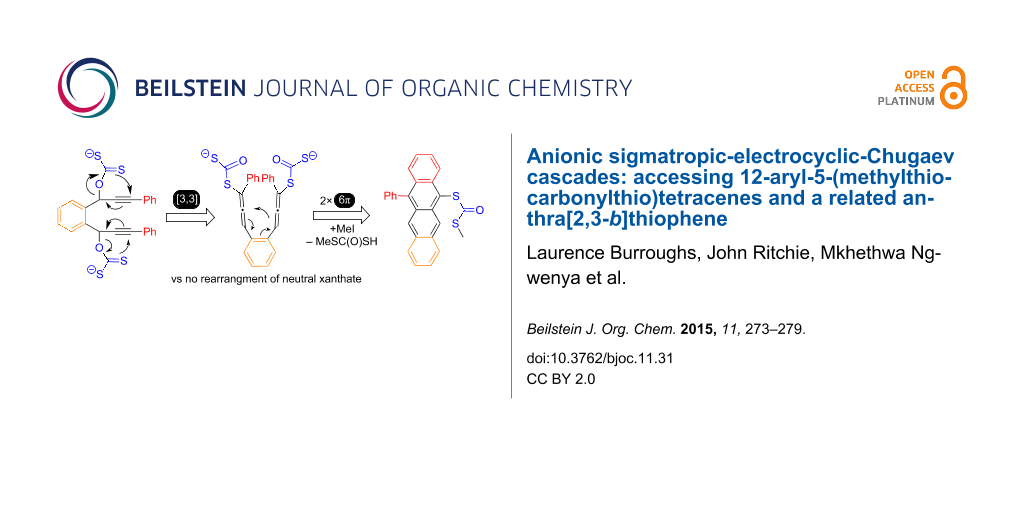
Abstract
1,4-Diols resulting from the double addition of ArCCLi (Ar = Ph, substituted phenyl, 2-thienyl) to ortho-C6H4(CHO)2 undergo cascades to tetracenes on simple admixture of LiHDMS, CS2 and MeI. Acene formation proceeds by [3,3]-sigmatropic rearrangement of xanthate anions followed by 6π electrocyclisations. The reactions are terminated by E2 or anionic Chugaev-type eliminations. Structural packing motifs and electronic properties are reported for the tetracenes.

Graphical Abstract
Introduction
In recent years polyacenes, especially tetra- and pentacenes, have been in the vanguard of new field effect and other organic electronic based devices [1,2]. Although the simple parent acenes have useful device characteristics in their own right, it is often desirable to be able to tune this performance by use of suitable substituted variants [3,4]. Unfortunately, attaining such derivatives rapidly through simple chemistry is often problematic [5,6]. Cross-coupling approaches (formally an excellent approach for acene library preparation) [7-13] are often hindered by the insolubility, or poor availability, of the parent haloacenes. Conversely, stepwise synthesis of a family of acene derivatives from various acyclic precursors is normally very step intensive. The prevalence of these issues in the synthesis of substituted tetracenes caused Lin [14], building on the anthracycline natural product work of Saá [15], to introduce a 1,2-bis-allene cascade approach for rapid access to tetracene sulfoxides in 2007 (Scheme 1).
![[1860-5397-11-31-i1]](/bjoc/content/inline/1860-5397-11-31-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Use of bis-allene intermediates 2 for rapid access to substituted tetracenes [14,16].
Scheme 1: Use of bis-allene intermediates 2 for rapid access to substituted tetracenes [14,16].
In 2012 Liu used a Pd-based strategy to provide 12-substituted 5-aryltetracenes [after final trapping with RB(OH)2)] [16]. Both of these reactions rely on the formation of bis-allenes 2, attained by Lin through 2,3-sigmatropic rearrangement of 1a [14] or by SN2’ carbonate displacement in 1b by Pd0(PPh3)2 in Liu’s case [16]. To circumvent reversibility of these pericyclic annulation strategies Lin relied on PhSOH elimination while Liu relied on ubiquitous palladium β-hydride steps leading to tetracenes 3 and 4. We are interested in very efficient routes to tetracene derivatives containing one or more thiolate (SH) groups for the use in highly electrically conducting organics. In this regard we were attracted by a single result in the early literature [17] showing that traces of allenes related to 2 (X = SCOSMe) were accessible via nominal [3,3]-sigmatropic rearrangements of xanthates. As the thiocarbamate products derived from these are predicted to be easily hydrolised to thiolates this potentially offers a simple route to a protected SH analogue of 3. Lin’s chemistry [14] cannot be used as no simple method to modify SOPh to SH is available. We proposed that use of starting material xanthate 1c should provide suitably protected 5-thiotetracene derivatives directly (Scheme 2).
