Abstract
A new approach towards the synthesis of multisubstituted thiophenes is elaborated based on Rh(II)-catalyzed domino reactions of acyclic diazoesters with α-cyanothioacetamides. It provides a way for the preparation of 5-amino-3-(alkoxycarbonylamino)thiophene-2-carboxylates, 2-(5-amino-2-methoxycarbonylthiophene-3-yl)aminomalonates and (2-cyano-5-aminothiophene-3-yl)carbamates with the preparative yields of up to 67%. It was also shown that α-cyanothioacetamides easily interact with dirhodium carboxylates to give rather stable 2:1 complexes, resulting in an evident decrease in the efficiency of the catalytic process at moderate temperatures (20–30 °C).

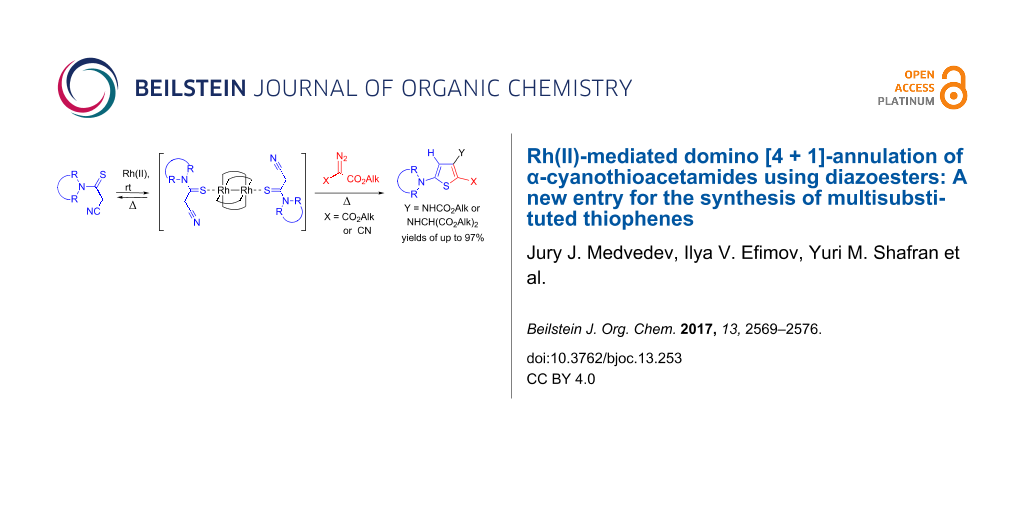
Graphical Abstract
Introduction
In recent years the diversified reactivity of metal carbenes, catalytically generated from diazocarbonyl compounds, has found wide application in organic synthesis [1-16]. A particular interest was attracted recently to domino reactions of diazo compounds with intermediate formation of ylides [7-21]. Thus, it was for example shown that ammonium or oxonium ylides generated in the course of intermolecular processes can be easily trapped by ketones, imines, α,β-unsaturated carbonyl compounds, activated multiple bonds, or other nucleophiles to furnish heterocyclic cores [22-32]. Similar intramolecular transformations of intermediate ylides with several nucleophilic reaction centers in the initial substrate, are also possible. The known examples of such reactions are for instance syntheses of multisubstituted indolines [23-25], pyrrolidines [26-29], dihydropyrroles [29], tetrahydrofurans [27,28], and 2,5-dihydrofurans [31,32], which proceed as intramolecular interaction of generated ammonium or oxonium ylides with carbonyl groups [23,27,28], C=C double [24-26], or C≡C triple [29-32] bonds in the structure of the initial molecule (Scheme 1).
![[1860-5397-13-253-i1]](/bjoc/content/inline/1860-5397-13-253-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General scheme for intramolecular heterocylization of intermediate X-ylides.
Scheme 1: General scheme for intramolecular heterocylization of intermediate X-ylides.
Intermolecular reaction of metal carbenes with thioamides usually generates thiocarbonyl ylides which leads for example to enaminones [14,33-35] or, in the reaction with N-phenyl maleimide, gives rise to formation of S-containing heterocycles by 1,3-dipolar cycloaddition [35]. However, to the best of our knowledge, there are no literature data on analogous intramolecular reactions of C=S ylides involving thiocarbonyl and any other nucleophilic group within the same molecule.
The main objective of our current research was to study Rh(II)-catalyzed reactions of diazocarbonyl compounds with α-cyanothioacetamides, bearing both thioamide and cyano groups in their structure. Based on the known literature findings [36-38] one might expect that a catalytic reaction of diazocarbonyl compounds with α-cyanothioacetamides would first of all affect the electron-rich sulfur atom of the C=S group, leading to the generation of intermediate thiocarbonyl ylides, which would further react intramolecularly with the cyano group to produce a heterocyclic structure. One cannot exclude an alternative route when the carbenoid interacts with the cyano group to yield an oxazole heterocycle [39-45]. Herein we present the first detailed results of this study.
Results and Discussion
To determine the scope and limitations of these reactions, several thioamides 1a–e of cyanoacetic acid (differing in the structure of substituents in the amino fragment) and diazoesters of three types: acyclic diazomalonates 2a,b, their cyclic analogue, 5-diazo-2,2-dimethyl-1,3-dioxane-4,6-dione (diazo Meldrum’s acid, 2c), as well as α-cyanodiazoacetic ester 2d were used in the study (Figure 1).
![[1860-5397-13-253-1]](/bjoc/content/figures/1860-5397-13-253-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Thioamides 1a–e, diazoesters 2a–d and Rh(II)-catalysts used in the project.
Figure 1: Thioamides 1a–e, diazoesters 2a–d and Rh(II)-catalysts used in the project.
Dirhodium carboxylates [Rh2(OAc)4, Rh2(Oct)4 and Rh2(Piv)4] which were found to be the most effective catalysts in reactions of diazo carbonyl compounds with different substrates [46,47], were employed in this research.
At first, we studied reactions of thioamides 1 with diazomalonates 2a,b, which usually display high reactivity in Rh-catalyzed transformations [26,48-51] (Table 1). Traditionally, similar catalytic reactions of diazomalonates occur under relatively mild conditions [26]. However, our initial attempts to carry out the processes in CH2Cl2 at room temperature or on heating with dirhodium tetraacetate or the even more active dirhodium tetraoctanoate, did not lead to a notable decomposition of diazoesters 2a,b. At the same time it was found that at elevated temperatures, for example on refluxing the diazoester 2a with thioacetamide 1a in the presence of Rh2(OAc)4 (2 mol %) in benzene solution during 5.5 h, the complete decomposition of 2a took place to produce 2,4-diaminothiophenes 3a and 4a in 51 and 35% yields, respectively (Table 1, entry 1). The products were separated and individually isolated by means of preparative chromatography on silica gel.
