Abstract
A visible-light-induced photoredox-catalyzed bromoetherification of alkenols is described. This approach, with CBr4 as the bromine source through generation of bromine in situ, provides a mild and operationally simple access to the synthesis of β-bromotetrahydrofurans and -tetrahydropyrans with high efficiency and regioselectivity.

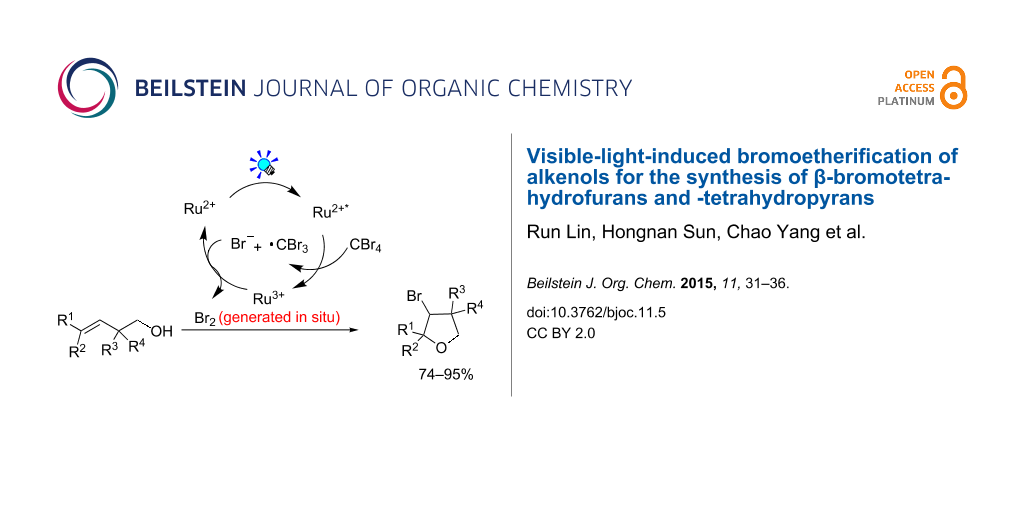
Graphical Abstract
Introduction
The halocyclization of alkenes provides an excellent synthetic method for halogenated heterocycles [1-3]. In recent years, haloaminocyclization [4,5], halolactonization [6,7] and haloetherification [8,9] of alkenes have received considerable attention from chemists, and various approaches have been made in this area. Initially, the classical synthetic pathway for bromocyclization proceeds utilizing bromine [10]. However, molecular bromine is hazardous and difficult to handle. Further research show that N-bromosuccinimide (NBS) is an effective alternative for the bromocyclization [11-14]. Furthermore, Wei Sun and co-workers disclosed an intriguing strategy to access the haloetherfication of alkenols with N-chlorosuccinimide (NCS), leading to the synthesis of β-chlorotetrahydrofurans [15]. Recently we have reported that visible-light-induced photoredox catalysis could serve as a more environmental-friendly alternative reaction system to obtain Br2 in situ from CBr4, an oxidative quencher of photoredox catalyst [16-22]. Thus, as part of difunctionalization of alkenes, with our continuous investigations on the photoredox catalytic reactions [16,23-27], herein we report our preliminary studies on visible-light-induced photoredox-catalyzed bromoetherification of alkenols using CBr4 as the bromine source.
Results and Discussion
Our initial studies were focused on the reaction of alkenol 1a as a model reaction for optimizing the reaction conditions. We were encouraged by the discovery that when 1a, CBr4 and Ru(bpy)3Cl2 were irradiated by blue LEDs in MeCN for 4 hours, trans-β-bromotetrahydrofuran 2a was obtained via 5-endo bromoetherification reaction, although the yield was only 31% (Table 1, entry 2). We have reported the bromoetherification of compound 1a as an example in our previous article [16]. However, considering the value of this strategy for the synthesis of β-bromotetrahydrofurans and -tetrahydropyrans, further research were carried out to optimize the reaction conditions. Moreover, the stereochemistry of the bromotetrahydrofurans compound 2a was misidentified before. Herein, the stereochemistry of the bromotetrahydrofurans compound 2a was determined by NOE spectra, for details see Supporting Information File 1. After a screening of selected solvents, we found solvents had a significant effect on the reaction efficiency (Table 1, entries 1–5). The reaction in DMSO led to the highest yield up to 94% (Table 1, entry 1). In addition, 2 equivalents of CBr4 were required for the efficient transformation (Table 1, entries 6 and 7). Furthermore, when the catalyst loading was reduced to even 1 mol %, the reaction also gave a comparable result (Table 1, entry 8). It should be pointed out that no reaction was observed in the absence of light or photocatalyst.