![[1860-5397-11-31-i2]](/bjoc/content/inline/1860-5397-11-31-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed access to aryl substituted 5-thiolatotetracene derivatives.
Scheme 2: Proposed access to aryl substituted 5-thiolatotetracene derivatives.
The required [3,3]-sigmatropic rearrangements and subsequent 6π elecrocyclisations of 1c have precise stereochemical requirements (Scheme 2). Only the meso diastereomer of 1c is predicted by Woodward–Hoffman analyses [18] to deliver anti-6 that is required for facile E2 elimination leading to the desired tetracene 7 under thermal conditions. However, the initially required 1c are typically attained as ca. 1:1 rac/meso mixtures and this might be expected to limit the potential yield of 7 to only 50% under simple heating (in the absence of other factors).
Houk has demonstrated that both electronic donor or acceptor and steric effects favour placing the larger/most electronically biased substituent ‘outwards’ in disrotatory 6π processes [19]. This might also depopulate 5 limiting the final yield of 7. However, the following factors suggested to us the viability of Scheme 2: (i) traces of allenes have been observed when preparing xanthates from propargylic alcohols [17]; (ii) the relative van der Waals volumes of SOPh, Ph, CS2− and C(=S)SMe (104.2, 76.9, 63.4, and 82.0 Å3, respectively [20]) and related electronic properties [σ(SOMe) +0.52, σ(Ph) +0.06, σ(SCOMe) +0.39 [21]] and the work of Lin [14] suggest that significant populations of intermediate 5, with ‘inward Ph’ should be accessible; (iii) even if a rac-diol is used in the cascade, the possibility of aromatisation of 6 through Chugaev [22] syn elimination. Finally the system of Scheme 2 provides a unique opportunity to probe if these rearrangements do indeed proceed from the neutral xanthates 1c or via the previously unprecedented 1d–2d–6d anionic cascades.
Results and Discussion
Investigation of the chemistry of Scheme 2 commenced with the preparation of the required diols 8 through simple acetylide addition to o-phthalaldehyde (60–91% yield, see Supporting Information File 1). All of the additions proceeded in high yield, but under all conditions tried, no strong bias to either the rac or meso diastereomer could be realised. The meso enriched diastereomer of 8a could be realised by treating rac/meso mixtures of bis-lithium alkoxide of 8a with freshly prepared anhydrous NBu4F (2 equivalents) [23] (Scheme 3). Acid quench of the resultant purple dianion leads to ca. 5:1 meso:rac 8a. We assign this transformation to an equilibrium between dialkoxide 9 and the benzylic anion 10. Intramolecular proton delivery via cyclic transition state 11 is proposed to favour the meso dialkoxide prior to protonic quench. Samples of rac enriched 8a were prepared from Sonogashira coupling of anti enriched 8j. The latter could be prepared directly from o-bromobenzaldehyde as shown (Scheme 3) with ca. 1:7 syn:anti enrichment by recrystallisation from CHCl3. The enantiotopic ArCH signals of rac-8a are split into separate signals upon treatment with Eu(facam)3 confirming it to be the C2 chiral diastereomer while no equivalent splitting in 1H NMR samples of 8a prepared from purple 11 (in line with it being the meso diastereomer). These assignments are in line with the finding of Saá [15]. The chemical shifts of the methine CHOH proton in rac-8a (δH 6.20) and meso-8a (δH 6.33) reflect an equivalent trend in diols 8b–f where two distinct sets of equivalent signals are seen δH 6.14–6.20 and δH 6.23–6.35. On this basis we assign the higher chemical shift signal to the meso diastereomer.