Table 1: Rh(II)-Catalyzed reactions of diazomalonates 2a,b with α-cyanothioacetamides 1a–e.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-253-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | reactants | catalyst | solvent, reaction time | yield, %a | ||
|---|---|---|---|---|---|---|
| 3 | 4 | total (3 + 4) | ||||
| 1b | 1a; 2a | Rh2(OAc)4 | PhH, 6 h | 3a, 51 (55) | 4a, 35 (36) | 86 (91) |
| 2b | 1a; 2a | Rh2(OAc)4 | PhMe, 3 h | 3a, 38 | 4a, 42 | 80 |
| 3c | 1a; 2a | Rh2(Oct)4 | PhH, 2 h | 3a, 35 | 4a, 23 | 58 |
| 4b | 1a; 2a | Rh2(Piv)4 | PhH, 2 h | 3a, 48 (58) | 4a, 27 (33) | 75 (91) |
| 5c | 1a; 2a | Rh2(Piv)4 | PhH, 1.5 h | 3a, 21 | 4a, 32 | 53 |
| 6b | 1a; 2b | Rh2(Piv)4 | PhH, 3 h | 3a’, 44 (67) | 4a’, 20 (30) | 64 (97) |
| 7b | 1b; 2a | Rh2(OAc)4 | PhMe, 8 hd | 3b, 27 | 4b, 25 | 52 |
| 8b | 1c; 2a | Rh2(OAc)4 | PhMe, 6 hd | 3c, 35 | 4c, 26 | 61 |
| 9b | 1d; 2a | Rh2(OAc)4 | PhH, 8 h | 3d, 30 | 4d, 24 | 54 |
| 10b | 1e; 2a | Rh2(OAc)4 | PhMe, 6 hd | 3e, 33 | 4e, 27 | 60 |
aIsolated yields are indicated in the table. The calculated yields based on reacted thioamide 1 are indicated in brackets. b1.2 equiv of diazo compound 2 were used. c2.1 equiv of diazo compound 2 were used. dReaction time in PhH was >20 h.
The same reaction with 2a in toluene solution (reflux at 110 °C) instead of benzene lead to a decrease of the yield of thiophene 3a (to 38%) in favour of thiophene 4a (42%, Table 1, entry 2). However, even under these fairly severe conditions, it took from 3 to 6 h to achieve a complete conversion of diazomalonate, which is unusual for Rh(II)-catalyzed reactions of this diazo compound [26].
Application of more active catalysts like dirhodium tetraoctanoate or tetrapivalate makes it possible to reduce the reaction time at reflux in benzene to 2 h (Table 1, entries 3 and 4). In the case of dirhodium tetraoctanoate, the yields of the compounds 3a and 4a comprised 35 and 23% (Table 1, entry 3), while when using dirhodium tetrapivalate they were 48 and 27%, respectively (Table 1, entry 4). Thus the total yield of the main reaction products 3a, 4a increased to 75% (91% based on the reacted thioamide 1a). However, a full conversion of thioamide 1a in these experiments was not achieved in spite of the 1.2-fold excess of diazomalonate 2a used in the process. Further increasing the amount of diazomalonate 2a (up to 2.1 equiv) in the reaction with rhodium tetrapivalate resulted in the enhancement of the yield of thiophene 4a (up to 32%), though the total yield of the main reaction products 3a + 4a therewith decreased to 53% (Table 1, entry 5).
Replacing diazomalonate 2a by diethyl diazomalonate (2b) did not essentially change the yields of the main products, 3a’ and 4a’ (44 and 20%, respectively; Table 1, entry 6).
Thus the experiments with diazomalonate 2a and thioacetamide 1a demonstrated that the most appropriate conditions for the catalytic reaction of a diazo carbonyl compound with thioamides, were the employment of an 1.2-fold excess of diazoester 2, Rh2(OAc)4 or Rh2(OPiv)4 as the catalysts, and performing the reaction at 80–110 °C.
To determine scope and limitations of the process, a series of experiments with diazomalonate 2a and thioamides 1b–e of different structure in the presence of Rh2(OAc)4 were carried out (Table 1, entries 7–10). It was found that within this series, the reactivity of thioamides depended significantly on the size of the substituent on the nitrogen atom of the thioamide group. Thus, decomposition of diazomalonate 2a in the presence of thioacetamide 1d bearing a bulky substituent occurred in boiling benzene within 8 h to afford thiophenes 3d and 4d in 30 and 24% yields (Table 1, entry 9). At the same time, thioamides 1b,c,e with less bulky alkyl groups under similar conditions (reflux in benzene) reacted with diazomalonate 2a much slower (Table 1; >20 h). That is why, to achieve satisfactory results, these reactions were carried out under reflux in toluene solution (110 °C; 6–8 h) giving rise to thiophenes 3b,c,e and 4b,c,e in 27–35% and 25–27% yields, respectively (Table 1, entries 7, 8 and 10).
The structures of reaction products 3 and 4 were established by a standard set of spectroscopic methods (1H, 13C NMR, HRMS) and, in the case of thiophenes 4a and 3b, was also confirmed by X-ray analysis (Figure 2). In the 1H NMR spectra of these compounds, the characteristic signals of NH protons are observed in the range of 9.84–9.68 and 7.96–7.80 ppm for 3a–e and 4a–e, respectively. The signals of thiophene H-4 and C-4 atoms in the 1H and 13C NMR spectra of compounds 3a–e and 4a–e are seen as singlets in the range of 6.95–6.58, 5.53–5.20 ppm and 97.7–93.7, 92.3–88.3 ppm, respectively.
![[1860-5397-13-253-2]](/bjoc/content/figures/1860-5397-13-253-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The structures of compounds 4a and 3b according to the data of X-ray analysis (Olex2 plot with 50% prohability level of ellipsoids).
Figure 2: The structures of compounds 4a and 3b according to the data of X-ray analysis (Olex2 plot with 50% ...
Attempts to extend the reaction under study to the cyclic analogue of diazomalonate, diazoisopropylidenemalonate 2c, were unsuccessful. Here, Rh2(OAc)4, Rh2(Piv)4 and Rh2(Oct)4 have been used as the catalysts, but with none of them a detectable conversion of the reagents was achieved. By and large this observation correlates with the literature data regarding relative inertness of the cyclic diazoester 2c in Rh(II)-catalyzed reactions in comparison with diazomalonates and the other diazo compounds [26,52,53].
The investigation of diazocyanoacetic ester 2d in reactions with thioamides 1, catalyzed by dirhodium pivalate, had shown that they produced the structural analogues of carboxylates 3, namely thiophenes 5. At the same time no concurrent formation of aminomalonates of type 4 in the reaction media was detected in these processes. Furthermore, it turned out that diazocyanoester 2d (in contrast to dialkyl diazomalonates 2a,b) easily decomposed in the presence of dirhodium pivalate in methylene chloride even at room temperature (Scheme 2).