Table 1: Survey on the photocatalytic bromoetherification of alkenols.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-5-i3.svg?max-width=637&scale=1.0)
|
|||
| Entry | Conditions | Time (h) | Yield (%)b |
|---|---|---|---|
| 1 | Standard conditionsa | 4 | 94 |
| 2 | CH3CN as solvent | 4 | 31 |
| 3 | DCM as solvent | 12 | 28 |
| 4 | THF as solvent | 24 | 71 |
| 5 | DMF as solvent | 22 | 77 |
| 6 | Only 1 equiv CBr4 was used | 6 | 76 |
| 7 | Only 1.5 equiv CBr4 was used | 5 | 88 |
| 8 | Only 1 mol % Ru(bpy)3Cl2 was used | 7 | 90 |
aStandard conditions: alkenol 1a (0.2 mmol, 1 equiv), CBr4 (0.4 mmol, 2 equiv), Ru(bpy)3Cl2 (0.006 mmol, 3 mol %) in dry DMSO (0.1 M) irradiated by blue LEDs (1 W); bisolated yield.
With the optimized reaction conditions in hand, various substituted butenols were subsequently investigated for the scope of the reaction. As shown in Table 2, electronically distinct styrenes ranging from electron-rich to electron-deficient provided good yields of the desired 5-endo bromoetherification products (Table 2, entries 1–16). Additionally, trisubstituted alkenols were also examined and showed high reactivity (Table 2, entries 17 and 18). The alkenol with geminal dimethyl substituent produced the expected 5-endo bromoetherification product in 90% yield (Table 2, entry 19).
Table 2: Photocatalytic bromoetherification of butenols.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-5-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yield (%)b |
|---|---|---|---|
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-5-i5.svg?max-width=637&scale=1.18182)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-5-i6.svg?max-width=637&scale=1.18182)
|
||
| 1 | R = 4-OMePh | 2a | 94 |
| 2 | R = 3-OMePh | 2b | 93 |
| 3 | R = 2-OMePh | 2c | 89 |
| 4 | R = Ph | 2d | 88 |
| 5 | R = 4-MePh | 2e | 90 |
| 6 | R = 3-MePh | 2f | 85 |
| 7 | R = 2-MePh | 2g | 84 |
| 8 | R = 4-BrPh | 2h | 90 |
| 9 | R = 3-BrPh | 2i | 87 |
| 10 | R = 2-BrPh | 2j | 86 |
| 11 | R = 4-FPh | 2k | 89 |
| 12 | R = 4-NO2Ph | 2l | 74 |
| 13 | R = 2,4-diOMePh | 2m | 93 |
| 14 | R = 2,5-diOMePh | 2n | 84 |
| 15 | R = 2-OMe-5-ClPh | 2o | 88 |
| 16 | R = 2-OMe-naphthalen-1-yl | 2p | 86 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-5-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-5-i8.svg?max-width=637&scale=1.0)
|
||
| 17 | R1 = 4-OTBDPSPh, R2 = Me | 2q | 87 |
| 18 | R1 = R2 = 4-OMePh | 2r | 83 |
| 19 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-5-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-5-i10.svg?max-width=637&scale=1.0)
2s |
90 |
aStandard conditions: butenol 1 (0.2 mmol, 1 equiv), CBr4 (0.4 mmol, 2 equiv), Ru(bpy)3Cl2 (0.006 mmol, 3 mol %) in dry DMSO (0.1 M) irradiated by blue LEDs (1W) for 4 h; bisolated yield.