![[1860-5397-11-31-i3]](/bjoc/content/inline/1860-5397-11-31-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Cascade optimisation (Table 1) was carried out using 8a in THF unless otherwise stated. Typically diol 8a (ca. 1:1 rac:meso) was deprotonated at an initial low temperature (T1), then treated sequentially with CS2 and MeI before finally being brought to a second higher temperature (T2) to facilitate aromatisation leading to 7a (see Supporting Information File 1 for full optimisation details). Simply allowing −78 °C solutions of the dialkoxide to warm slowly to ambient temperature in the presence of excess CS2/MeI provided small amounts of tetracene 7a (Table 1, run 1). Formation of the xanthate is favoured at −30 °C but this is slowed at −50 °C (Table 1, runs 2 and 3). The IR νmax [cm−1] (rac-1c: 1035; meso-1c: 1036) of run 2 are consistent with the isolation of xanthate [17].
Table 1: Optimisation of yield of tetracene 7a.a
| Run | Base | rac:mesob | T1 [°C] | T2 [°C] | 1c [%]c | 7a [%]c |
|---|---|---|---|---|---|---|
| 1 | NaH | 1:1.1 | −78 | 22 | 23 | 9 |
| 2 | NaH | 1:1.1 | −30 | 40 | 95 | – |
| 3 | NaH | 1:1.1 | −50 | 40 | 45 | 30 |
| 4 | NaH | 1:1.1 | 0 | 60 | 30 | 43 |
| 5 | LiHDMS | 1:1.1 | −50 | 40 | 39 | 38 |
| 6 | LiHDMS | 1:1.1 | 0 | 60 | 30b | 60 |
| 7 | KHDMS | 1:1.1 | 0 | 60 | 9 | 50 |
| 8 | LiHDMS | 1:4 | 0 | 60 | 5b | 38 |
| 9 | LiHDMS | 8:1 | 0 | 60 | 5b | 89 |
aUsing 8a (0.45 mmol) in THF (5.0 mL), with base (2.0 equiv), CS2 (3.0 equiv), MeI (8.0 equiv), see Supporting Information File 1 for details. bDetermined by NMR spectroscopy. cIsolated yields, except where noted.
All attempts to convert the neutral xanthate 1c (either rac or meso from Table 1, run 2) to tetracene 7a under thermal or photochemical conditions failed. Either 1c was recovered, or it slowly decomposed under forcing conditions (>200 °C; or 180–365 nm Hg lamp). Exceptionally, traces of 7a were detected in reactions eletro-catalysed by Bauld’s catalyst (tris(4-bromophenyl)ammoniumyl hexachloroantimonate) [24] at 25 °C but these showed very poor chemoselectivity. Conversely, rapid one-pot heating of a mixture of all the reaction components maximises the yield of 7a (Table 1, runs 3–6). These results very strongly suggest unprecedented anionic [3,3]-sigmatropic rearrangement starting from 1d; another addition to the body of evidence for the importance of charge upon sigmatropic rearrangements [25,26]. In the subsequent cascade the second 6π electrocyclisation appears rate limiting. The yield of 7a in run 6 (60%) indicates conversion via syn-6d (unprecedented anionic Chugaev elimination) is possible to some extent. If only E2 termination of the cascade was possible (i.e., via anti-6) a maximum yield of 52% 7a should be realised from the 1:1.1 rac:meso sample of 8a used. This idea is strongly supported by runs 8 and 9 and the observation that replacing MeI with other alkylating agents (EtBr, BnBr) resulted in only traces of tetracenes. Of the bases screened (see Supporting Information File 1), LiHDMS was superior, only its potassium analogue gave comparable performance (Table 1, run 7).
In all reactions of Table 2 there is some unrecovered material. One common byproduct is an intensely red compound detected at high Rf (0.82, 4:1 pentane/CH2Cl2) in TLC analyses conducted under argon. The very high air sensitivity of this compound prevents its characterisation but it is tentatively ascribed to a mixture (12, Scheme 4) of hydroquinone and its monomethylether on the basis of partial 1H NMR spectrum and ESI mass spectra.
