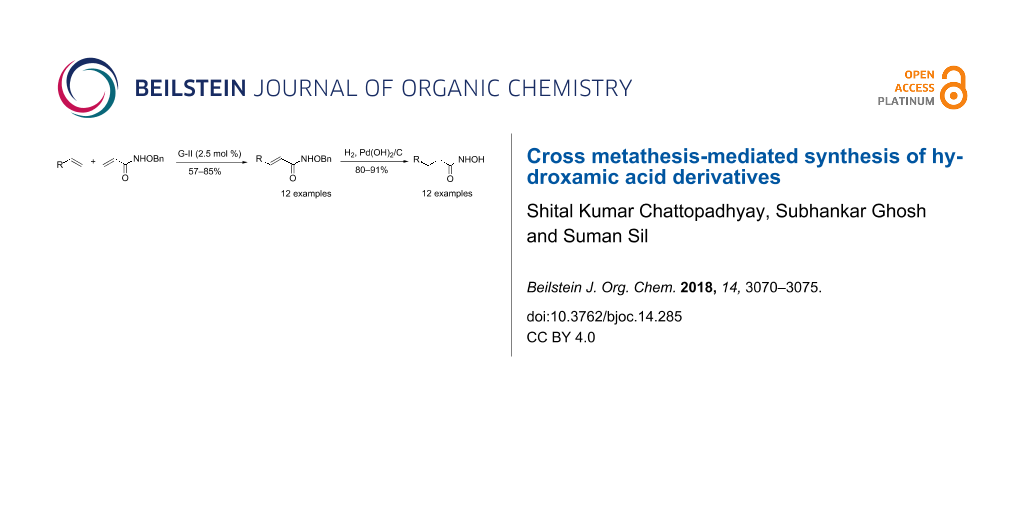
Abstract
An alternative synthesis of α,ß-unsaturated hydroxamates via cross metathesis between a class-I olefin and N-benzyloxyacrylamide is reported. The reaction proceeds better in the presence of Grubbs’ second generation catalyst within short time and in good yields (57–85%) with a range of substrates. Subsequent hydrogenation of each of the CM products delivers the title compounds in moderate to very good yield (70–89%). An important demonstration of the protocol is the preparation of the unusual amino acid component of the bioactive cyclic peptide Chap-31.

Graphical Abstract
Introduction
Cross-metathesis reactions (CM) have rapidly grown [1-3] to be a reliable method for the preparation of functionalized alkenes and derivatives thereof. Intricacies regarding the electronic nature of olefins, their substitution patterns and steric demands are more or less settled through the works of many workers in many reports [4-7]. Yet, a number of new reports describing the CM-mediated synthesis of functionalized alkenes of various kinds continue to appear. For example, cross metathesis with acrylates [8-10], α,ß-unsaturated acid chlorides [11], acrylamides [12-14], vinyl sulfones [15], vinylphosphine oxides [16], vinyl phosphonates [17], enones [18], and nitrile functionalities [19,20] have been shown to yield shorter routes to compounds of interest as well as for green chemical applications [21,22].
Hydroxamates belong to a class of valuable biologically relevant compounds of proven record of utility. For example, the hydroxamate SAHA (1, Figure 1) [23] and the didehydrohydroxamate TSA (2) [24], display useful anticancer properties through inhibition of histone deacetylase enzymes (HDAc) and are used as FDA-approved drugs. Similarly, the cyclic peptide Chap-31 (3) [25] with a terminal hydroxamic acid residue has shown promising anticancer activity. Access to such derivatives usually involves the preparation of the corresponding acid and subsequent amide bond formation with hydroxylamines. Although this two-step protocol is widely used, a direct access to α,ß-unsaturated and saturated hydroxamates from cross metathesis of alkenes may prove to be of advantage. In continuation of our earlier studies [26,27] on HDAC inhibitors, we herein report a direct access to α,ß-unsaturated hydroxamates through cross-metathesis reaction.
![[1860-5397-14-285-1]](/bjoc/content/figures/1860-5397-14-285-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some bioactive molecules containing hydroxamate functionality.
