,3-Bis[(2,2,5,7,8-pentamethylchroman-6-yl)-2-imidazolidinylidene]dichloro(o-isopropoxyphenylmethylene)ruthenium (9)
In a Schlenk flask imidazolinium salt 14 (80 mg, 0.16 mmol) was dried under vacuum at 80 °C for 1 h. The flask was then cooled to rt and dry toluene (3 mL) was added under argon atmosphere. To the resulting suspension potassium tert-amylate (1.7 M in toluene, 94 µL, 0.16 mmol) was added and the reaction mixture was stirred for 20 min at rt. Then a solution of Hoveyda–Grubbs 1st generation catalyst (96 mg, 0.16 mmol) in toluene (3 mL) was added and the reaction was carried out at 65 °C for 2 h. The reaction mixture was purified by column chromatography (hexane/ethyl acetate 10:1) to give a green solid (86 mg, 68% yield) Mp 162–164 °C; IR (KBr) ν: 2973, 2925, 2854, 1475, 1457, 1274 cm−1; 1H NMR (400 MHz, CDCl3) δ 16.74 (s, 1H, Ru=CH-), 7.47, 6.94, 6.84 and 6.78 (4m, 4H, -CHAr), 4.89 (sept., J = 6.1 Hz, 1H, -CHiPr), 4.16 (s, 4H, -N-CH2CH2-N-), 2.69 (m, 4H, H-4), 2.42, 2.29 and 2.20 (3s (2 br s and 1s), 18H, 3s H-5a, 7a, 8b), 1.88 (t, J = 6.5 Hz, 4H, H-3), 1.44 (s, 12H, H-2a, 2b), 1.28 (d, J = 6.0 Hz, 6H, CH3 iPr) ppm; 13C NMR (100 MHz, CDCl3) δ 297.5 (CH=Ru), 211.7 (CNHC), 152.1 (IVCAr), 145.4 (IVCAr), 135.0 (IVCAr weak), 133.5 (IVCAr weak), 132.2 (IVCAr weak), 129.1 (IIICHAr), 129.0 (IVCweak), 123.3 (IVCAr weak), 122.9 (IIICHAr), 122.4 (IIICHAr), 117.1 (IVCAr weak), 112.8 (IIICHAr), 74.7 (CHIPr), 73.3 (Cchroman-2), 53.1 and 51.8 (N-(CH2)2-N), 33.1 (Cchroman-3), 27.9 (Cchroman-2a and 2b), 25.7 (CH3 IPr), 21.2 (Cchroman-4), 18.3 (CH3 chroman weak), 15.2 (CH3 chroman weak), 12.0 (CH3 chroman) ppm; EIMS m/z: 796 (10), 794 (10), 610 (14), 572 (7), 473 (41), 472 (100), 416 (31), 243 (7), 181 (8), 108 (12), 69 (14), 44 (14%); HRMS (EI): [M]+ calcd for C41H54Cl2N2O3102Ru: 794.2549, found: 794.2571. Some signals in 13C NMR spectrum are weak (see text “Synthesis of the catalyst”).
Supporting Information
| Supporting Information File 1: Experimental procedures of the testing of the new catalyst and copies of 1H and 13C NMR spectra of new compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
Acknowledgements
The authors acknowledge financial support from the Polish National Science Centre (UMO-2011/02/A/ST5/00459). All syntheses were performed in the Centre of Synthesis and Analysis BioNanoTechno of University of Bialystok. The equipment of the Centre was funded by the EU, as a part of the Operational Program Development of Eastern Poland 2007-2013, project: POPW.01.03.00-20-034/09-00.