Table 2: Preparation of derivatives.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-31-i6.svg?max-width=637&scale=1.0)
|
||||
| Compound | Precursor diol (8) rac:meso ratio | R1 | R2 | Yield (7) [%] |
|---|---|---|---|---|
| 7a | 1.6:1.0 | H | H | 85 |
| 7b | 1.9:1.0 | OMe | OMe | 47 |
| 7c | 4.8:1.0 | CF3 | H | 56 |
| 7d | 1.8:1.0 | OMe | H | >99 |
| 7e | 1.6:1.0 | F | H | 29 |
| 7f | 1.0:1.1 | H | CF3 | 44 |
| 7g | 1.0:1.2 | H | OMe | 38 |
| 7h | 1.0:1.6 | H | t-Bu | 22 |
| 7i | 1.0:1.0 | – | – | 38 |
| 7j | 1.0:2.0b | – | – | 50 |
aFrom diol precursor (0.45 mmol) in THF (5.0 mL), with LiHDMS (2.0 equiv), CS2 (3.0 equiv), MeI (8.0 equiv), isolated yields. bEquivalent anti:syn ratio for 8j.
![[1860-5397-11-31-i4]](/bjoc/content/inline/1860-5397-11-31-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The poorly performing runs of Table 1 also show a broad isolable red band (Rf ca. 0.18, 4:1 pentane/CH2Cl2) whose 1H and 13C NMR and MS data were intractable. For example, the 1H NMR spectrum shows only a broad envelope of signals at δH 7.95–7.20; a number of very similar isomeric species seem to be present. To cast light on these issues model alcohol 13 was treated with NaH/CS2/MeI at −78 °C and the mixture allowed to come to ambient temperature. This resulted in the smooth formation of allene 14. In particular, the presence of allenic and C=O signals in the 13C NMR spectrum at 212.1 and 188.5 ppm and the absence of any alkyne C≡C resonances in the region δC 80–90 are indicative of this transformation. All attempts to isolate 14 resulted only in the rapid formation of red oils whose mass spectra show molecular ions at (14)n (n = 1–3). Attempted direct crystallisation provided only trace amounts of cyclobutene 15 which is otherwise unstable in solution (see Supporting Information File 1). Its extensive decomposition prevented other characterisation. Based on this model system, it is proposed that intermolecular [2 + 2] reactions of bis-allenes, similar to results in other reported allenic rearrangements to rubrene [27,28], 2c,d result in the formation of numereous stereoisomeric oligomers resulting in the broad uninformative NMR spectra in the cascade byproducts. The structure of 15 is unremarkable except for the presence of a highly elongated C–C bond (1.63 Å) brought on through the steric congestion of the adjacent quaternary centers. A similar situation has been reported [29].
The use of the optimal conditions provided a series of acene derivatives (Table 2). All reactions resulted in chromatographically stable red microcrystalline solids. As anionic Chugaev elimination appeared the preferred aromatisation route from the studies of Table 1 (compare runs 6, 8 and 9), preparations of 7a–c strongly benefit from higher rac:meso ratios that increase the population of the equivalent syn-6 intermediates (Scheme 2). Steric congestion in the anion Chugaev transition state appears to favour this as all these compounds are isolated in good to excellent yields. Conversely 7e–h are isolated in lower yields due to a combination of higher meso content in 8e–h (leading less efficient E2 elimination) and lower steric promotion in the anion Chugaev elimination. Steric, rather than electronic, factors seem to affect the reaction most as evidenced by the quantitative yield of 7d compared to 7b (47%), 7c (56%), 7f (44%) and 7g (38%). The decreasing yields suggest that meta substitution promotes the 6π cyclisation while para electronic affects are minor and unhelpful according to the observed trend. Increasing the reaction temperature, in attempts to facilitate E2 elimination, was generally not useful as this led only to increased amounts of inert xanthates through sulfur alkylation. However, in the case of 7h this approach did allow us to reach 50 ± 4% yields (range for 6 runs).
Compounds 7a, d, f–h and j were subjected to single-crystal X-ray crystallography. This confirmed the molecular connectivity but more importantly allowed insight into their crystal packing features (Scheme 5 and Supporting Information File 1) across the family of structures. Pairs of 7a associate with slip-stack pairing (Cπ···Cπ 3.51–3.72 Å). Each of these (7a)2 ‘dimers’ is linked to the next through π contacts to the xanthate methyl (Cπ···MeS 3.38 Å). The ‘gaps’ in the columns are filled by an additional motif (Cπ···Cπ 3.32–3.59 Å) almost perpendicular to the stacking. In 7d a lattice of (7d)n chains propagates through C(11)π···MeS (3.39 Å) contacts. Adjacent chains overlap to produce the partial brickwork stack motif showing Cπ···Cπ 3.51–3.60 Å between the most electron rich and deficient aryl rings. Offset stacking ribbons are found in 7f (i.e., graphic ‘a’ is above ‘b’, etc.). The closest contacts are C···Cedge at 3.82–3.96 Å and Cπ···F–CF2 3.2 Å. Perpendicular ribbons propagate through the crystal linked by inter-digitated xanthates or CF3 groups. Structure 7f is the only one of the di/trisubstituted family not to show local C2 symmetry in intermolecular paring of the tetracenes. The structures of 7g,h (Scheme 5) are closely related to those of 7d and 7a, respectively. Finally, the least substituted tetracene 7j forms ribbons of herringbone structures.