Figure 1: Some bioactive molecules containing hydroxamate functionality.
Results and Discussion
It is known that a CM reaction between a class-I olefin and a class-II olefin proceeds better in the presence of 2nd generation catalysts. Accordingly, CM between 1-decene (4, R = C7H15) and N-benzyloxyacrylamide 5 (Scheme 1) was attempted with Grubbs’ second generation catalyst [(1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(phenylmethylene) (tricyclohexylphosphine)ruthenium, G-II]. After some experimentation, it was found that the reaction proceeds quickly in refluxing dichloromethane to provide the CM product 6 (R = C7H15) in 81% yield. The yield of 6 was improved to 84% when Hoveyda–Grubbs 2nd generation catalyst [1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(o-isopropoxyphenylmethylene)ruthenium] (HG-II) was used under identical conditions. Hydrogenation of the later in the presence of Pd(OH)2/C proceeded uneventfully resulting in the saturation of the double bond as well as concommitant deprotection of the O-benzyl group. The one-pot CM-hydrogenation sequence using the same ruthenium catalyst has recently found applications [28-30]. However, similar attempts in our case, i.e., direct conversion of 4 + 5 → 7 proved to be problematic and conversion to the desired product was not observed under the attempted conditions. An intractable mixture of compounds was the result.
![[1860-5397-14-285-i1]](/bjoc/content/inline/1860-5397-14-285-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cross metathesis between a class-I alkene and N-benzyloxyacryl amide.
Scheme 1: Cross metathesis between a class-I alkene and N-benzyloxyacryl amide.
Having established the conditions for stepwise CM and hydrogenation reactions, we extended the study to other substrates (Table 1). For example, the yields of the two steps for dodecene forming 6b and 7b were more or less similar with those for decene when either of the 2nd-generation catalysts was used. However, analogous reaction with bromobutene 4c as CM partner proceeded with some compromise in yield with G-II. Moreover, HG-II in this case proved to be less successful. Similarly, the allylbenzene derivatives 4d–f reacted with more or less similar ease with G-II to produce the corresponding CM products 6d–f, respectively. Considerable isomerization (1:1 by 1H NMR) of the CM-product 6d to the corresponding styrene derivative was noticed when HG-II was used in place of G-II. 6e behaved similarly. Reaction with the styrene derivative 4g resulted in low conversion to the CM product 6g (57%). Styrene derivatives, belonging to class-I olefins according to Grubbs’ generalizations [31], are indeed known to be a sluggish partner in CM reactions, with homodimerization to stilbene being a recurring problem.
Table 1: Hydroxamates prepared.
| Entry | Alkene 4 | CM product 6 (% yield) | reduction product 7 (% yield) |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-285-i2.svg?max-width=637&scale=1.0)
4a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-285-i3.svg?max-width=637&scale=1.0)
6a (81) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-285-i4.svg?max-width=637&scale=1.0)
7a (85) |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-285-i5.svg?max-width=637&scale=1.0)
4b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-285-i6.svg?max-width=637&scale=1.0)
6b (85) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-285-i7.svg?max-width=637&scale=1.0)
7b (83) |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-285-i8.svg?max-width=637&scale=1.0)
4c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-285-i9.svg?max-width=637&scale=1.0)
6c (72) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-285-i10.svg?max-width=637&scale=1.0)
7c (70) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-285-i11.svg?max-width=637&scale=1.0)
4d |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-285-i12.svg?max-width=637&scale=1.0)
6d (77) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-285-i13.svg?max-width=637&scale=1.0)
7d (89) |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-285-i14.svg?max-width=637&scale=1.0)
4e |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-285-i15.svg?max-width=637&scale=1.0)
6e (72) |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-285-i16.svg?max-width=637&scale=1.0)
7e (85) |