References
-
Fürstner, A. Chem. Commun. 2011, 47, 6505–6511. doi:10.1039/c1cc10464k
Return to citation in text: [1] -
Nguyen, S. T.; Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1992, 114, 3974–3975. doi:10.1021/ja00036a053
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. doi:10.1021/ja202818v
Return to citation in text: [1] -
Keitz, B. K.; Endo, K.; Patel, P. R.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2012, 134, 693–699. doi:10.1021/ja210225e
Return to citation in text: [1] -
Rosebrugh, L. E.; Herbert, M. B.; Marx, V. M.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 1276–1279. doi:10.1021/ja311916m
Return to citation in text: [1] -
Occhipinti, G.; Hansen, F. R.; Törnroos, K. W.; Jensen, V. R. J. Am. Chem. Soc. 2013, 135, 3331–3334. doi:10.1021/ja311505v
Return to citation in text: [1] -
Khan, R. K. M.; O'Brien, R. V.; Torker, S.; Li, B.; Hoveyda, A. H. J. Am. Chem. Soc. 2012, 134, 12774–12779. doi:10.1021/ja304827a
Return to citation in text: [1] -
Buchmeiser, M. R. Chem. Rev. 2009, 109, 303–321. doi:10.1021/cr800207n
Return to citation in text: [1] -
Klučiar, M.; Grela, K.; Mauduit, M. Dalton Trans. 2013, 42, 7354–7358. doi:10.1039/c2dt32856a
Return to citation in text: [1] -
Kośnik, W.; Grela, K. Dalton Trans. 2013, 42, 7463–7467. doi:10.1039/c3dt33010a
Return to citation in text: [1] -
Śledź, P.; Mauduit, M.; Grela, K. Chem. Soc. Rev. 2008, 37, 2433–2442. doi:10.1039/b711482f
Return to citation in text: [1] -
Szczepaniak, G.; Kosiński, K.; Grela, K. Green Chem. 2014, 16, 4474–4492. doi:10.1039/C4GC00705K
Return to citation in text: [1] -
Pastva, J.; Skowerski, K.; Czarnocki, S. J.; Žilková, N.; Čejka, J.; Bastl, Z.; Balcar, H. ACS Catal. 2014, 4, 3227–3236. doi:10.1021/cs500796u
Return to citation in text: [1] -
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q
Return to citation in text: [1] -
Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g
Return to citation in text: [1] -
Grela, K.; Harutyunyan, S.; Michrowska, A. Angew. Chem., Int. Ed. 2002, 41, 4038–4040. doi:10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0
Return to citation in text: [1] [2] -
Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882
Return to citation in text: [1] [2] -
Abbas, M.; Leitgeb, A.; Slugovc, C. Synlett 2013, 24, 1193–1196. doi:10.1055/s-0033-1338425
Return to citation in text: [1] -
Abbas, M.; Slugovc, C. Tetrahedron Lett. 2011, 52, 2560–2562. doi:10.1016/j.tetlet.2011.03.038
Return to citation in text: [1] -
Hryniewicka, A.; Morzycki, J. W.; Siergiejczyk, L.; Witkowski, S.; Wójcik, J.; Gryff-Keller, A. Aust. J. Chem. 2009, 62, 1363–1370. doi:10.1071/CH08443
Return to citation in text: [1] [2] -
Hryniewicka, A.; Kozłowska, A.; Witkowski, S. J. Organomet. Chem. 2012, 701, 87–92. doi:10.1016/j.jorganchem.2011.12.024
Return to citation in text: [1] [2] -
Hryniewicka, A.; Morzycki, J. W.; Witkowski, S. J. Organomet. Chem. 2010, 695, 1265–1270. doi:10.1016/j.jorganchem.2010.02.025
Return to citation in text: [1] [2] -