![[1860-5397-11-31-i5]](/bjoc/content/inline/1860-5397-11-31-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Stacking motifs in 7a, d, f–h and j. Y = COSMe.
Scheme 5: Stacking motifs in 7a, d, f–h and j. Y = COSMe.
Estimates of the HOMO–LUMO data for 7 were taken from UV and CV measurements (see Table 3 and Supporting Information File 1), as well as by DFT calculations. Tetracenes 7d and 7f show the widest range in HOMO–LUMO perturbation while Eg opt. is ca. 0.4 eV lower that Eg calcd. across the series. We could not attain the reduction potentials of 7 but from the onset of oxidation data we could estimate the HOMO energy levels in 7 and these followed the same trend as EHOMO calcd. Preliminary testing of vacuum deposited thin polycrystalline films (ca. 800 nm) of 7a and 7j showed dielectric behaviour (σ <10−10 S cm−1) indicating that additional derivitisation and radical cation salt formation is required for the attainment of high electrical conductivity, as in the case of tetrathiotetracene [30].
Table 3: Electro-optic properties of 7a–j.
| Compound | E½ (ox.) [V]a | HOMO/ LUMO calcd. [eV]b | νmax (vis) [nm]c | Eg opt. [eV]d | Eg calcd. [eV]b |
|---|---|---|---|---|---|
| 7a | +0.52 | −5.16/−2.51 | 287 | 2.27 | 2.65 |
| 7b | +0.22 | −5.21/−2.57 | 297 | 2.23 | 2.63 |
| 7c | +1.07 | −5.23/−2.93 | 297 | 2.18 | 2.60 |
| 7d | +0.29 | −4.89/−2.36 | 296 | 2.09 | 2.55 |
| 7e | +0.65 | −5.58/−2.96 | 286 | 2.27 | 2.62 |
| 7f | +0.76 | −5.61/−2.98 | 290 | 2.24 | 2.63 |
| 7g | +0.43 | −5.20/−2.69 | 290 | 2.28 | 2.51 |
| 7h | +0.55 | −5.33/−2.69 | 289 | 2.26 | 2.65 |
| 7i | +0.60 | −5.27/−2.32 | 283 | 2.49 | 2.95 |
| 7j | +0.62 | −5.10/−2.40 | 283 | 2.36 | 2.70 |
aBy cyclic voltammetry, referenced against Fc/Fc+. bDFT: Calculated with the B3LYP-6-31G(d,p) basis set using Gaussian 09 Rev.D.01. cIn CH2Cl2. dDetermined from the onset (Tauc) of the lowest energy visible absorption band.
Conclusion
Typical [3,3]-sigmatropic rearrangements of xanthates are normally considered to proceed via neutral species (such as 1c). The tetracenes 7 herein are not formed this way, instead the evidence here strongly suggests that the required [3,3]-6π–6π electrocyclic cascades takes place via anionic xanthate species before final capping with methyl iodide. Final aromatisation through E2 or the anionic equivalent of the Chugaev reaction are also both viable. As neutral Chugaev reactions normally require very high temperatures this alternative approach is attractive as only moderate temperatures are required (60–80 °C). This procedure allows rapid access to mono sulfur-containing acenes, and is applicable to small scale library synthesis. Only low cost reagents are required and otherwise difficult to synthesise hindered 1,3,4,12-tetrasubstituted species can be made straightforwardly.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterisation data, X-ray structures, data for the DFT calculations, and NMR spectra. | ||
| Format: PDF | Size: 7.8 MB | Download |
Acknowledgements
This project has received funding from the European Union’s Seventh Programme for research, technological development and demonstration under grant agreement No 308768. We are grateful to Prof. Martins Rutkis (Institute of Solid State Physics, University of Latvia) for the measurements on 7a/h. DK thanks the Higher Education Council of Pakistan for a Fellowship. LB, JR and SW acknowledge support by the EPSRC UK NSCCS Computational Service. We thank Dr. Darren Walsh (University of Nottingham) for the help with the electrochemical studies. We thank European Thermodynamics for their involvement in the programme (JR) and use of the I19 Diamond Facility [31] is acknowledged.
