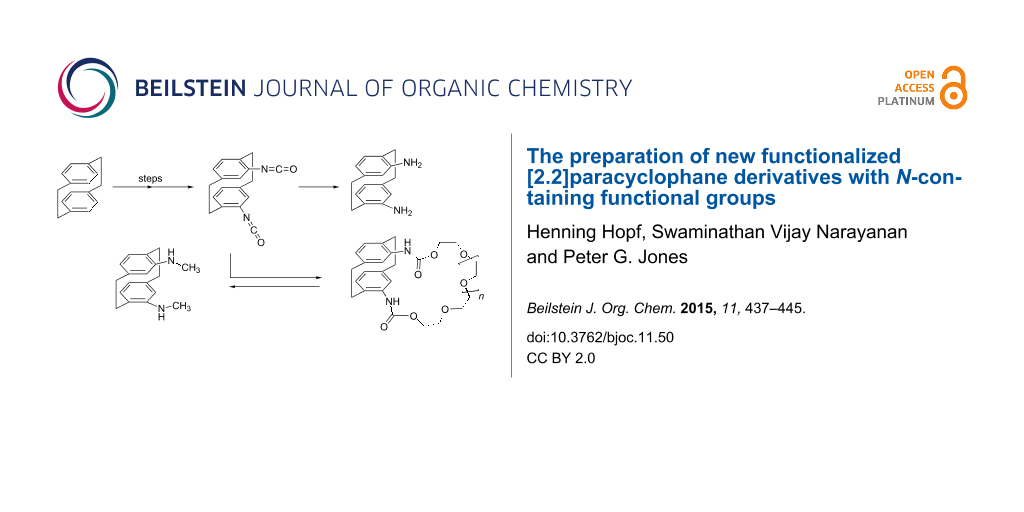
Abstract
The two isomeric bis(isocyanates) 4,12- and 4,16-di-isocyanato[2.2]paracyclophane, 16 and 28, have been prepared from their corresponding diacids by simple routes. The two isomers are versatile intermediates for the preparation of various cyclophanes bearing substituents with nitrogen-containing functional groups, e.g., the pseudo-ortho diamine 8, the bis secondary amine 23, and the crownophanes 18 and 19. Several of these new cyclophane derivatives (18, 19, 22, 26, 28) have been characterized by X-ray structural analysis.

Graphical Abstract
Introduction
Although hundreds of mono- und disubstituted derivatives of [2.2]paracyclophane [3,4] have been described since its initial preparation [5], relatively little is known about more highly substituted and highly functionalized [2.2]paracyclophanes, which are displayed in general form in formula 1 in Figure 1.
![[1860-5397-11-50-1]](/bjoc/content/figures/1860-5397-11-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: A selection of highly substituted/functionalized [2.2]paracyclophanes.
Figure 1: A selection of highly substituted/functionalized [2.2]paracyclophanes.
In this general structure we distinguish between bridge substituents [Sb]m, where the number of substituents m can lie between 1 and 8, and substituents that are anchored in the benzene rings, [Sar]n, n again ranging from 1 to 8. Of course, these two types of substituents will display different reactivities since they are bonded to differently hybridized carbon atoms. In a way, the rigid cyclophane carbon framework may be regarded as a hexadecavalent “superatom” that can bind up to 16 substituents in a geometrically clearly defined way. If both bridges and rings are fully substituted (1, m = 8, n = 8) we are dealing with an elongated, American football-type of carbon core to which 16 substituents are bound in a pincushion-like fashion. If only 8 substituents are anchored on one side of the cyclophane molecule, all pointing in the same direction, a structure results that bears functional groups on the one half of the molecule whereas the other consists of aliphatic and aromatic C–H bonds only. It is very likely that these geometries will have a strong influence on chemical and physical properties (e.g., solubility behavior) of the respective derivatives. Clearly, many other arrangements of important functional groups are possible, depending on the number of substituents (Sb and Sar) and their locations. So far the most thoroughly studied, more than singly substituted system is the pseudo-geminally disubstituted derivative in which the two (identical or non-identical) substituents are directly above/below each other (see below).