To further demonstrate the general value of this strategy, a number of longer-chain pentenols were prepared and submitted to the optimized reaction conditions. As can be seen in Table 3, various styrenes were reacted efficiently to form the substituted tetrahydropyrans in high yield via 6-endo bromoetherification (Table 3, entries 1 and 2). Furthermore, not only primary alcohols but also secondary alcohols were tolerated using the reaction conditions albeit a mixture of 6-endo and 5-exo bromoetherification products obtained (Table 3, entries 3 and 4). Interestingly, for terminal alkene, the 5-exo bromoetherification product was achieved in 84% yield (Table 3, entry 5).
Table 3: Photocatalytic bromoetherification of pentenolsa.
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-5-i11.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yield (%)b |
|---|---|---|---|
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-5-i12.svg?max-width=637&scale=1.18182)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-5-i13.svg?max-width=637&scale=1.18182)
|
||
| 1 | R = 4-OMe | 2t | 91 |
| 2 | R = 4-Me | 2u | 87 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-5-i14.svg?max-width=637&scale=1.18182)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-5-i15.svg?max-width=637&scale=1.18182)
|
||
| 3 | R = Me | 2v:3v = 1:1.1 | 74 |
| 4 | R = Ph | 2w:3w = 1:1.3 | 80 |
| 5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-5-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-5-i17.svg?max-width=637&scale=1.0)
3x |
84 |
aStandard conditions: pentenol 1 (0.2 mmol, 1 equiv), CBr4 (0.4 mmol, 2 equiv), Ru(bpy)3Cl2 (0.006 mmol, 3 mol %) in dry DMSO (0.1 M) irradiated by blue LEDs (1 W) for 4 hours; bisolated yield.
To add more credence to the involvement of bromine in this protocol, a control experiment was conducted by reaction of alkenol 1a with liquid bromine in DMSO which led to trans-β-bromotetrahydrofuran 2a in 95% yield (Scheme 1). Such a result is in accordance with the case of 1a reacted under the standard reaction conditions of this protocol.
![[1860-5397-11-5-i1]](/bjoc/content/inline/1860-5397-11-5-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Control experiment with liquid bromine for bromoethrification of alkenols.
Scheme 1: Control experiment with liquid bromine for bromoethrification of alkenols.
Based upon the above results, the mechanism is proposed as shown in Scheme 2. Firstly, oxidative quenching of the visible-light-induced excited state Ru(bpy)32+* by CBr4, generates Br− along with the Ru(bpy)33+ complex. Then bromine was generated in situ through the oxidation of Br− by Ru(bpy)33+ [16], sequential reaction with alkene 1a forms the three-membered bromonium intermediate 4 [28]. Finally, intramolecular nucleophilic cyclization furnishes the desired product β-bromotetrahydrofuran 2a.
![[1860-5397-11-5-i2]](/bjoc/content/inline/1860-5397-11-5-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the photocatalytic bromoetherification of alkenols.
Scheme 2: Proposed mechanism for the photocatalytic bromoetherification of alkenols.
Conclusion
In summary, we have developed a mild and operationally simple method for the bromoetherification of alkenols with CBr4 as the bromine source, utilizing visible-light-induced phototedox catalysis. The reaction proceeds with high efficiency and regioselectivity for the synthesis of β-bromotetrahydrofurans and -tetrahydropyranes.
Experimental
General procedure for the photocatalytic bromoetherification of alkenols
To a 10 mL round bottom flask equipped with a magnetic stir bar were added alkenols 1 (0.2 mmol), CBr4 (132 mg, 0.4 mmol), Ru(bpy)3Cl2 (4.6 mg, 0.006 mmol) and dry DMSO (2 mL). The mixture was irradiated with blue LEDs (1 W) at room temperature without being degassed for 4 hours. Then water was added and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated. The residue was purified by flash column chromatography to give the final products 2.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra for products. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
French, A. N.; Bissmire, S.; Wirth, T. Chem. Soc. Rev. 2004, 33, 354–362. doi:10.1039/b310389g
Return to citation in text: [1] -
Rodríguez, F.; Fañanás, F. J. In Handbook of Cyclization Reactions; Ma, S., Ed.; Wiley-VCH: New York, 2010; Vol. 4, pp 951–990.