Burton, G. W.; Doba, T.; Gabe, E. J.; Hughes, L.; Lee, F. L.; Prasad, L.; Ingold, K. U. J. Am. Chem. Soc. 1985, 107, 7053–7065. doi:10.1021/ja00310a049
Return to citation in text: [1] [2] [3] -
Burton, G. W.; Ingold, K. U. Acc. Chem. Res. 1986, 19, 194–201. doi:10.1021/ar00127a001
Return to citation in text: [1] [2] [3] -
Smith, L. I.; Renfrow, W. B., Jr.; Opie, J. W. J. Am. Chem. Soc. 1942, 64, 1082–1084. doi:10.1021/ja01257a020
Return to citation in text: [1] -
Dean, F. M.; Matkin, D. A.; Orabi, M. O. A. J. Chem. Soc., Perkin Trans. 1 1981, 1437–1442. doi:10.1039/p19810001437
Return to citation in text: [1] [2] -
Mahdavian, E.; Sangsura, S.; Landry, G.; Eytina, J.; Salvatore, B. A. Tetrahedron Lett. 2009, 50, 19–21. doi:10.1016/j.tetlet.2008.10.056
Return to citation in text: [1] -
Smith, L. I.; Hoehn, H. H.; Ungnade, H. E. J. Org. Chem. 1939, 4, 351–357. doi:10.1021/jo01215a019
Return to citation in text: [1] -
Suzuki, H.; Manabe, H.; Inouye, M. Chem. Lett. 1985, 14, 1671–1674. doi:10.1246/cl.1985.1671
Return to citation in text: [1] -
Choi, T.-L.; Lee, C. W.; Chatterjee, A. K.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 10417–10418. doi:10.1021/ja016386a
Return to citation in text: [1] [2] -
Funk, T. W.; Efskind, J.; Grubbs, R. H. Org. Lett. 2005, 7, 187–190. doi:10.1021/ol047929z
Return to citation in text: [1] -
Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::AID-ANIE1277>3.0.CO;2-E
Return to citation in text: [1] -
Engelhardt, F. C.; Schmitt, M. J.; Taylor, R. E. Org. Lett. 2001, 3, 2209–2212. doi:10.1021/ol016061z
Return to citation in text: [1] -
Wojtkielewicz, A.; Maj, J.; Morzycki, J. W. Tetrahedron Lett. 2009, 50, 4734–4737. doi:10.1016/j.tetlet.2009.06.032
Return to citation in text: [1] -
Wojtkielewicz, A.; Maj, J.; Dzieszkowska, A.; Morzycki, J. W. Tetrahedron 2011, 67, 6868–6875. doi:10.1016/j.tet.2011.06.086
Return to citation in text: [1] -
Venuti, M. C. Synthesis 1982, 61–63. doi:10.1055/s-1982-29701
Return to citation in text: [1] -
Hensle, E. M.; Tobis, J.; Tiller, J. C.; Bannwarth, W. J. Fluorine Chem. 2008, 129, 968–973. doi:10.1016/j.jfluchem.2008.05.024
Return to citation in text: [1] -
Stępień, D. K.; Cyrański, M. K.; Dobrzycki, Ł.; Wałejko, P.; Baj, A.; Witkowski, S.; Paradowska, K.; Wawer, I. J. Mol. Struct. 2014, 1076, 512–517. doi:10.1016/j.molstruc.2014.08.003
Return to citation in text: [1] [2] -
Hryniewicka, A.; Misztalewska, I.; Czajkowska-Szczykowska, D.; Urbańczyk-Lipkowska, Z.; Morzycki, J. W.; Witkowski, S. Tetrahedron 2014, 70, 6810–6816. doi:10.1016/j.tet.2014.07.056
Return to citation in text: [1] [2]
| 32. | Funk, T. W.; Efskind, J.; Grubbs, R. H. Org. Lett. 2005, 7, 187–190. doi:10.1021/ol047929z |
| 33. | Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::AID-ANIE1277>3.0.CO;2-E |
| 34. | Engelhardt, F. C.; Schmitt, M. J.; Taylor, R. E. Org. Lett. 2001, 3, 2209–2212. doi:10.1021/ol016061z |
| 35. | Wojtkielewicz, A.; Maj, J.; Morzycki, J. W. Tetrahedron Lett. 2009, 50, 4734–4737. doi:10.1016/j.tetlet.2009.06.032 |