| 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-285-i17.svg?max-width=637&scale=1.0)
4f |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-285-i18.svg?max-width=637&scale=1.0)
6f (78) |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-285-i19.svg?max-width=637&scale=1.0)
7f (80) |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-285-i20.svg?max-width=637&scale=1.0)
4g |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-285-i21.svg?max-width=637&scale=1.0)
6g (57) |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-285-i22.svg?max-width=637&scale=1.0)
7g (83) |
| 8 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-285-i23.svg?max-width=637&scale=1.0)
4h |
![[Graphic 23]](/bjoc/content/inline/1860-5397-14-285-i24.svg?max-width=637&scale=1.0)
6h (79) |
![[Graphic 24]](/bjoc/content/inline/1860-5397-14-285-i25.svg?max-width=637&scale=1.0)
7h (81) |
| 9 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-14-285-i26.svg?max-width=637&scale=1.0)
4i |
![[Graphic 26]](/bjoc/content/inline/1860-5397-14-285-i27.svg?max-width=637&scale=1.0)
6i (73) |
![[Graphic 27]](/bjoc/content/inline/1860-5397-14-285-i28.svg?max-width=637&scale=1.0)
7i (78) |
| 10 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-14-285-i29.svg?max-width=637&scale=1.0)
4j |
![[Graphic 29]](/bjoc/content/inline/1860-5397-14-285-i30.svg?max-width=637&scale=1.0)
6j (70) |
![[Graphic 30]](/bjoc/content/inline/1860-5397-14-285-i31.svg?max-width=637&scale=1.0)
7j (75) |
| 11 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-14-285-i32.svg?max-width=637&scale=1.0)
4k |
![[Graphic 32]](/bjoc/content/inline/1860-5397-14-285-i33.svg?max-width=637&scale=1.0)
6k (78) |
![[Graphic 33]](/bjoc/content/inline/1860-5397-14-285-i34.svg?max-width=637&scale=1.0)
7k (86) |
| 12 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-14-285-i35.svg?max-width=637&scale=1.0)
4l |
![[Graphic 35]](/bjoc/content/inline/1860-5397-14-285-i36.svg?max-width=637&scale=1.0)
6l (78) |
![[Graphic 36]](/bjoc/content/inline/1860-5397-14-285-i37.svg?max-width=637&scale=1.0)
7l (84) |
Alkenes 4h–j containing a benzyl ester functionality at two, three and four carbons apart, respectively, participated in the reaction nearly equally well to give the corresponding CM products 6h–j. Hydrogenation of each of these compounds separately led to the corresponding saturated hydroxamic acid derivatives 7h–j with concommittant cleavage of the terminal benzyl ester functionality. In an extension to the synthesis of the unusual amino acid component of the important anticancer cyclic peptide compound Chap-31, we attempted the cross-metathesis reaction of N-benzyloxyacryl amide 5 with the homoallylglycine derivative 4k (Table 1, entry 11) and the bis-homoallyl glycine derivative 4l (Table 1, entry 12) [32], separately. Fortunately, both the reactions proceeded well and the desired amino acid derivatives 7k and 7l were obtained in good yields after hydrogenation.
Conclusion
In conclusion, we have developed a direct access to functionalized hydroxamic acid derivatives using a cross-metathesis reaction between N-benzyloxyacylamide and a range of terminal alkenes. The products include hydroxamic acid derivatives with a long alkyl chain, aromatic and heteroaromatic cores, halogen residue, carboxylic acid moiety at the terminal relevant position for drug discovery. Moreover, an alternate preparation of the amino acid component of the important cyclic peptide Chap-31 may encourage the preparation of cyclic peptide based HDAC inhibitors. The developed methodology may hence complement the existing literature on the preparation of such class of compounds and may find applications.
Experimental
General procedure for cross metathesis
![[Graphic 37]](/bjoc/content/inline/1860-5397-14-285-i38.svg?max-width=637&scale=1.18182)
Grubbs’ second generation catalyst G-II (10 mg, 2 mol %), was added to a stirred solution of the olefin 4a (158 mg, 1.13 mmol), and olefin 5 (100 mg, 0.56 mmol), in anhydrous and degassed CH2Cl2 (3 mL) at rt and the reaction mixture was heated to reflux for 6 h under argon atmosphere. The reaction mixture was allowed to cool to room temperature and then concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/ethyl acetate 60:40) to provide the CM product (E)-N-benzyloxy)undec-2-enamide (6a, 133 mg, 81%) as a colourless viscous liquid.