References
-
Cicoira, F.; Santato, C., Eds. Organic Electronics: Emerging Concepts and Technologies; Wiley-VCH: Weinheim, Germany, 2013.
Return to citation in text: [1] -
So, F., Ed. Organic Electronics: Materials, Processing, Devices and Applications; CRC Press: Boca Raton, USA, 2010.
Return to citation in text: [1] -
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Takimiya, K.; Osaka, I.; Nakano, M. Chem. Mater. 2014, 26, 587–593. doi:10.1021/cm4021063
Return to citation in text: [1] -
Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4946. doi:10.1021/cr030666m
Return to citation in text: [1] -
Qu, H.; Chi, C. Curr. Org. Chem. 2010, 14, 2070–2108. doi:10.2174/138527210793351580
Return to citation in text: [1] -
Müller, A. M.; Avlasevich, Y. S.; Schoeller, W. W.; Müllen, K.; Bardeen, C. J. J. Am. Chem. Soc. 2007, 129, 14240–14250. doi:10.1021/ja073173y
Return to citation in text: [1] -
Yagodkin, E.; Douglas, C. J. Tetrahedron Lett. 2010, 51, 3037–3040. doi:10.1016/j.tetlet.2010.03.121
Return to citation in text: [1] -
Papagni, A.; Trombini, C.; Lombardo, M.; Bergantin, S.; Chams, A.; Chiarucci, M.; Miozzo, L.; Parravicini, M. Organometallics 2011, 30, 4325–4329. doi:10.1021/om2003943
Return to citation in text: [1] -
Gu, X.; Luhman, W. A.; Yagodkin, E.; Holmes, R. J.; Douglas, C. J. Org. Lett. 2012, 14, 1390–1393. doi:10.1021/ol300098p
Return to citation in text: [1] -
McGarry, K. A.; Xie, W.; Sutton, C.; Risko, C.; Wu, Y.; Young, V. G., Jr.; Brédas, J.-L.; Frisbie, C. D.; Douglas, C. J. Chem. Mater. 2013, 25, 2254–2263. doi:10.1021/cm400736s
Return to citation in text: [1] -
Mamada, M.; Katagiri, H.; Sakanoue, T.; Tokito, S. Cryst. Growth Des. 2015, 15, 442–448. doi:10.1021/cg501519a
Return to citation in text: [1] -
Okamoto, T.; Nakahara, K.; Saeki, A.; Seki, S.; Oh, J. H.; Akkerman, H. B.; Bao, Z.; Matsuo, Y. Chem. Mater. 2011, 23, 1646–1649. doi:10.1021/cm200356y
Return to citation in text: [1] -
Lin, Y.-C.; Lin, C.-H. Org. Lett. 2007, 9, 2075–2078. doi:10.1021/ol070467f
Return to citation in text: [1] [2] [3] [4] [5] -
Rodríguez, D.; Castedo, L.; Domínguez, D.; Saá, C. Org. Lett. 2003, 5, 3119–3121. doi:10.1021/ol035168e
Return to citation in text: [1] [2] -
Chen, M.; Chen, Y.; Liu, Y. Chem. Commun. 2012, 48, 12189–121191. doi:10.1039/c2cc36700a
Return to citation in text: [1] [2] [3] -
Tomita, K.; Nagano, M. Chem. Pharm. Bull. 1968, 16, 1911–1917. doi:10.1248/cpb.16.1911
Return to citation in text: [1] [2] [3] -
Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811
Return to citation in text: [1] -
Evenseck, J. D.; Thomas, B. E., IV; Spellmeyer, D. C.; Houk, K. N. J. Org. Chem. 1995, 60, 7134–7141. doi:10.1021/jo00127a016
Return to citation in text: [1] -
Zhao, Y. H.; Abraham, M. H.; Zissimos, A. M. J. Org. Chem. 2003, 68, 7368–7373. doi:10.1021/jo034808o