To the best of our knowledge the first fully functionalized [2.2]paracyclophane was the fluorocarbon perfluoro[2.2]paracyclophane (2) first prepared by Dolbier and co-workers [6]. The corresponding perchloro and perbromo derivatives are apparently unknown.
Other notable oligofluoro hydrocarbons are the octafluoro derivative 3 with eight fluorine substituents in Sb positions [7-10], and its isomer 4 with the eight fluorine substituents attached to the two benzene rings [11,12].
These paracyclophane derivatives are of interest as substrates for the preparation of highly and/or perfluorinated parylene derivatives, the polymer obtained by flash vacuum pyrolysis of the respective paracyclophane precursors. Among the highly alkyl-substituted [2.2]paracyclophanes the octamethyl compound 5 is the most highly substituted derivative presently known [13,14].
Finally, various ethynylated derivatives 6 have been described by us recently [15], compounds that, inter alia, are of interest in materials science [16-18].
Among the oligo- or polysubstituted paracyclophanes with preparative potential, we think that those bearing nitrogen-containing functional groups, although known for many years [19], deserve more attention. A case in point is the (achiral) pseudo-geminal diamine 7 (Figure 2) [20], which has been used as a reusable spacer for “topochemical reaction control in solution” [21].
![[1860-5397-11-50-2]](/bjoc/content/figures/1860-5397-11-50-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A selection of [2.2]paracyclophanes carrying several nitrogen-containing substituents.
Figure 2: A selection of [2.2]paracyclophanes carrying several nitrogen-containing substituents.
We are interested in generalizing the methods described for the preparation of 7 and in the present contribution to concentrate on the preparation of the pseudo-ortho-diamine 8, which is not only chiral but also offers numerous possibilities for further transformations. Related to the investigation of 8 are compounds of type 9, in which the N-carrying functional groups are now attached to the pseudo-para positions. With identical atoms/groups X these compounds are of course achiral because of their inversion symmetry. A specific example of this type of derivative would be that with X = CO, i.e., the bis(isocyanate) (see below).
Once we have developed high-yielding routes to 8 and 9, we plan to apply them to the preparation of the isomeric tetraamines 10 and 11 from the corresponding, already available tetraacids [22,23].
Results and Discussion
Preparation and reactivity of 4,12-diamino[2.2]paracyclophane (8, pseudo-ortho-diamine 8)
The diamine 8 was obtained by the sequence summarized in Scheme 1.
![[1860-5397-11-50-i1]](/bjoc/content/inline/1860-5397-11-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The preparation of 4,12-diamino[2.2]paracyclophane (8).
Scheme 1: The preparation of 4,12-diamino[2.2]paracyclophane (8).
The crucial intermediate in this route is the pseudo-ortho-diacid 14, which has been prepared several times before [24-27] by various routes.
We prefer to start with the parent hydrocarbon [2.2]paracyclophane (12), convert this to the dibromide 13, metalate it to the corresponding dilithio derivative, from which 14 is finally obtained by a CO2 quench [25,26]. On treatment of 14 with oxalyl chloride in anhydrous dichloromethane in the presence of catalytic amounts of DMF in dichloromethane (10% solution) at room temperature [27], the expected bis(acyl chloride) was obtained in quantitative yield and characterized by the usual spectroscopic methods (see Supporting Information File 1). Subjecting this intermediate to sodium azide treatment in acetone at room temperature provided the isolable azidocarbonyl derivative 15 in 90% yield; for its analytical data see Supporting Information File 1. Compound 15 can even be purified by silica gel column chromatography although it was noted that Curtius rearrangement to the bis(isocyanate) 16 slowly set in. Hence 15 was employed in the Curtius step (toluene, reflux) without further purification and at about 95 °C the evolution of nitrogen gas could be clearly seen. The bis(isocyanate) 16 was isolated in quantitative yield and characterized by its spectroscopic and analytical data (see Supporting Information File 1). Finally, when 16 was first refluxed in ethanol and the resulting solution (presumably containing the corresponding bis(urethane) is subsequently treated with an aqueous solution of potassium hydroxide (20%), 4,12-diamino[2.2]paracyclophane (8) was obtained in quantitative yield. Since we had converted its pseudo-geminal isomer 7 by sodium hypochlorite oxidation into the corresponding azo compound [20] we also attempted this transformation for 8.