IR (neat): 3183, 3064, 2926, 2855, 1669, 1683 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H, NH,), 7.38–7.29 (m, 5H, ArH), 6.74–6.67 (m, 1H, C3-H), 5.72 (d, J = 15.2 Hz, 1H, C2-H), 4.80 (s, 2H, OCH2-), 2.08 (q, J = 6.8 Hz, 2H, C4-H), 1.33 (brs, 3H, CH2), 1.12 (s, 12H, CH2), 0.81 (t, J = 6.8 Hz, 3H, C11-H3); 13C NMR (100 MHz, DMSO-d6) δ 163.4 (CO), 144.3 (C3), 136.4 (ArC), 129.2 (ArCH), 128.7 (ArCH), 121.1 (C2), 77.4 (OCH2), 31.8 (C4), 31.7 (C5), 29.3 (CH2), 29.1 (CH2), 29.0 (CH2), 28.2 (CH2), 22.6 (CH2), 14.3 (C11); HRMS (TOF MS ES+) m/z: [M + Na]+ calcd for C18H27NNaO2, 312.1939; found, 312.1956.
General procedure for hydrogenation
![[Graphic 38]](/bjoc/content/inline/1860-5397-14-285-i39.svg?max-width=637&scale=1.18182)
CM product 6a (50 mg, 0.17 mmol) was taken in a MeOH (3 mL) containing 1 drop of TFA [33]. Then Pd(OH)2 (10 mg) was added and the solution was degassed several times. Hydrogen gas was let in and the resulting heterogeneous mixture was vigorously stirred at atmospheric pressure for 2 h. It was filtered through Celite, the filter cake was washed with methanol (5 mL) and the combined filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3/MeOH 97:3) to provide the product N-hydroxyundecanamide 7a (85%) as a colorless solid.
Mp 85 °C; IR (neat): 3259, 3058, 2956, 1663, 1624 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H, NH), 8.93 (brs, 1H, OH), 1.92 (t, J = 7.2 Hz, 2H, C2-H), 1.44 (m, 2H, C3-H), 1.19 (s, 14H, 7× CH2), 0.81 (t, J = 6.8 Hz, 3H, C11-H); 13C NMR (100 MHz, DMSO-d6) δ 170.4 (CO), 32.6 (C2), 31.6 (C3), 29.3 (CH2), 29.3 (CH2), 29.1 (CH2), 28.9 (CH2), 25.5 (CH2), 22.5 (CH2), 14.3 (C11); HRMS (TOF MS ES+) m/z: [M + Na]+ calcd for C11H23NNaO2, 224.1626; found, 224.1638.
Supporting Information
| Supporting Information File 1: Analytical data of all new compounds as well as copies of their 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 6.6 MB | Download |
References
-
O'Leary, D. J.; O'Neil, G. W. Cross-Metathesis. In Handbook of Metathesis; Grubbs, R. H.; Wenzel, A. G.; O'Leary, D. J.; Khosravi, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; Vol. 2, pp 171–294. doi:10.1002/9783527674107.ch16
Return to citation in text: [1] -
Zukowska, K.; Grela, K. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 5, pp 1257–1301. doi:10.1016/b978-0-08-097742-3.00527-9
Return to citation in text: [1] -
Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900–1923. doi:10.1002/anie.200200556
Return to citation in text: [1] -
Zukowska, K.; Grela, K. Cross Metathesis. In Olefin Metathesis-Theory and Practice; Grela, K., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2014; pp 37–83. doi:10.1002/9781118711613.ch2
Return to citation in text: [1] -
Chatterjee, A. K. Olefin Cross-Metathesis. In Handbook of Metathesis: Catalyst Development, 1st ed.; Grubbs, R. H., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003; Vol. 10, pp 246–295. doi:10.1002/9783527619481.ch20
Return to citation in text: [1] -
Montgomery, T. P.; Johns, A. M.; Grubbs, R. H. Catalysts 2017, 7, 87. doi:10.3390/catal7030087
Return to citation in text: [1] -
Ogba, O. M.; Warner, N. C.; O’ Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510–4544. doi:10.1039/c8cs00027a
Return to citation in text: [1] -
Yu, E. C.; Johnson, B. M.; Townsend, E. M.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2016, 55, 13210–13214. doi:10.1002/anie.201608087
See for a recent reference.