Return to citation in text: [1] -
McDaniel, D. H.; Brown, H. C. J. Org. Chem. 1958, 23, 420–427. doi:10.1021/jo01097a026
Return to citation in text: [1] -
Li, J. J. Chugaev elimination. Name Reactions; Springer-Verlag: Heidelberg, Germany, 2009; pp 110–111. doi:10.1007/978-3-642-01053-8_52
Return to citation in text: [1] -
Sun, H.; DiMagno, S. G. J. Am. Chem. Soc. 2005, 127, 2050–2051. doi:10.1021/ja0440497
Return to citation in text: [1] -
Bellville, D. J.; Wirth, D. D.; Bauld, N. L. J. Am. Chem. Soc. 1981, 103, 718–720. doi:10.1021/ja00393a061
Return to citation in text: [1] -
Lutz, R. P. Chem. Rev. 1984, 84, 205–247. doi:10.1021/cr00061a001
Return to citation in text: [1] -
Denmark, S. E.; Marlin, J. E.; Rajendra, G. J. Org. Chem. 2013, 78, 66–82. doi:10.1021/jo301919e
Return to citation in text: [1] -
Rigaudy, J.; Capdevielle, P. Tetrahedron 1977, 33, 767–773. doi:10.1016/0040-4020(77)80190-7
Return to citation in text: [1] -
Braga, D.; Jaafari, A.; Miozzo, L.; Moret, M.; Rizzato, S.; Papagni, A.; Yassar, A. Eur. J. Org. Chem. 2011, 4160–4169. doi:10.1002/ejoc.201100033
Return to citation in text: [1] -
http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Casian, A.; Stockholm, J. G.; Dusciac, V.; Nicic, V. J. Nanoelectron. Optoelectron. 2009, 4, 95–100. doi:10.1166/jno.2009.1008
Return to citation in text: [1] -
Nowell, X. H.; Barnett, S. A.; Christensen, K. E.; Teat, S. J.; Allan, D. R. J. Synchrotron Radiat. 2012, 19, 435–441. doi:10.1107/S0909049512008801
Return to citation in text: [1]
| 31. | Nowell, X. H.; Barnett, S. A.; Christensen, K. E.; Teat, S. J.; Allan, D. R. J. Synchrotron Radiat. 2012, 19, 435–441. doi:10.1107/S0909049512008801 |
| 1. | Cicoira, F.; Santato, C., Eds. Organic Electronics: Emerging Concepts and Technologies; Wiley-VCH: Weinheim, Germany, 2013. |
| 2. | So, F., Ed. Organic Electronics: Materials, Processing, Devices and Applications; CRC Press: Boca Raton, USA, 2010. |
| 17. | Tomita, K.; Nagano, M. Chem. Pharm. Bull. 1968, 16, 1911–1917. doi:10.1248/cpb.16.1911 |
| 7. | Müller, A. M.; Avlasevich, Y. S.; Schoeller, W. W.; Müllen, K.; Bardeen, C. J. J. Am. Chem. Soc. 2007, 129, 14240–14250. doi:10.1021/ja073173y |
| 8. | Yagodkin, E.; Douglas, C. J. Tetrahedron Lett. 2010, 51, 3037–3040. doi:10.1016/j.tetlet.2010.03.121 |
| 9. | Papagni, A.; Trombini, C.; Lombardo, M.; Bergantin, S.; Chams, A.; Chiarucci, M.; Miozzo, L.; Parravicini, M. Organometallics 2011, 30, 4325–4329. doi:10.1021/om2003943 |
| 10. | Gu, X.; Luhman, W. A.; Yagodkin, E.; Holmes, R. J.; Douglas, C. J. Org. Lett. 2012, 14, 1390–1393. doi:10.1021/ol300098p |
| 11. | McGarry, K. A.; Xie, W.; Sutton, C.; Risko, C.; Wu, Y.; Young, V. G., Jr.; Brédas, J.-L.; Frisbie, C. D.; Douglas, C. J. Chem. Mater. 2013, 25, 2254–2263. doi:10.1021/cm400736s |