![[1860-5397-13-253-i2]](/bjoc/content/inline/1860-5397-13-253-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Rh(II)-Catalyzed reactions of α-diazocyanoacetic ester 2d with α-cyanothioacetamides 1a–e.
Scheme 2: Rh(II)-Catalyzed reactions of α-diazocyanoacetic ester 2d with α-cyanothioacetamides 1a–e.
The catalytic reactions of diazocyanoester 2d with the thioamides 1a–e gave rise to thiophenes 5b–e in the yields of up to 51%. The structure of the isolated thiophenes 5a–e was established by a regular set of spectroscopic data, whereupon the structure of compound 5c was in addition confirmed by X-ray analysis (Figure 3).
![[1860-5397-13-253-3]](/bjoc/content/figures/1860-5397-13-253-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The structure of thiophene 5c according to the data of X-ray analysis (Olex2 plot with 50% probability level of ellipsoids).
Figure 3: The structure of thiophene 5c according to the data of X-ray analysis (Olex2 plot with 50% probabil...
In the 1H NMR spectra of these compounds, the characteristic signals of NH protons are observed in the range of 7.15–7.09 ppm for 5a–e. The signals of thiophene H-4 and C-4 atoms in the 1H and 13C spectra of the compounds 5a–e are seen as the singlets at the range of 6.40–6.78 and 96.8–92.8 ppm, respectively.
Hence it was established that the main products of diazoesters 2a,b,d in reactions with thioacetamides 1a–e, catalyzed by rhodium complexes, were sulfur-containing heterocycles, 5-amino-3-(alkoxycarbonylamino)thiophene-2-carboxylates 3, 2-(5-amino-2-methoxycarbonylthiophen-3-yl)aminomalonates 4 and (2-cyano-5-aminothiophen-3-yl)carbamates 5, which could be formally referred to as the derivatives of carbamates 3, 5 and heteroaromatic amines 4.
To elucidate the reasons for the low reactivity of diazoesters 2a,b with thioamides 1 in the considered catalytic processes, the interaction of thioacetamides 1a–e with Rh2(Piv)4 and Rh2(Oct)4 was studied (Scheme 3). The reaction was carried out in methylene chloride at room temperature using a 2-fold excess of thioacetamides 1. It produced almost quantitatively dark green adducts 6a–e, 6b’–e’, which structure was confirmed by analytical methods, X-ray analysis for complex 6e (Figure 4), and by analogy with literature data [54,55]. According to the literature findings, dirhodium(II) tetracarboxylates can form 1:2 adducts with such neutral ligands as thioamides [54,55] and thioesters [56-58]. The reactions give adducts with up to quantitative yields which is an evidence of high reactivity of rhodium complexes in these processes [54,55]. This literature data brought us to the suggestion that low reactivity of diazoesters 2 toward thioamides 1 in the presence of Rh(II)-catalysts is caused by binding the two latter chemicals to furnish the adducts of the type 6 where both axial active sites of the catalyst are inaccessible for further interaction with diazoesters 2.
![[1860-5397-13-253-i3]](/bjoc/content/inline/1860-5397-13-253-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Interaction of thioacetamide 1e with dirhodium pivalate to produce complex 6e.
Scheme 3: Interaction of thioacetamide 1e with dirhodium pivalate to produce complex 6e.
![[1860-5397-13-253-4]](/bjoc/content/figures/1860-5397-13-253-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The structure of the complex 6e according to the data of X-ray analysis (Olex2 plot with 50% probability level of ellipsoids).
Figure 4: The structure of the complex 6e according to the data of X-ray analysis (Olex2 plot with 50% probab...
To verify this assumption, a separate experiment was performed by the example of dimethyl diazomalonate (2a) decomposition with the obtained Rh-complex 6e. And it was demonstrated that under these conditions the same (as with Rh2L4) thiophenes 3e and 4e were formed. In this connection it is believed that at the elevated temperatures (80–110 °C) a partial dissociation of these complexes into original components 1 and Rh-catalyst takes place that directs the whole process into the ‘carbenoid channel’, as illustrated in Scheme 4. Initially generated from diazoester 2 carbenoid A attacks the sulfur atom of thioamide 1 to give the key intermediate S-ylide B [36-38,59,60], which is stabilized by ‘thioamide resonance’ [36-38]. The anion center of S-ylide B then attacks the carbon atom of the cyano group leading to intermediate C [61] which further turns into (imino)dihydrothiophene D through intra- or intermolecular transfer of a proton from the activated CH2 group. And finally a 1,3-shift of the alkoxycarbonyl group [62] in intermediate D completes the process to furnish thiophenes 3 or 5.
![[1860-5397-13-253-i4]](/bjoc/content/inline/1860-5397-13-253-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The assumed mechanism for the formation of thiophenes 3, 5.
Scheme 4: The assumed mechanism for the formation of thiophenes 3, 5.
Principally, thiophenes 3 and 5 could be derived from the S-ylide B in a somewhat different way, as for instance: coordination E of cyano group in S-ylide with a rhodium catalyst [63] gives rise to zwitterion F with a negative charge located on the rhodium atom, followed by recovery of the catalyst, proton transfer and, finally, 1,3-shift of alkoxycarbonyl group in the intermediate imine D to produce thiophenes 3 and 5.
However, this pathway seems to be less probable, since intramolecular cyclization B → C should have lower activation energy relative to intermolecular interaction of C=S ylide with the rhodium catalyst.
Within the adopted general scheme, the occurrence of thiophenes 4 could be rationalized by partial hydrolysis of carbamates 3 under the reaction conditions with the initial formation of the primary heteroaromatic amines 7. The latter then interact with carbenoids A, to produce thiophenes 4 through an ordinary N–H insertion process [6-13] (Scheme 5).
![[1860-5397-13-253-i5]](/bjoc/content/inline/1860-5397-13-253-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The plausible mechanism for the formation of thiophenes 4.
Scheme 5: The plausible mechanism for the formation of thiophenes 4.
To the best of our knowledge, the discovered processes are the first examples of intramolecular reactions of thiocarbonyl ylides with cyano groups, acting as an electrophile, with subsequent 1,3-migration of the alkoxycarbonyl moiety to produce the corresponding thiophenes. These reactions represent a new facile one-pot preparative method for the synthesis of 2,4-(diamino)thiophenes from the available reagents. The known findings on the synthesis of (diamino)thiophenes are limited to several articles on the transformations of 2-chlorothioacet-amides [64,65] and reactions of oxathioles [66] or 5-methylthiophenes [67] with amines.
Bearing a potential practical application of these compounds in mind, it is worthy to note that the structures comprising a 2,4-(diamino)thiophene fragment possess photosensitivity and hence could be used in OLED devices [68]. Also these compounds display high nucleophilicity in reactions with electrophiles which are often accompanied by a variety of thiophene heterocycle transformations, and hence could be successfully used for their further functionalization [69].