Return to citation in text: [1] -
Biermann, U.; Meier, M. A. R.; Butte, W.; Metzger, J. O. Eur. J. Lipid Sci. Technol. 2011, 113, 39–45. doi:10.1002/ejlt.201000109
Return to citation in text: [1] -
Bailey, G. A.; Fogg, D. E. J. Am. Chem. Soc. 2015, 137, 7318–7321. doi:10.1021/jacs.5b04524
Return to citation in text: [1] -
Ferrié, L.; Bouzbouz, S.; Cossy, J. Org. Lett. 2009, 11, 5446–5448. doi:10.1021/ol9021386
Return to citation in text: [1] -
Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::aid-anie1277>3.0.co;2-e
Return to citation in text: [1] -
Guan, J.; Hachey, M.; Puri, L.; Howieson, V.; Saliba, K. J.; Auclair, K. Beilstein J. Org. Chem. 2016, 12, 963–968. doi:10.3762/bjoc.12.95
See for a recent report of CM with N-alkylated acrylamides.
Return to citation in text: [1] -
Boufroura, H.; Mauduit, M.; Drège, E.; Joseph, D. J. Org. Chem. 2013, 78, 2346–2354. doi:10.1021/jo302435a
See for CM with Weinreb’s amide.
Return to citation in text: [1] -
Michrowska, A.; Bieniek, M.; Kim, M.; Klajn, R.; Grela, K. Tetrahedron 2003, 59, 4525–4531. doi:10.1016/s0040-4020(03)00682-3
Return to citation in text: [1] -
Demchuk, O. M.; Pietrusiewicz, K. M.; Michrowska, A.; Grela, K. Org. Lett. 2003, 5, 3217–3220. doi:10.1021/ol035011m
Return to citation in text: [1] -
Malla, R. K.; Ridenour, J. N.; Spilling, C. D. Beilstein J. Org. Chem. 2014, 10, 1933–1941. doi:10.3762/bjoc.10.201
Return to citation in text: [1] -
Abbas, M.; Leitgeb, A.; Slugovc, C. Synlett 2013, 24, 1193–1196. doi:10.1055/s-0033-1338425
Return to citation in text: [1] -
Bidange, J.; Fischmeister, C.; Bruneau, C.; Dubois, J.-L.; Couturier, J.-L. Monatsh. Chem. 2015, 146, 1107–1113. doi:10.1007/s00706-015-1480-1
See for a detailed study of CM with acrylonitrile.
Return to citation in text: [1] -
Bruneau, C.; Fischmeister, C.; Miao, X.; Malacea, R.; Dixneuf, P. H. Eur. J. Lipid Sci. Technol. 2010, 112, 3–9. doi:10.1002/ejlt.200900105
Return to citation in text: [1] -
Bilel, H.; Hamdi, N.; Zagrouba, F.; Fischmeister, C.; Bruneau, C. RSC Adv. 2012, 2, 9584. doi:10.1039/c2ra21638h
Return to citation in text: [1] -
Miao, X.; Malacea, R.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H. Green Chem. 2011, 13, 2911. doi:10.1039/c1gc15569e
Return to citation in text: [1] -
Mann, B. S.; Johnson, J. R.; Cohen, M. H.; Justice, R.; Pazdur, R. Oncologist 2007, 12, 1247–1252. doi:10.1634/theoncologist.12-10-1247
Return to citation in text: [1] -
Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. J. Biol. Chem. 1990, 265, 17174.
Return to citation in text: [1] -
Komatsu, Y.; Tomizaki, K.-Y.; Tsukamoto, M.; Kato, T.; Nishino, N.; Sato, S.; Yamori, T.; Tsuruo, T.; Furumai, R.; Yoshida, M.; Horinouchi, S.; Hayashi, H. Cancer Res. 2001, 61, 4459.