Return to citation in text: [1] -
Denmark, S. E.; Kuester, W. E.; Burk, M. T. Angew. Chem., Int. Ed. 2012, 51, 10938–10953. doi:10.1002/anie.201204347
Return to citation in text: [1] -
Amjad, M.; Knight, D. W. Tetrahedron Lett. 2006, 47, 2825–2828. doi:10.1016/j.tetlet.2006.02.017
Return to citation in text: [1] -
Cui, J.; Jia, Q.; Feng, R.-Z.; Liu, S.-S.; He, T.; Zhang, C. Org. Lett. 2014, 16, 1442–1445. doi:10.1021/ol500238k
Return to citation in text: [1] -
Dowle, M. D.; Davies, D. I. Chem. Soc. Rev. 1979, 8, 171–197. doi:10.1039/cs9790800171
Return to citation in text: [1] -
Ranganathan, S.; Muraleedharan, K. M.; Vaish, N. K.; Jayaraman, N. Tetrahedron 2004, 60, 5273–5308. doi:10.1016/j.tet.2004.04.014
Return to citation in text: [1] -
Cardillo, G.; Orena, M. Tetrahedron 1990, 46, 3321–3408. doi:10.1016/S0040-4020(01)81510-6
Return to citation in text: [1] -
Montaña, A. M.; Batalla, C.; Barcia, J. A. Curr. Org. Chem. 2009, 13, 919–938. doi:10.2174/138527209788452135
Return to citation in text: [1] -
Staninets, V. I.; Shilov, E. A. Russ. Chem. Rev. 1971, 40, 272–283. doi:10.1070/RC1971v040n03ABEH001918
Return to citation in text: [1] -
Cook, C.-h.; Cho, Y.-s.; Jew, S.-s.; Jung, Y.-H. Arch. Pharmacal Res. 1985, 8, 39–41. doi:10.1007/BF02897564
Return to citation in text: [1] -
Feldman, K. S.; Crawford Mechem, C.; Nader, L. J. Am. Chem. Soc. 1982, 104, 4011–4012. doi:10.1021/ja00378a042
Return to citation in text: [1] -
Crich, D.; Sartillo-Piscil, F.; Quintero-Cortes, L.; Wink, D. J. J. Org. Chem. 2001, 67, 3360–3364. doi:10.1021/jo016388d
Return to citation in text: [1] -
Huang, D.; Wang, H.; Xue, F.; Guan, H.; Li, L.; Peng, X.; Shi, Y. Org. Lett. 2011, 13, 6350–6353. doi:10.1021/ol202527g
Return to citation in text: [1] -
Zeng, X.; Miao, C.; Wang, S.; Xia, C.; Sun, W. Chem. Commun. 2013, 49, 2418–2420. doi:10.1039/c2cc38436a
Return to citation in text: [1] -
Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Beilstein J. Org. Chem. 2014, 10, 622–627. doi:10.3762/bjoc.10.53
Return to citation in text: [1] [2] [3] [4] -
Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056
Return to citation in text: [1] -
Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223
Return to citation in text: [1] -
Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W. Chem. Commun. 2012, 48, 2337–2339. doi:10.1039/c2cc17130a
Return to citation in text: [1] -
Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Lin, R.; Xia, W. J. Org. Chem. 2012, 77, 6302–6306. doi:10.1021/jo300796j
Return to citation in text: [1] -
Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Org. Lett. 2013, 15, 624–627. doi:10.1021/ol303437m
Return to citation in text: [1] -
Guo, L.; Yang, C.; Zheng, L.; Xia, W. Org. Biomol. Chem. 2013, 11, 5787–5792. doi:10.1039/c3ob41245h
Return to citation in text: [1] -
Sun, H.; Yang, C.; Lin, R.; Xia, W. Adv. Synth. Catal. 2014, 356, 2775–2780. doi:10.1002/adsc.201400476
Return to citation in text: [1] -
Roberts, I.; Kimball, G. E. J. Am. Chem. Soc. 1937, 59, 947–948. doi:10.1021/ja01284a507
Return to citation in text: [1]
| 1. | French, A. N.; Bissmire, S.; Wirth, T. Chem. Soc. Rev. 2004, 33, 354–362. doi:10.1039/b310389g |