The intended, chiral azo compound 17 was, however, not produced. Presumably the two amino functions are too far apart for intramolecular bridging and the strain of the desired 17 would be too high for a successful ring closure.
Reacting 16 with tetraethylene glycol (TEG) under high dilution conditions (toluene, reflux, 7 d) provides the crownophane 18 in 68% yield (Scheme 2). Likewise, replacement of TEG by pentaethylene glycol (PEG) resulted in the formation of the next higher homolog 19 (73%). Both compounds were readily characterized by their analytical data (see Supporting Information File 1) and also by an X-ray structural analysis of 18.
![[1860-5397-11-50-i2]](/bjoc/content/inline/1860-5397-11-50-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of cyclic and acyclic urethanes from 4,12-diisocyanato[2.2]paracyclophane (16).
Scheme 2: Preparation of cyclic and acyclic urethanes from 4,12-diisocyanato[2.2]paracyclophane (16).
Several features are common to all structures presented in this paper. The usual features of cyclophane strain include the following. The bridge C–C single bonds are lengthened to 1.58–1.60 Å, the bridge sp3-bond angles are widened to ca. 112°, and the bridgehead angles within the six-membered rings are narrowed to 116–118°. These rings display a flattened boat conformation (deviations of bridgehead atoms 0.14–0.17 Å) and are mutually parallel (interplanar angles ≤ 1°, calculated without the bridgehead carbons).
In the structure of 18 (Figure 3a), the large macrocyclic ring displays an unsymmetrical configuration (e.g., the four O–C–C–O torsion angles starting from O2 are ap, ≈ –90°, +sc, –sc; for detailed values, see the deposited material). This is at least in part connected with the transannular hydrogen bond N2–H02···O1, with H···O 2.07(2) Å and N–H···O 156(2)°. The other NH group is involved in a rather non-linear intermolecular hydrogen bond N1–H01···O4 [H···O 2.28(2) Å, N–H···O 133(2)°], which, together with the "weak" interaction C2–H2A···O3 [H···O 2.47 Å, C–H···O 173°], links the molecules via a 21 screw axis to form columns parallel to the a-axis (Figure 3b).
![[1860-5397-11-50-3]](/bjoc/content/figures/1860-5397-11-50-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a, above): The molecule of compound 18 in the crystal; ellipsoids represent 50% probability levels. The intramolecular hydrogen bond is indicated by a dashed line. (b, below): Packing diagram of 18 viewed parallel to the c axis in the region z ≈ 0. Intermolecular hydrogen bonds (one classical, one "weak") are shown as thick dashed lines. H atoms not involved in these interactions are omitted for clarity.
Figure 3: (a, above): The molecule of compound 18 in the crystal; ellipsoids represent 50% probability levels...
It is a well-known phenomenon, already observed by Cram and Reich in the early days of cyclophane chemistry, that on heating, equilibria are set up between various [2.2]cyclophane isomers [28]. For example, on heating pseudo-ortho-disubstituted derivatives, these are converted into their pseudo-para-diastereomers.
Carrying out the same experiment with 18, several days of heating in refluxing triglyme, i.e., at 216 °C, did not, however, furnish the corresponding pseudo-para-crownophane, but provided a dark brown intractable material.
Changing the trapping agent for 16 to butane-1,4-diol (toluene, reflux, 20 h) led to bis(urethane diol) 20 (43%). In the hope that this would react with a second equivalent of 16 and yield the cyclic 2:2-adduct 21, we added more of the bis(isocyanate). Under similar conditions to those above, only polymeric material was obtained, however. We hence assume that intermolecular oligo-/polymerization is favored over the intramolecular process.