Conclusion
In summary, the investigation of Rh(II)-catalyzed domino reactions of acyclic diazoesters with α-cyanothioacetamides lead to elaboration of a new approach for the synthesis of multisubstituted thiophenes. These reactions could be applied for the preparation of 5-amino-3-(alkoxycarbonylamino)thiophen-2-carboxylates 3 (yields up to 67%), 2-(5-amino-2-methoxycarbonylthiophen-3-yl)aminomalonates 4 (up to 36%) and (2-cyano-5-aminothiophen-3-yl)carbamates 5 (up to 51%). It was also shown that α-cyanothioacetamides react with dirhodium carboxylates to form rather stable 2:1 complexes, that somewhat decreases the efficiency of the catalytic process at moderate temperatures (20–30 °C).
Acknowledgments
The authors express their gratitude to the SPbSU resource centers: "Center for Magnetic Resonance", "Chemical Analysis and Materials Research Centre", "Resource Education Center" and "Research Centre for X-Ray Diffraction Methods of Investigation". I.V.E. gratefully acknowledge generous financial support from "Russian Foundation for Basic Research" (RFBR, # 16-33-50162 mol_nr). V.A.B. thanks Russian Foundation for Basic Research (project 17-03-00641) for financial support in part of the synthesis of cyanothioacetamides.
References
-
Chanthamath, S.; Iwasa, S. Acc. Chem. Res. 2016, 49, 2080–2090. doi:10.1021/acs.accounts.6b00070
Return to citation in text: [1] -
Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A. B. Chem. Rev. 2003, 103, 977–1050. doi:10.1021/cr010007e
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2008, 64, 7041–7095. doi:10.1016/j.tet.2008.04.079
Return to citation in text: [1] -
Maas, G. Chem. Soc. Rev. 2004, 33, 183–190. doi:10.1039/b309046a
Return to citation in text: [1] -
Singh, V. K.; Gupta, A. D.; Sekar, G. Synthesis 1997, 137–149. doi:10.1055/s-1997-1172
Return to citation in text: [1] -
Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; McKervey, M. A. Chem. Rev. 2015, 115, 9981–10080. doi:10.1021/acs.chemrev.5b00121
Return to citation in text: [1] [2] -
Gillingham, D.; Fei, N. Chem. Soc. Rev. 2013, 42, 4918–4931. doi:10.1039/c3cs35496b
Return to citation in text: [1] [2] [3] -
Burtoloso, A. C. B.; Santiago, J. V.; Bernardim, B.; Talero, A. G. Curr. Org. Synth. 2015, 12, 650–659. doi:10.2174/157017941205150821153658
Return to citation in text: [1] [2] [3] -
Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2012, 45, 1365–1377. doi:10.1021/ar300051u
Return to citation in text: [1] [2] [3] -
Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217
Return to citation in text: [1] [2] [3] -
Collet, F.; Dodd, R. H.; Dauban, P. Chem. Commun. 2009, 5061–5074. doi:10.1039/B905820F
Return to citation in text: [1] [2] [3] -
Moody, C. J. Angew. Chem., Int. Ed. 2007, 46, 9148–9150. doi:10.1002/anie.200703016
Return to citation in text: [1] [2] [3] -
Xu, B.; Zhu, S.-F.; Zhang, Z.-C.; Yu, Z.-X.; Ma, Y.; Zhou, Q.-L. Chem. Sci. 2014, 5, 1442–1448. doi:10.1039/c3sc52807c
Return to citation in text: [1] [2] [3] -
Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263–309. doi:10.1021/cr00003a001
Return to citation in text: [1] [2] [3] -
Zhang, Z.; Wang, J. Tetrahedron 2008, 64, 6577–6605. doi:10.1016/j.tet.2008.04.074
Return to citation in text: [1] [2] -
Ando, W. Acc. Chem. Res. 1977, 10, 179–185. doi:10.1021/ar50113a005
Return to citation in text: [1] [2] -
Jia, P.; Huang, Y. Org. Lett. 2016, 18, 2475–2478. doi:10.1021/acs.orglett.6b01045
Return to citation in text: [1] -
Alamsetti, S. K.; Spanka, M.; Schneider, C. Angew. Chem., Int. Ed. 2016, 55, 2392–2396. doi:10.1002/anie.201509247
Return to citation in text: [1] -
Gao, F.; Huang, Y. Adv. Synth. Catal. 2014, 356, 2422–2428. doi:10.1002/adsc.201400176
Return to citation in text: [1] -
Xie, P.; Wang, L.; Yang, L.; Li, E.; Ma, J.; Huang, Y.; Chen, R. J. Org. Chem. 2011, 76, 7699–7705. doi:10.1021/jo2008737
Return to citation in text: [1] -
Cremonesi, G.; Dalla Croce, P.; Fontana, F.; La Rosa, C. Heterocycles 2007, 73, 873–876. doi:10.3987/COM-07-S(U)30
Return to citation in text: [1] -
Guo, X.; Hu, W. Acc. Chem. Res. 2013, 46, 2427–2440. doi:10.1021/ar300340k
Return to citation in text: [1] -
Jing, C.; Xing, D.; Hu, W. Chem. Commun. 2014, 50, 951–953. doi:10.1039/C3CC48067D
Return to citation in text: [1] [2] [3] -
Yadagiri, D.; Reddy, A. C. S.; Anbarasan, P. Chem. Sci. 2016, 7, 5934–5938. doi:10.1039/C6SC01075J
Return to citation in text: [1] [2] [3] -
Jiang, L.; Xu, R.; Kang, Z.; Feng, Y.; Sun, F.; Hu, W. J. Org. Chem. 2014, 79, 8440–8446. doi:10.1021/jo501282h
Return to citation in text: [1] [2] [3] -
Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Jing, C.; Xing, D.; Gao, L.; Li, J.; Hu, W. Chem. – Eur. J. 2015, 21, 19202–19207. doi:10.1002/chem.201503621
Return to citation in text: [1] [2] [3] [4] -
Nicolle, S. M.; Lewis, W.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749–3753. doi:10.1002/anie.201511433
Return to citation in text: [1] [2] [3] [4] -
Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 12962–12967. doi:10.1002/anie.201507122
Return to citation in text: [1] [2] [3] [4] -
Urabe, F.; Miyamoto, S.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Org. Lett. 2014, 16, 1004–1007. doi:10.1021/ol403746r
Return to citation in text: [1] [2] -
Shi, T.; Guo, X.; Teng, S.; Hu, W. Chem. Commun. 2015, 51, 15204–15207. doi:10.1039/C5CC05000F
Return to citation in text: [1] [2] [3] -
Wang, J.; Yao, X.; Wang, T.; Han, J.; Zhang, J.; Zhang, X.; Wang, P.; Zhang, Z. Org. Lett. 2015, 17, 5124–5127. doi:10.1021/acs.orglett.5b02663
Return to citation in text: [1] [2] [3] -
Koduri, N. D.; Scott, H.; Hileman, B.; Cox, J. D.; Coffin, M.; Glicksberg, L.; Hussaini, S. R. Org. Lett. 2012, 14, 440–443. doi:10.1021/ol202812d
Return to citation in text: [1] -
Koduri, N. D.; Wang, Z.; Cannell, G.; Cooley, K.; Lemma, T. M.; Miao, K.; Nguyen, M.; Frohock, B.; Castaneda, M.; Scott, H.; Albinescu, D.; Hussaini, S. R. J. Org. Chem. 2014, 79, 7405–7414. doi:10.1021/jo5011312
Return to citation in text: [1] -
Padwa, A.; Kinder, F. R.; Nadler, W. R.; Zhi, L. Heterocycles 1993, 35, 367–383. doi:10.3987/COM-92-S29
Return to citation in text: [1] [2] -
Mloston, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532.