| 2. | Rodríguez, F.; Fañanás, F. J. In Handbook of Cyclization Reactions; Ma, S., Ed.; Wiley-VCH: New York, 2010; Vol. 4, pp 951–990. |
| 3. | Denmark, S. E.; Kuester, W. E.; Burk, M. T. Angew. Chem., Int. Ed. 2012, 51, 10938–10953. doi:10.1002/anie.201204347 |
| 10. | Staninets, V. I.; Shilov, E. A. Russ. Chem. Rev. 1971, 40, 272–283. doi:10.1070/RC1971v040n03ABEH001918 |
| 8. | Cardillo, G.; Orena, M. Tetrahedron 1990, 46, 3321–3408. doi:10.1016/S0040-4020(01)81510-6 |
| 9. | Montaña, A. M.; Batalla, C.; Barcia, J. A. Curr. Org. Chem. 2009, 13, 919–938. doi:10.2174/138527209788452135 |
| 6. | Dowle, M. D.; Davies, D. I. Chem. Soc. Rev. 1979, 8, 171–197. doi:10.1039/cs9790800171 |
| 7. | Ranganathan, S.; Muraleedharan, K. M.; Vaish, N. K.; Jayaraman, N. Tetrahedron 2004, 60, 5273–5308. doi:10.1016/j.tet.2004.04.014 |
| 4. | Amjad, M.; Knight, D. W. Tetrahedron Lett. 2006, 47, 2825–2828. doi:10.1016/j.tetlet.2006.02.017 |
| 5. | Cui, J.; Jia, Q.; Feng, R.-Z.; Liu, S.-S.; He, T.; Zhang, C. Org. Lett. 2014, 16, 1442–1445. doi:10.1021/ol500238k |
| 16. | Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Beilstein J. Org. Chem. 2014, 10, 622–627. doi:10.3762/bjoc.10.53 |
| 23. | Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W. Chem. Commun. 2012, 48, 2337–2339. doi:10.1039/c2cc17130a |
| 24. | Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Lin, R.; Xia, W. J. Org. Chem. 2012, 77, 6302–6306. doi:10.1021/jo300796j |
| 25. | Sun, H.; Yang, C.; Gao, F.; Li, Z.; Xia, W. Org. Lett. 2013, 15, 624–627. doi:10.1021/ol303437m |
| 26. | Guo, L.; Yang, C.; Zheng, L.; Xia, W. Org. Biomol. Chem. 2013, 11, 5787–5792. doi:10.1039/c3ob41245h |
| 27. | Sun, H.; Yang, C.; Lin, R.; Xia, W. Adv. Synth. Catal. 2014, 356, 2775–2780. doi:10.1002/adsc.201400476 |
| 16. | Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Beilstein J. Org. Chem. 2014, 10, 622–627. doi:10.3762/bjoc.10.53 |
| 16. | Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Beilstein J. Org. Chem. 2014, 10, 622–627. doi:10.3762/bjoc.10.53 |
| 17. | Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056 |
| 18. | Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687 |
| 19. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 20. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 21. | Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f |
| 22. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 28. | Roberts, I.; Kimball, G. E. J. Am. Chem. Soc. 1937, 59, 947–948. doi:10.1021/ja01284a507 |
| 15. | Zeng, X.; Miao, C.; Wang, S.; Xia, C.; Sun, W. Chem. Commun. 2013, 49, 2418–2420. doi:10.1039/c2cc38436a |
| 11. | Cook, C.-h.; Cho, Y.-s.; Jew, S.-s.; Jung, Y.-H. Arch. Pharmacal Res. 1985, 8, 39–41. doi:10.1007/BF02897564 |
| 12. | Feldman, K. S.; Crawford Mechem, C.; Nader, L. J. Am. Chem. Soc. 1982, 104, 4011–4012. doi:10.1021/ja00378a042 |
| 13. | Crich, D.; Sartillo-Piscil, F.; Quintero-Cortes, L.; Wink, D. J. J. Org. Chem. 2001, 67, 3360–3364. doi:10.1021/jo016388d |
| 14. | Huang, D.; Wang, H.; Xue, F.; Guan, H.; Li, L.; Peng, X.; Shi, Y. Org. Lett. 2011, 13, 6350–6353. doi:10.1021/ol202527g |
| 16. | Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Beilstein J. Org. Chem. 2014, 10, 622–627. doi:10.3762/bjoc.10.53 |
© 2015 Lin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)