In order to make the crownophane 18 resemble a “real” crown ether more closely [29], we next tried to reduce its carbonyl groups to methylene units by lithium aluminum hydride (Scheme 3).
![[1860-5397-11-50-i3]](/bjoc/content/inline/1860-5397-11-50-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: LiAlH4-reduction of crownophane 18.
Scheme 3: LiAlH4-reduction of crownophane 18.
Interestingly, the process (reduction in refluxing THF for 7 h) caused a complete destruction of the newly created heterocyclic ring and provided the aldehyde 22 together with the bis secondary amine 23, the latter in acceptable yields. Although it was not tested specifically, it is likely that 22 is the precursor for 23.
We assume that the reduction of 18 starts as usual by attack of hydride at the urethane carbonyl group. Because of the inherent strain in the tetrahedral intermediate thus generated, the polyether bridge functions as a leaving group to form an aldehyde intermediate. This is subsequently reduced a second time to the N-CH3 moiety. In compound 22 one formyl group is still present whereas in 23 this has been lost completely. Both compounds were characterized by their spectroscopic data (Supporting Information File 1), and for 22 we could additionally determine the solid-state structure by X-ray structural analysis (Figure 4a). The orientation of the side chains in 22 is described by the torsion angles C14–C15–N1–C17 131.2(2)°, C15–N1–C17–O −1.3(2)° (the standard trans-amide geometry) and C6–C5–N2–C18 176.7(1)°. Both NH groups are involved in classical intermolecular hydrogen bonds. The interaction N2–H02···O [H···O 2.23(2) Å, N–H···O 164(2)°] forms inversion-symmetric dimeric units (easily cognizable in the Figure), which are in turn linked through N1–H01···O [H···O 2.02(2) Å, N–H···O 168(2)°, 21 screw axis parallel to b] to form a layer structure parallel to the bc-plane (Figure 4b). A very short C–H···π contact involving the formyl hydrogen, C17–H17···Cent (C12,13,15,16)] [normalized H···π 2.50 Å, C–H···π 174°] through the same screw operator supports the hydrogen bonding system but for clarity is not drawn explicitly in Figure 4b.
![[1860-5397-11-50-4]](/bjoc/content/figures/1860-5397-11-50-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a, above): The molecule of compound 22 in the crystal; ellipsoids represent 30% probability levels. (b, below): Packing diagram of 22 viewed perpendicular to the bc-plane. Classical hydrogen bonds are shown as thick dashed lines. H atoms not involved in these interactions are omitted for clarity.
Figure 4: (a, above): The molecule of compound 22 in the crystal; ellipsoids represent 30% probability levels...
Preparation of 4,16-bis(isocyanato)[2.2]paracyclophane (28, pseudo-para-bis(isocyanate) 28)
With the successful route to 8 and 16 in hand, we next turned our attention to the preparation of several new pseudo-para-substituted [2.2]paracyclophane derivatives carrying N-functional groups. As shown in Scheme 4, the crucial substrate 25 was prepared from the pseudo-para-dibromide 24 [24,29]. Treating 25 with thionyl chloride in DMF yielded the bis(acyl chloride) 26 in quantitative yield. In contrast to most other acyl chlorides, 26 is a stable compound with a high melting point (210 °C), which could easily be recrystallized from dichloromethane/pentane to furnish single crystals suitable for X-structural analysis.
![[1860-5397-11-50-i4]](/bjoc/content/inline/1860-5397-11-50-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The preparation of several derivatives of 4,16-dicarboxy[2.2]paracyclophane (25) carrying N-containing functional groups.
Scheme 4: The preparation of several derivatives of 4,16-dicarboxy[2.2]paracyclophane (25) carrying N-contain...
The molecule of 26 (Figure 5) possesses crystallographic inversion symmetry, consistent with its pseudo-para form. We have reported the structure of the pseudo-gem isomer [20]. The cell constants b and c of 26, 11.5213 and 7.5417 Å, respectively, suggest that the packing should display the "7,11"-pattern previously noted by us as being common to a large number of simple cyclophane derivatives; it involves layers of hexagonally arranged molecules, generally with C–H···π contacts [20]. Compound 26 does indeed display the standard pattern, but the sole H···π contact (from H5) is rather long at 2.89 Å (normalized) and indeed there are few other notable intermolecular contacts. The same "7,11"-packing is displayed by compound 28, for which an explicit packing diagram is presented (see below).