Return to citation in text: [1] [2] [3] -
Huisgen, R.; Fulka, C.; Kalwinsch, I.; Li, X.; Mloston, G.; Moran, J. R.; Proebstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701
Return to citation in text: [1] [2] [3] -
Mloston, G.; Heimgartner, H. Curr. Org. Chem. 2011, 15, 675–693. doi:10.2174/138527211794518961
Return to citation in text: [1] [2] [3] -
Chen, J.; Shao, Y.; Ma, L.; Ma, M.; Wan, X. Org. Biomol. Chem. 2016, 14, 10723–10732. doi:10.1039/C6OB02037B
Return to citation in text: [1] -
Austeri, M.; Rix, D.; Zeghida, W.; Lacour, J. Org. Lett. 2011, 13, 1394–1397. doi:10.1021/ol2000815
Return to citation in text: [1] -
Zibinsky, M.; Fokin, V. V. Org. Lett. 2011, 13, 4870–4872. doi:10.1021/ol201949h
Return to citation in text: [1] -
Billedeau, R. J.; Klein, K. R.; Kaplan, D.; Lou, Y. Org. Lett. 2013, 15, 1421–1423. doi:10.1021/ol400062w
Return to citation in text: [1] -
Karad, S. N.; Liu, R.-S. Angew. Chem., Int. Ed. 2014, 53, 5444–5448. doi:10.1002/anie.201403015
Return to citation in text: [1] -
Cai, A.-J.; Zheng, Y.; Ma, J.-A. Chem. Commun. 2015, 51, 8946–8949. doi:10.1039/C5CC02749G
Return to citation in text: [1] -
Loy, N. S. Y.; Choi, S.; Kim, S.; Park, C.-M. Chem. Commun. 2016, 52, 7336–7339. doi:10.1039/C6CC01742H
Return to citation in text: [1] -
Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998.
Return to citation in text: [1] -
Hansen, J.; Davies, H. M. L. Coord. Chem. Rev. 2008, 252, 545–555. doi:10.1016/j.ccr.2007.08.019
Return to citation in text: [1] -
Chan, W.-W.; Lo, S.-F.; Zhou, Z.; Yu, W.-Y. J. Am. Chem. Soc. 2012, 134, 13565–13568. doi:10.1021/ja305771y
Return to citation in text: [1] -
Li, H.; Hsung, R. P. Org. Lett. 2009, 11, 4462–4465. doi:10.1021/ol901860b
Return to citation in text: [1] -
Nair, V.; Nair, S. M.; Mathai, S.; Liebscher, J.; Ziemer, B.; Narsimulu, K. Tetrahedron Lett. 2004, 45, 5759–5762. doi:10.1016/j.tetlet.2004.05.062
Return to citation in text: [1] -
Liu, Y.; Shao, X.; Zhang, P.; Lu, L.; Shen, Q. Org. Lett. 2015, 17, 2752–2755. doi:10.1021/acs.orglett.5b01170
Return to citation in text: [1] -
Lee, Y.-R.; Choi, J.-H. Bull. Korean Chem. Soc. 2006, 27, 503–507. doi:10.5012/bkcs.2006.27.4.503
Return to citation in text: [1] -
Sharma, S.; Han, S. H.; Han, S.; Ji, W.; Oh, J.; Lee, S.-Y.; Oh, J. S.; Jung, Y. H.; Kim, I. S. Org. Lett. 2015, 17, 2852–2855. doi:10.1021/acs.orglett.5b01298
Return to citation in text: [1] -
Faraglia, G.; Graziani, R.; Volponi, L.; Casellato, U. Inorg. Chim. Acta 1988, 148, 159–168. doi:10.1016/S0020-1693(00)87496-4
Return to citation in text: [1] [2] [3] -
Faraglia, G.; Volponi, L.; Sitran, S. Thermochim. Acta 1988, 132, 217–227. doi:10.1016/0040-6031(88)87112-0
Return to citation in text: [1] [2] [3] -
Clark, R. J. H.; Hemlpeman, A. J.; Dawes, H. M.; Hursthouse, M. B.; Flint, C. D. J. Chem. Soc., Dalton Trans. 1985, 1775–1780. doi:10.1039/dt9850001775
Return to citation in text: [1] -
Kitchens, J.; Bear, J. L. J. Inorg. Nucl. Chem. 1969, 31, 2415–2421. doi:10.1016/0022-1902(69)80572-5
Return to citation in text: [1] -
Kitchens, J.; Bear, J. L. J. Inorg. Nucl. Chem. 1970, 32, 49–58. doi:10.1016/0022-1902(70)80448-1
Return to citation in text: [1] -
Mloston, G.; Heimgartner, H. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A. J., Ed.; Wiley & Sons: New York, 2003.
Return to citation in text: [1] -
Gendek, T.; Mlostoń, G.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2002, 85, 451–463. doi:10.1002/1522-2675(200202)85:2<451::AID-HLCA451>3.0.CO;2-9
Return to citation in text: [1] -
Bokach, N. A.; Kukushkin, V. Y. Russ. Chem. Rev. 2005, 74, 153–170. doi:10.1070/RC2005v074n02ABEH000979
Return to citation in text: [1] -
Koch, R.; Finnerty, J. J.; Murali, S.; Wentrup, C. J. Org. Chem. 2012, 77, 1749–1759. doi:10.1021/jo2023069
Return to citation in text: [1] -
Cmoch, P. J. Mol. Struct. 2009, 919, 348–355. doi:10.1016/j.molstruc.2008.10.003
Return to citation in text: [1] -
Chupp, J. P. J. Heterocycl. Chem. 1970, 7, 285–289. doi:10.1002/jhet.5570070206
Return to citation in text: [1] -
Rolfs, A.; Liebscher, J. J. Chem. Soc., Chem. Commun. 1994, 1437–1438. doi:10.1039/c39940001437
Return to citation in text: [1] -
Hirai, K.; Ishiba, T. Chem. Pharm. Bull. 1972, 20, 2384–2393. doi:10.1248/cpb.20.2384
Return to citation in text: [1] -
Rehwald, M.; Gewald, K.; Böttcher, G. Heterocycles 1997, 45, 493–500. doi:10.3987/COM-96-7702
Return to citation in text: [1] -
Ofuku, K.; Kita, H.; Otsu, S.; Kagawa, N. Semiconductor for photoelectric conversion material, photoelectric conversion element, and solar battery. JP patent JP 2004207224, July 22, 2004.