| 5. | Keitz, B. K.; Endo, K.; Patel, P. R.; Herbert, M. B.; Grubbs, R. H. J. Am. Chem. Soc. 2012, 134, 693–699. doi:10.1021/ja210225e |
| 6. | Rosebrugh, L. E.; Herbert, M. B.; Marx, V. M.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 1276–1279. doi:10.1021/ja311916m |
| 7. | Occhipinti, G.; Hansen, F. R.; Törnroos, K. W.; Jensen, V. R. J. Am. Chem. Soc. 2013, 135, 3331–3334. doi:10.1021/ja311505v |
| 8. | Khan, R. K. M.; O'Brien, R. V.; Torker, S.; Li, B.; Hoveyda, A. H. J. Am. Chem. Soc. 2012, 134, 12774–12779. doi:10.1021/ja304827a |
| 21. | Hryniewicka, A.; Morzycki, J. W.; Siergiejczyk, L.; Witkowski, S.; Wójcik, J.; Gryff-Keller, A. Aust. J. Chem. 2009, 62, 1363–1370. doi:10.1071/CH08443 |
| 40. | Hryniewicka, A.; Misztalewska, I.; Czajkowska-Szczykowska, D.; Urbańczyk-Lipkowska, Z.; Morzycki, J. W.; Witkowski, S. Tetrahedron 2014, 70, 6810–6816. doi:10.1016/j.tet.2014.07.056 |
| 4. | Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525–8527. doi:10.1021/ja202818v |
| 23. | Hryniewicka, A.; Morzycki, J. W.; Witkowski, S. J. Organomet. Chem. 2010, 695, 1265–1270. doi:10.1016/j.jorganchem.2010.02.025 |
| 40. | Hryniewicka, A.; Misztalewska, I.; Czajkowska-Szczykowska, D.; Urbańczyk-Lipkowska, Z.; Morzycki, J. W.; Witkowski, S. Tetrahedron 2014, 70, 6810–6816. doi:10.1016/j.tet.2014.07.056 |
| 3. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 21. | Hryniewicka, A.; Morzycki, J. W.; Siergiejczyk, L.; Witkowski, S.; Wójcik, J.; Gryff-Keller, A. Aust. J. Chem. 2009, 62, 1363–1370. doi:10.1071/CH08443 |
| 22. | Hryniewicka, A.; Kozłowska, A.; Witkowski, S. J. Organomet. Chem. 2012, 701, 87–92. doi:10.1016/j.jorganchem.2011.12.024 |
| 23. | Hryniewicka, A.; Morzycki, J. W.; Witkowski, S. J. Organomet. Chem. 2010, 695, 1265–1270. doi:10.1016/j.jorganchem.2010.02.025 |
| 39. | Stępień, D. K.; Cyrański, M. K.; Dobrzycki, Ł.; Wałejko, P.; Baj, A.; Witkowski, S.; Paradowska, K.; Wawer, I. J. Mol. Struct. 2014, 1076, 512–517. doi:10.1016/j.molstruc.2014.08.003 |
| 2. | Nguyen, S. T.; Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1992, 114, 3974–3975. doi:10.1021/ja00036a053 |
| 24. | Burton, G. W.; Doba, T.; Gabe, E. J.; Hughes, L.; Lee, F. L.; Prasad, L.; Ingold, K. U. J. Am. Chem. Soc. 1985, 107, 7053–7065. doi:10.1021/ja00310a049 |
| 25. | Burton, G. W.; Ingold, K. U. Acc. Chem. Res. 1986, 19, 194–201. doi:10.1021/ar00127a001 |
| 39. | Stępień, D. K.; Cyrański, M. K.; Dobrzycki, Ł.; Wałejko, P.; Baj, A.; Witkowski, S.; Paradowska, K.; Wawer, I. J. Mol. Struct. 2014, 1076, 512–517. doi:10.1016/j.molstruc.2014.08.003 |
| 17. | Grela, K.; Harutyunyan, S.; Michrowska, A. Angew. Chem., Int. Ed. 2002, 41, 4038–4040. doi:10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0 |
| 18. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 16. | Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179. doi:10.1021/ja001179g |
| 19. | Abbas, M.; Leitgeb, A.; Slugovc, C. Synlett 2013, 24, 1193–1196. doi:10.1055/s-0033-1338425 |