Return to citation in text: [1] -
Mukherjee, J.; Sil, S.; Chattopadhyay, S. K. Beilstein J. Org. Chem. 2015, 11, 2487–2492. doi:10.3762/bjoc.11.270
Return to citation in text: [1] -
Mukherjee, J. P.; Sil, S.; Chattopadhyay, S. K. Tetrahedron Lett. 2016, 57, 739–742. doi:10.1016/j.tetlet.2016.01.005
Return to citation in text: [1] -
Schmidt, B.; Pohler, M. Org. Biomol. Chem. 2003, 1, 2512. doi:10.1039/b303441k
Return to citation in text: [1] -
Miao, X.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H.; Dubois, J.-L.; Couturier, J.-L. ChemSusChem 2012, 5, 1410–1414. doi:10.1002/cssc.201200086
Return to citation in text: [1] -
Skowerski, K.; Białecki, J.; Czarnocki, S. J.; Żukowska, K.; Grela, K. Beilstein J. Org. Chem. 2016, 12, 5–15. doi:10.3762/bjoc.12.2
Return to citation in text: [1] -
Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882
Return to citation in text: [1] -
Chattopadhyay, S. K.; Sil, S.; Mukherjee, J. P. Beilstein J. Org. Chem. 2017, 13, 2153–2156. doi:10.3762/bjoc.13.214
Return to citation in text: [1] -
Pahari, A. K.; Mukherjee, J. P.; Chattopadhyay, S. K. Tetrahedron 2014, 70, 7185–7191. doi:10.1016/j.tet.2014.07.045
Return to citation in text: [1]
| 33. | Pahari, A. K.; Mukherjee, J. P.; Chattopadhyay, S. K. Tetrahedron 2014, 70, 7185–7191. doi:10.1016/j.tet.2014.07.045 |
| 31. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 32. | Chattopadhyay, S. K.; Sil, S.; Mukherjee, J. P. Beilstein J. Org. Chem. 2017, 13, 2153–2156. doi:10.3762/bjoc.13.214 |
| 1. | O'Leary, D. J.; O'Neil, G. W. Cross-Metathesis. In Handbook of Metathesis; Grubbs, R. H.; Wenzel, A. G.; O'Leary, D. J.; Khosravi, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; Vol. 2, pp 171–294. doi:10.1002/9783527674107.ch16 |
| 2. | Zukowska, K.; Grela, K. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 5, pp 1257–1301. doi:10.1016/b978-0-08-097742-3.00527-9 |
| 3. | Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900–1923. doi:10.1002/anie.200200556 |
| 12. | Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::aid-anie1277>3.0.co;2-e |
| 13. |
Guan, J.; Hachey, M.; Puri, L.; Howieson, V.; Saliba, K. J.; Auclair, K. Beilstein J. Org. Chem. 2016, 12, 963–968. doi:10.3762/bjoc.12.95
See for a recent report of CM with N-alkylated acrylamides. |
| 14. |
Boufroura, H.; Mauduit, M.; Drège, E.; Joseph, D. J. Org. Chem. 2013, 78, 2346–2354. doi:10.1021/jo302435a
See for CM with Weinreb’s amide. |
| 26. | Mukherjee, J.; Sil, S.; Chattopadhyay, S. K. Beilstein J. Org. Chem. 2015, 11, 2487–2492. doi:10.3762/bjoc.11.270 |
| 27. | Mukherjee, J. P.; Sil, S.; Chattopadhyay, S. K. Tetrahedron Lett. 2016, 57, 739–742. doi:10.1016/j.tetlet.2016.01.005 |
| 11. | Ferrié, L.; Bouzbouz, S.; Cossy, J. Org. Lett. 2009, 11, 5446–5448. doi:10.1021/ol9021386 |
| 28. | Schmidt, B.; Pohler, M. Org. Biomol. Chem. 2003, 1, 2512. doi:10.1039/b303441k |