| 12. | Mamada, M.; Katagiri, H.; Sakanoue, T.; Tokito, S. Cryst. Growth Des. 2015, 15, 442–448. doi:10.1021/cg501519a |
| 13. | Okamoto, T.; Nakahara, K.; Saeki, A.; Seki, S.; Oh, J. H.; Akkerman, H. B.; Bao, Z.; Matsuo, Y. Chem. Mater. 2011, 23, 1646–1649. doi:10.1021/cm200356y |
| 20. | Zhao, Y. H.; Abraham, M. H.; Zissimos, A. M. J. Org. Chem. 2003, 68, 7368–7373. doi:10.1021/jo034808o |
| 5. | Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4946. doi:10.1021/cr030666m |
| 6. | Qu, H.; Chi, C. Curr. Org. Chem. 2010, 14, 2070–2108. doi:10.2174/138527210793351580 |
| 18. | Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811 |
| 3. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 4. | Takimiya, K.; Osaka, I.; Nakano, M. Chem. Mater. 2014, 26, 587–593. doi:10.1021/cm4021063 |
| 19. | Evenseck, J. D.; Thomas, B. E., IV; Spellmeyer, D. C.; Houk, K. N. J. Org. Chem. 1995, 60, 7134–7141. doi:10.1021/jo00127a016 |
| 17. | Tomita, K.; Nagano, M. Chem. Pharm. Bull. 1968, 16, 1911–1917. doi:10.1248/cpb.16.1911 |
| 16. | Chen, M.; Chen, Y.; Liu, Y. Chem. Commun. 2012, 48, 12189–121191. doi:10.1039/c2cc36700a |
| 14. | Lin, Y.-C.; Lin, C.-H. Org. Lett. 2007, 9, 2075–2078. doi:10.1021/ol070467f |
| 16. | Chen, M.; Chen, Y.; Liu, Y. Chem. Commun. 2012, 48, 12189–121191. doi:10.1039/c2cc36700a |
| 15. | Rodríguez, D.; Castedo, L.; Domínguez, D.; Saá, C. Org. Lett. 2003, 5, 3119–3121. doi:10.1021/ol035168e |
| 16. | Chen, M.; Chen, Y.; Liu, Y. Chem. Commun. 2012, 48, 12189–121191. doi:10.1039/c2cc36700a |
| 22. | Li, J. J. Chugaev elimination. Name Reactions; Springer-Verlag: Heidelberg, Germany, 2009; pp 110–111. doi:10.1007/978-3-642-01053-8_52 |
| 21. | McDaniel, D. H.; Brown, H. C. J. Org. Chem. 1958, 23, 420–427. doi:10.1021/jo01097a026 |
| 30. | Casian, A.; Stockholm, J. G.; Dusciac, V.; Nicic, V. J. Nanoelectron. Optoelectron. 2009, 4, 95–100. doi:10.1166/jno.2009.1008 |
| 25. | Lutz, R. P. Chem. Rev. 1984, 84, 205–247. doi:10.1021/cr00061a001 |
| 26. | Denmark, S. E.; Marlin, J. E.; Rajendra, G. J. Org. Chem. 2013, 78, 66–82. doi:10.1021/jo301919e |
| 27. | Rigaudy, J.; Capdevielle, P. Tetrahedron 1977, 33, 767–773. doi:10.1016/0040-4020(77)80190-7 |
| 28. | Braga, D.; Jaafari, A.; Miozzo, L.; Moret, M.; Rizzato, S.; Papagni, A.; Yassar, A. Eur. J. Org. Chem. 2011, 4160–4169. doi:10.1002/ejoc.201100033 |
| 17. | Tomita, K.; Nagano, M. Chem. Pharm. Bull. 1968, 16, 1911–1917. doi:10.1248/cpb.16.1911 |
| 24. | Bellville, D. J.; Wirth, D. D.; Bauld, N. L. J. Am. Chem. Soc. 1981, 103, 718–720. doi:10.1021/ja00393a061 |
| 23. | Sun, H.; DiMagno, S. G. J. Am. Chem. Soc. 2005, 127, 2050–2051. doi:10.1021/ja0440497 |
| 15. | Rodríguez, D.; Castedo, L.; Domínguez, D.; Saá, C. Org. Lett. 2003, 5, 3119–3121. doi:10.1021/ol035168e |
© 2015 Burroughs et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)