Return to citation in text: [1] -
Chupp, J. P. J. Heterocycl. Chem. 1972, 9, 1033–1038. doi:10.1002/jhet.5570090513
Return to citation in text: [1]
| 62. | Koch, R.; Finnerty, J. J.; Murali, S.; Wentrup, C. J. Org. Chem. 2012, 77, 1749–1759. doi:10.1021/jo2023069 |
| 63. | Cmoch, P. J. Mol. Struct. 2009, 919, 348–355. doi:10.1016/j.molstruc.2008.10.003 |
| 6. | Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; McKervey, M. A. Chem. Rev. 2015, 115, 9981–10080. doi:10.1021/acs.chemrev.5b00121 |
| 7. | Gillingham, D.; Fei, N. Chem. Soc. Rev. 2013, 42, 4918–4931. doi:10.1039/c3cs35496b |
| 8. | Burtoloso, A. C. B.; Santiago, J. V.; Bernardim, B.; Talero, A. G. Curr. Org. Synth. 2015, 12, 650–659. doi:10.2174/157017941205150821153658 |
| 9. | Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2012, 45, 1365–1377. doi:10.1021/ar300051u |
| 10. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 11. | Collet, F.; Dodd, R. H.; Dauban, P. Chem. Commun. 2009, 5061–5074. doi:10.1039/B905820F |
| 12. | Moody, C. J. Angew. Chem., Int. Ed. 2007, 46, 9148–9150. doi:10.1002/anie.200703016 |
| 13. | Xu, B.; Zhu, S.-F.; Zhang, Z.-C.; Yu, Z.-X.; Ma, Y.; Zhou, Q.-L. Chem. Sci. 2014, 5, 1442–1448. doi:10.1039/c3sc52807c |
| 1. | Chanthamath, S.; Iwasa, S. Acc. Chem. Res. 2016, 49, 2080–2090. doi:10.1021/acs.accounts.6b00070 |
| 2. | Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A. B. Chem. Rev. 2003, 103, 977–1050. doi:10.1021/cr010007e |
| 3. | Pellissier, H. Tetrahedron 2008, 64, 7041–7095. doi:10.1016/j.tet.2008.04.079 |
| 4. | Maas, G. Chem. Soc. Rev. 2004, 33, 183–190. doi:10.1039/b309046a |
| 5. | Singh, V. K.; Gupta, A. D.; Sekar, G. Synthesis 1997, 137–149. doi:10.1055/s-1997-1172 |
| 6. | Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; McKervey, M. A. Chem. Rev. 2015, 115, 9981–10080. doi:10.1021/acs.chemrev.5b00121 |
| 7. | Gillingham, D.; Fei, N. Chem. Soc. Rev. 2013, 42, 4918–4931. doi:10.1039/c3cs35496b |
| 8. | Burtoloso, A. C. B.; Santiago, J. V.; Bernardim, B.; Talero, A. G. Curr. Org. Synth. 2015, 12, 650–659. doi:10.2174/157017941205150821153658 |
| 9. | Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2012, 45, 1365–1377. doi:10.1021/ar300051u |
| 10. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 11. | Collet, F.; Dodd, R. H.; Dauban, P. Chem. Commun. 2009, 5061–5074. doi:10.1039/B905820F |
| 12. | Moody, C. J. Angew. Chem., Int. Ed. 2007, 46, 9148–9150. doi:10.1002/anie.200703016 |
| 13. | Xu, B.; Zhu, S.-F.; Zhang, Z.-C.; Yu, Z.-X.; Ma, Y.; Zhou, Q.-L. Chem. Sci. 2014, 5, 1442–1448. doi:10.1039/c3sc52807c |
| 14. | Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263–309. doi:10.1021/cr00003a001 |
| 15. | Zhang, Z.; Wang, J. Tetrahedron 2008, 64, 6577–6605. doi:10.1016/j.tet.2008.04.074 |
| 16. | Ando, W. Acc. Chem. Res. 1977, 10, 179–185. doi:10.1021/ar50113a005 |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 27. | Jing, C.; Xing, D.; Gao, L.; Li, J.; Hu, W. Chem. – Eur. J. 2015, 21, 19202–19207. doi:10.1002/chem.201503621 |
| 28. | Nicolle, S. M.; Lewis, W.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749–3753. doi:10.1002/anie.201511433 |
| 29. | Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 12962–12967. doi:10.1002/anie.201507122 |
| 39. | Chen, J.; Shao, Y.; Ma, L.; Ma, M.; Wan, X. Org. Biomol. Chem. 2016, 14, 10723–10732. doi:10.1039/C6OB02037B |
| 40. | Austeri, M.; Rix, D.; Zeghida, W.; Lacour, J. Org. Lett. 2011, 13, 1394–1397. doi:10.1021/ol2000815 |
| 41. | Zibinsky, M.; Fokin, V. V. Org. Lett. 2011, 13, 4870–4872. doi:10.1021/ol201949h |
| 42. | Billedeau, R. J.; Klein, K. R.; Kaplan, D.; Lou, Y. Org. Lett. 2013, 15, 1421–1423. doi:10.1021/ol400062w |
| 43. | Karad, S. N.; Liu, R.-S. Angew. Chem., Int. Ed. 2014, 53, 5444–5448. doi:10.1002/anie.201403015 |
| 44. | Cai, A.-J.; Zheng, Y.; Ma, J.-A. Chem. Commun. 2015, 51, 8946–8949. doi:10.1039/C5CC02749G |
| 45. | Loy, N. S. Y.; Choi, S.; Kim, S.; Park, C.-M. Chem. Commun. 2016, 52, 7336–7339. doi:10.1039/C6CC01742H |
| 23. | Jing, C.; Xing, D.; Hu, W. Chem. Commun. 2014, 50, 951–953. doi:10.1039/C3CC48067D |
| 24. | Yadagiri, D.; Reddy, A. C. S.; Anbarasan, P. Chem. Sci. 2016, 7, 5934–5938. doi:10.1039/C6SC01075J |
| 25. | Jiang, L.; Xu, R.; Kang, Z.; Feng, Y.; Sun, F.; Hu, W. J. Org. Chem. 2014, 79, 8440–8446. doi:10.1021/jo501282h |
| 46. | Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998. |
| 47. | Hansen, J.; Davies, H. M. L. Coord. Chem. Rev. 2008, 252, 545–555. doi:10.1016/j.ccr.2007.08.019 |
| 22. | Guo, X.; Hu, W. Acc. Chem. Res. 2013, 46, 2427–2440. doi:10.1021/ar300340k |
| 23. | Jing, C.; Xing, D.; Hu, W. Chem. Commun. 2014, 50, 951–953. doi:10.1039/C3CC48067D |