| 29. | Miao, X.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H.; Dubois, J.-L.; Couturier, J.-L. ChemSusChem 2012, 5, 1410–1414. doi:10.1002/cssc.201200086 |
| 30. | Skowerski, K.; Białecki, J.; Czarnocki, S. J.; Żukowska, K.; Grela, K. Beilstein J. Org. Chem. 2016, 12, 5–15. doi:10.3762/bjoc.12.2 |
| 8. |
Yu, E. C.; Johnson, B. M.; Townsend, E. M.; Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2016, 55, 13210–13214. doi:10.1002/anie.201608087
See for a recent reference. |
| 9. | Biermann, U.; Meier, M. A. R.; Butte, W.; Metzger, J. O. Eur. J. Lipid Sci. Technol. 2011, 113, 39–45. doi:10.1002/ejlt.201000109 |
| 10. | Bailey, G. A.; Fogg, D. E. J. Am. Chem. Soc. 2015, 137, 7318–7321. doi:10.1021/jacs.5b04524 |
| 24. | Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. J. Biol. Chem. 1990, 265, 17174. |
| 4. | Zukowska, K.; Grela, K. Cross Metathesis. In Olefin Metathesis-Theory and Practice; Grela, K., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2014; pp 37–83. doi:10.1002/9781118711613.ch2 |
| 5. | Chatterjee, A. K. Olefin Cross-Metathesis. In Handbook of Metathesis: Catalyst Development, 1st ed.; Grubbs, R. H., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003; Vol. 10, pp 246–295. doi:10.1002/9783527619481.ch20 |
| 6. | Montgomery, T. P.; Johns, A. M.; Grubbs, R. H. Catalysts 2017, 7, 87. doi:10.3390/catal7030087 |
| 7. | Ogba, O. M.; Warner, N. C.; O’ Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510–4544. doi:10.1039/c8cs00027a |
| 25. | Komatsu, Y.; Tomizaki, K.-Y.; Tsukamoto, M.; Kato, T.; Nishino, N.; Sato, S.; Yamori, T.; Tsuruo, T.; Furumai, R.; Yoshida, M.; Horinouchi, S.; Hayashi, H. Cancer Res. 2001, 61, 4459. |
| 18. | Abbas, M.; Leitgeb, A.; Slugovc, C. Synlett 2013, 24, 1193–1196. doi:10.1055/s-0033-1338425 |
| 21. | Bilel, H.; Hamdi, N.; Zagrouba, F.; Fischmeister, C.; Bruneau, C. RSC Adv. 2012, 2, 9584. doi:10.1039/c2ra21638h |
| 22. | Miao, X.; Malacea, R.; Fischmeister, C.; Bruneau, C.; Dixneuf, P. H. Green Chem. 2011, 13, 2911. doi:10.1039/c1gc15569e |
| 17. | Malla, R. K.; Ridenour, J. N.; Spilling, C. D. Beilstein J. Org. Chem. 2014, 10, 1933–1941. doi:10.3762/bjoc.10.201 |
| 23. | Mann, B. S.; Johnson, J. R.; Cohen, M. H.; Justice, R.; Pazdur, R. Oncologist 2007, 12, 1247–1252. doi:10.1634/theoncologist.12-10-1247 |
| 16. | Demchuk, O. M.; Pietrusiewicz, K. M.; Michrowska, A.; Grela, K. Org. Lett. 2003, 5, 3217–3220. doi:10.1021/ol035011m |
| 15. | Michrowska, A.; Bieniek, M.; Kim, M.; Klajn, R.; Grela, K. Tetrahedron 2003, 59, 4525–4531. doi:10.1016/s0040-4020(03)00682-3 |
| 19. |
Bidange, J.; Fischmeister, C.; Bruneau, C.; Dubois, J.-L.; Couturier, J.-L. Monatsh. Chem. 2015, 146, 1107–1113. doi:10.1007/s00706-015-1480-1
See for a detailed study of CM with acrylonitrile. |
| 20. | Bruneau, C.; Fischmeister, C.; Miao, X.; Malacea, R.; Dixneuf, P. H. Eur. J. Lipid Sci. Technol. 2010, 112, 3–9. doi:10.1002/ejlt.200900105 |
© 2018 Chattopadhyay et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)