| 20. | Abbas, M.; Slugovc, C. Tetrahedron Lett. 2011, 52, 2560–2562. doi:10.1016/j.tetlet.2011.03.038 |
| 38. | Hensle, E. M.; Tobis, J.; Tiller, J. C.; Bannwarth, W. J. Fluorine Chem. 2008, 129, 968–973. doi:10.1016/j.jfluchem.2008.05.024 |
| 15. | Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q |
| 36. | Wojtkielewicz, A.; Maj, J.; Dzieszkowska, A.; Morzycki, J. W. Tetrahedron 2011, 67, 6868–6875. doi:10.1016/j.tet.2011.06.086 |
| 9. | Buchmeiser, M. R. Chem. Rev. 2009, 109, 303–321. doi:10.1021/cr800207n |
| 10. | Klučiar, M.; Grela, K.; Mauduit, M. Dalton Trans. 2013, 42, 7354–7358. doi:10.1039/c2dt32856a |
| 11. | Kośnik, W.; Grela, K. Dalton Trans. 2013, 42, 7463–7467. doi:10.1039/c3dt33010a |
| 12. | Śledź, P.; Mauduit, M.; Grela, K. Chem. Soc. Rev. 2008, 37, 2433–2442. doi:10.1039/b711482f |
| 13. | Szczepaniak, G.; Kosiński, K.; Grela, K. Green Chem. 2014, 16, 4474–4492. doi:10.1039/C4GC00705K |
| 14. | Pastva, J.; Skowerski, K.; Czarnocki, S. J.; Žilková, N.; Čejka, J.; Bastl, Z.; Balcar, H. ACS Catal. 2014, 4, 3227–3236. doi:10.1021/cs500796u |
| 18. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 27. | Dean, F. M.; Matkin, D. A.; Orabi, M. O. A. J. Chem. Soc., Perkin Trans. 1 1981, 1437–1442. doi:10.1039/p19810001437 |
| 24. | Burton, G. W.; Doba, T.; Gabe, E. J.; Hughes, L.; Lee, F. L.; Prasad, L.; Ingold, K. U. J. Am. Chem. Soc. 1985, 107, 7053–7065. doi:10.1021/ja00310a049 |
| 25. | Burton, G. W.; Ingold, K. U. Acc. Chem. Res. 1986, 19, 194–201. doi:10.1021/ar00127a001 |
| 22. | Hryniewicka, A.; Kozłowska, A.; Witkowski, S. J. Organomet. Chem. 2012, 701, 87–92. doi:10.1016/j.jorganchem.2011.12.024 |
| 26. | Smith, L. I.; Renfrow, W. B., Jr.; Opie, J. W. J. Am. Chem. Soc. 1942, 64, 1082–1084. doi:10.1021/ja01257a020 |
| 31. | Choi, T.-L.; Lee, C. W.; Chatterjee, A. K.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 10417–10418. doi:10.1021/ja016386a |
| 31. | Choi, T.-L.; Lee, C. W.; Chatterjee, A. K.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 10417–10418. doi:10.1021/ja016386a |
| 24. | Burton, G. W.; Doba, T.; Gabe, E. J.; Hughes, L.; Lee, F. L.; Prasad, L.; Ingold, K. U. J. Am. Chem. Soc. 1985, 107, 7053–7065. doi:10.1021/ja00310a049 |
| 25. | Burton, G. W.; Ingold, K. U. Acc. Chem. Res. 1986, 19, 194–201. doi:10.1021/ar00127a001 |
| 17. | Grela, K.; Harutyunyan, S.; Michrowska, A. Angew. Chem., Int. Ed. 2002, 41, 4038–4040. doi:10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0 |
| 29. | Smith, L. I.; Hoehn, H. H.; Ungnade, H. E. J. Org. Chem. 1939, 4, 351–357. doi:10.1021/jo01215a019 |
| 30. | Suzuki, H.; Manabe, H.; Inouye, M. Chem. Lett. 1985, 14, 1671–1674. doi:10.1246/cl.1985.1671 |
| 27. | Dean, F. M.; Matkin, D. A.; Orabi, M. O. A. J. Chem. Soc., Perkin Trans. 1 1981, 1437–1442. doi:10.1039/p19810001437 |
| 28. | Mahdavian, E.; Sangsura, S.; Landry, G.; Eytina, J.; Salvatore, B. A. Tetrahedron Lett. 2009, 50, 19–21. doi:10.1016/j.tetlet.2008.10.056 |
© 2015 Hryniewicka et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)