| 24. | Yadagiri, D.; Reddy, A. C. S.; Anbarasan, P. Chem. Sci. 2016, 7, 5934–5938. doi:10.1039/C6SC01075J |
| 25. | Jiang, L.; Xu, R.; Kang, Z.; Feng, Y.; Sun, F.; Hu, W. J. Org. Chem. 2014, 79, 8440–8446. doi:10.1021/jo501282h |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 27. | Jing, C.; Xing, D.; Gao, L.; Li, J.; Hu, W. Chem. – Eur. J. 2015, 21, 19202–19207. doi:10.1002/chem.201503621 |
| 28. | Nicolle, S. M.; Lewis, W.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749–3753. doi:10.1002/anie.201511433 |
| 29. | Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 12962–12967. doi:10.1002/anie.201507122 |
| 30. | Urabe, F.; Miyamoto, S.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Org. Lett. 2014, 16, 1004–1007. doi:10.1021/ol403746r |
| 31. | Shi, T.; Guo, X.; Teng, S.; Hu, W. Chem. Commun. 2015, 51, 15204–15207. doi:10.1039/C5CC05000F |
| 32. | Wang, J.; Yao, X.; Wang, T.; Han, J.; Zhang, J.; Zhang, X.; Wang, P.; Zhang, Z. Org. Lett. 2015, 17, 5124–5127. doi:10.1021/acs.orglett.5b02663 |
| 35. | Padwa, A.; Kinder, F. R.; Nadler, W. R.; Zhi, L. Heterocycles 1993, 35, 367–383. doi:10.3987/COM-92-S29 |
| 69. | Chupp, J. P. J. Heterocycl. Chem. 1972, 9, 1033–1038. doi:10.1002/jhet.5570090513 |
| 7. | Gillingham, D.; Fei, N. Chem. Soc. Rev. 2013, 42, 4918–4931. doi:10.1039/c3cs35496b |
| 8. | Burtoloso, A. C. B.; Santiago, J. V.; Bernardim, B.; Talero, A. G. Curr. Org. Synth. 2015, 12, 650–659. doi:10.2174/157017941205150821153658 |
| 9. | Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2012, 45, 1365–1377. doi:10.1021/ar300051u |
| 10. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 11. | Collet, F.; Dodd, R. H.; Dauban, P. Chem. Commun. 2009, 5061–5074. doi:10.1039/B905820F |
| 12. | Moody, C. J. Angew. Chem., Int. Ed. 2007, 46, 9148–9150. doi:10.1002/anie.200703016 |
| 13. | Xu, B.; Zhu, S.-F.; Zhang, Z.-C.; Yu, Z.-X.; Ma, Y.; Zhou, Q.-L. Chem. Sci. 2014, 5, 1442–1448. doi:10.1039/c3sc52807c |
| 14. | Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263–309. doi:10.1021/cr00003a001 |
| 15. | Zhang, Z.; Wang, J. Tetrahedron 2008, 64, 6577–6605. doi:10.1016/j.tet.2008.04.074 |
| 16. | Ando, W. Acc. Chem. Res. 1977, 10, 179–185. doi:10.1021/ar50113a005 |
| 17. | Jia, P.; Huang, Y. Org. Lett. 2016, 18, 2475–2478. doi:10.1021/acs.orglett.6b01045 |
| 18. | Alamsetti, S. K.; Spanka, M.; Schneider, C. Angew. Chem., Int. Ed. 2016, 55, 2392–2396. doi:10.1002/anie.201509247 |
| 19. | Gao, F.; Huang, Y. Adv. Synth. Catal. 2014, 356, 2422–2428. doi:10.1002/adsc.201400176 |
| 20. | Xie, P.; Wang, L.; Yang, L.; Li, E.; Ma, J.; Huang, Y.; Chen, R. J. Org. Chem. 2011, 76, 7699–7705. doi:10.1021/jo2008737 |
| 21. | Cremonesi, G.; Dalla Croce, P.; Fontana, F.; La Rosa, C. Heterocycles 2007, 73, 873–876. doi:10.3987/COM-07-S(U)30 |
| 36. | Mloston, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532. |
| 37. | Huisgen, R.; Fulka, C.; Kalwinsch, I.; Li, X.; Mloston, G.; Moran, J. R.; Proebstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701 |
| 38. | Mloston, G.; Heimgartner, H. Curr. Org. Chem. 2011, 15, 675–693. doi:10.2174/138527211794518961 |
| 23. | Jing, C.; Xing, D.; Hu, W. Chem. Commun. 2014, 50, 951–953. doi:10.1039/C3CC48067D |
| 27. | Jing, C.; Xing, D.; Gao, L.; Li, J.; Hu, W. Chem. – Eur. J. 2015, 21, 19202–19207. doi:10.1002/chem.201503621 |
| 28. | Nicolle, S. M.; Lewis, W.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749–3753. doi:10.1002/anie.201511433 |
| 29. | Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 12962–12967. doi:10.1002/anie.201507122 |
| 30. | Urabe, F.; Miyamoto, S.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Org. Lett. 2014, 16, 1004–1007. doi:10.1021/ol403746r |
| 31. | Shi, T.; Guo, X.; Teng, S.; Hu, W. Chem. Commun. 2015, 51, 15204–15207. doi:10.1039/C5CC05000F |
| 32. | Wang, J.; Yao, X.; Wang, T.; Han, J.; Zhang, J.; Zhang, X.; Wang, P.; Zhang, Z. Org. Lett. 2015, 17, 5124–5127. doi:10.1021/acs.orglett.5b02663 |
| 67. | Rehwald, M.; Gewald, K.; Böttcher, G. Heterocycles 1997, 45, 493–500. doi:10.3987/COM-96-7702 |
| 31. | Shi, T.; Guo, X.; Teng, S.; Hu, W. Chem. Commun. 2015, 51, 15204–15207. doi:10.1039/C5CC05000F |
| 32. | Wang, J.; Yao, X.; Wang, T.; Han, J.; Zhang, J.; Zhang, X.; Wang, P.; Zhang, Z. Org. Lett. 2015, 17, 5124–5127. doi:10.1021/acs.orglett.5b02663 |
| 14. | Padwa, A.; Hornbuckle, S. F. Chem. Rev. 1991, 91, 263–309. doi:10.1021/cr00003a001 |
| 33. | Koduri, N. D.; Scott, H.; Hileman, B.; Cox, J. D.; Coffin, M.; Glicksberg, L.; Hussaini, S. R. Org. Lett. 2012, 14, 440–443. doi:10.1021/ol202812d |
| 34. | Koduri, N. D.; Wang, Z.; Cannell, G.; Cooley, K.; Lemma, T. M.; Miao, K.; Nguyen, M.; Frohock, B.; Castaneda, M.; Scott, H.; Albinescu, D.; Hussaini, S. R. J. Org. Chem. 2014, 79, 7405–7414. doi:10.1021/jo5011312 |
| 35. | Padwa, A.; Kinder, F. R.; Nadler, W. R.; Zhi, L. Heterocycles 1993, 35, 367–383. doi:10.3987/COM-92-S29 |
| 68. | Ofuku, K.; Kita, H.; Otsu, S.; Kagawa, N. Semiconductor for photoelectric conversion material, photoelectric conversion element, and solar battery. JP patent JP 2004207224, July 22, 2004. |
| 27. | Jing, C.; Xing, D.; Gao, L.; Li, J.; Hu, W. Chem. – Eur. J. 2015, 21, 19202–19207. doi:10.1002/chem.201503621 |
| 28. | Nicolle, S. M.; Lewis, W.; Hayes, C. J.; Moody, C. J. Angew. Chem., Int. Ed. 2016, 55, 3749–3753. doi:10.1002/anie.201511433 |
| 64. | Chupp, J. P. J. Heterocycl. Chem. 1970, 7, 285–289. doi:10.1002/jhet.5570070206 |
| 65. | Rolfs, A.; Liebscher, J. J. Chem. Soc., Chem. Commun. 1994, 1437–1438. doi:10.1039/c39940001437 |
| 29. | Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 12962–12967. doi:10.1002/anie.201507122 |
| 24. | Yadagiri, D.; Reddy, A. C. S.; Anbarasan, P. Chem. Sci. 2016, 7, 5934–5938. doi:10.1039/C6SC01075J |
| 25. | Jiang, L.; Xu, R.; Kang, Z.; Feng, Y.; Sun, F.; Hu, W. J. Org. Chem. 2014, 79, 8440–8446. doi:10.1021/jo501282h |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 66. | Hirai, K.; Ishiba, T. Chem. Pharm. Bull. 1972, 20, 2384–2393. doi:10.1248/cpb.20.2384 |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 48. | Chan, W.-W.; Lo, S.-F.; Zhou, Z.; Yu, W.-Y. J. Am. Chem. Soc. 2012, 134, 13565–13568. doi:10.1021/ja305771y |
| 49. | Li, H.; Hsung, R. P. Org. Lett. 2009, 11, 4462–4465. doi:10.1021/ol901860b |
| 50. | Nair, V.; Nair, S. M.; Mathai, S.; Liebscher, J.; Ziemer, B.; Narsimulu, K. Tetrahedron Lett. 2004, 45, 5759–5762. doi:10.1016/j.tetlet.2004.05.062 |
| 51. | Liu, Y.; Shao, X.; Zhang, P.; Lu, L.; Shen, Q. Org. Lett. 2015, 17, 2752–2755. doi:10.1021/acs.orglett.5b01170 |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 36. | Mloston, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532. |
| 37. | Huisgen, R.; Fulka, C.; Kalwinsch, I.; Li, X.; Mloston, G.; Moran, J. R.; Proebstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701 |
| 38. | Mloston, G.; Heimgartner, H. Curr. Org. Chem. 2011, 15, 675–693. doi:10.2174/138527211794518961 |
| 61. | Bokach, N. A.; Kukushkin, V. Y. Russ. Chem. Rev. 2005, 74, 153–170. doi:10.1070/RC2005v074n02ABEH000979 |
| 54. | Faraglia, G.; Graziani, R.; Volponi, L.; Casellato, U. Inorg. Chim. Acta 1988, 148, 159–168. doi:10.1016/S0020-1693(00)87496-4 |
| 55. | Faraglia, G.; Volponi, L.; Sitran, S. Thermochim. Acta 1988, 132, 217–227. doi:10.1016/0040-6031(88)87112-0 |
| 36. | Mloston, G.; Heimgartner, H. Pol. J. Chem. 2000, 74, 1503–1532. |
| 37. | Huisgen, R.; Fulka, C.; Kalwinsch, I.; Li, X.; Mloston, G.; Moran, J. R.; Proebstl, A. Bull. Soc. Chim. Belg. 1984, 93, 511–532. doi:10.1002/bscb.19840930701 |
| 38. | Mloston, G.; Heimgartner, H. Curr. Org. Chem. 2011, 15, 675–693. doi:10.2174/138527211794518961 |
| 59. | Mloston, G.; Heimgartner, H. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A. J., Ed.; Wiley & Sons: New York, 2003. |
| 60. | Gendek, T.; Mlostoń, G.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2002, 85, 451–463. doi:10.1002/1522-2675(200202)85:2<451::AID-HLCA451>3.0.CO;2-9 |
| 54. | Faraglia, G.; Graziani, R.; Volponi, L.; Casellato, U. Inorg. Chim. Acta 1988, 148, 159–168. doi:10.1016/S0020-1693(00)87496-4 |
| 55. | Faraglia, G.; Volponi, L.; Sitran, S. Thermochim. Acta 1988, 132, 217–227. doi:10.1016/0040-6031(88)87112-0 |
| 56. | Clark, R. J. H.; Hemlpeman, A. J.; Dawes, H. M.; Hursthouse, M. B.; Flint, C. D. J. Chem. Soc., Dalton Trans. 1985, 1775–1780. doi:10.1039/dt9850001775 |
| 57. | Kitchens, J.; Bear, J. L. J. Inorg. Nucl. Chem. 1969, 31, 2415–2421. doi:10.1016/0022-1902(69)80572-5 |
| 58. | Kitchens, J.; Bear, J. L. J. Inorg. Nucl. Chem. 1970, 32, 49–58. doi:10.1016/0022-1902(70)80448-1 |
| 26. | Medvedev, J. J.; Galkina, O. S.; Klinkova, A. A.; Giera, D. S.; Hennig, L.; Schneider, C.; Nikolaev, V. A. Org. Biomol. Chem. 2015, 13, 2640–2651. doi:10.1039/C4OB02454K |
| 52. | Lee, Y.-R.; Choi, J.-H. Bull. Korean Chem. Soc. 2006, 27, 503–507. doi:10.5012/bkcs.2006.27.4.503 |
| 53. | Sharma, S.; Han, S. H.; Han, S.; Ji, W.; Oh, J.; Lee, S.-Y.; Oh, J. S.; Jung, Y. H.; Kim, I. S. Org. Lett. 2015, 17, 2852–2855. doi:10.1021/acs.orglett.5b01298 |
| 54. | Faraglia, G.; Graziani, R.; Volponi, L.; Casellato, U. Inorg. Chim. Acta 1988, 148, 159–168. doi:10.1016/S0020-1693(00)87496-4 |
| 55. | Faraglia, G.; Volponi, L.; Sitran, S. Thermochim. Acta 1988, 132, 217–227. doi:10.1016/0040-6031(88)87112-0 |
© 2017 Medvedev et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)