![[1860-5397-11-50-5]](/bjoc/content/figures/1860-5397-11-50-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The molecule of compound 26 in the crystal; ellipsoids represent 50% probability levels. Only the asymmetric unit is numbered.
Figure 5: The molecule of compound 26 in the crystal; ellipsoids represent 50% probability levels. Only the a...
Subjection of 26 to nucleophilic substitution with aqueous sodium azide yielded the bis(keto azide) 27 in 76% yield as a colorless amorphous solid. The compound is readily characterized by its typical azide and carbonyl absorption bands in the vibrational spectral (see Supporting Information File 1 for complete spectroscopic data). The Curtius degradation of 27 provided the desired bis(isocyanate) 28 readily, though in a disappointing isolated yield of 30% only. Since the yield of the crude product was three times as much, we assume that much of the material is lost during chromatography on silica gel. The compound was characterized by its spectroscopic and analytical data and also by the determination of its solid state structure by X-ray diffraction [30].
The molecule of 28 (Figure 6a) displays no formal crystallographic inversion symmetry, but is approximately inversion-symmetric (rmsd 0.02 Å). Again, we have reported the structure of the corresponding pseudo-gem isomer [20]. A search of the Cambridge database [31] revealed, perhaps surprisingly, that there are only two other X-ray structure determinations of organic isocyanates with the NCO group bonded to a phenyl ring. The dimensions of the isocyanate group observed for 28 [Cring–N 1.408(2), 1.419(2), N–C 1.194(3), 1.192(3), C–O 1.173(2), 1.174(2) Å; Cring–N–O 140.0(2), 136.9(2), N–C–O 172.6(2), 172.5(2)°] are closely similar to the values observed previously. As for 26, the cell constants a 7.2803 and b 11.4247 Å suggest that the packing should display the "7,11"-pattern, and this is indeed the case (Figure 6b); the relevant contacts are H16···Cent(C4,5,7,8) 2.75 and H8···Cent(C12,13,15,16) 2.81 Å. Some borderline C–H···O contacts are also observed (see deposited material).
![[1860-5397-11-50-6]](/bjoc/content/figures/1860-5397-11-50-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a, above): The molecule of compound 28 in the crystal; ellipsoids represent 50% probability levels. (b, below): Packing diagram of 28 viewed parallel to the c-axis in the region z ≈ ¼, showing C–H···π contacts as dashed lines.
Figure 6: (a, above): The molecule of compound 28 in the crystal; ellipsoids represent 50% probability levels...
Of course, compound 28 offers many possibilities for further elaboration. In a first attempt we reacted it with decane-1,10-diol under high dilution conditions in the hope of introducing a bridge between a “lower” and an “upper” deck of a cyclophane, a bridging situation unknown so far. Rather than the intended bis(urethane) 29, we obtained only intractable material, presumably a mixture of polyurethanes resulting from the α,ω-diol and the bis(isocyanate). It seems reasonable to assume that the future tether reacts once with 28 but then has no time to reorient itself to place the remaining OH function close to the remaining NCO moiety. The subsequent intermolecular pathway is much more likely than the intramolecular cyclization to 29.
Conclusion
Having developed different, mostly high-yielding routes to various diamino[2.2]paracyclophanes and related compounds [30], we are now in a position to prepare the tetramines 10 and 11 as the next steps to novel polyfunctionalized cyclophanes.
Supporting Information
| Supporting Information File 1: Experimental and characterization data. | ||
| Format: PDF | Size: 401.1 KB | Download |
References
-
Narayanan, S. V. Syntheses of Functionalized [2.2]Paracyclophanes – Structure and Reactivity Studies. Ph.D. Thesis, TU Braunschweig, Germany, 2005.
The present publication is largely based on this Ph.D. Thesis.
-
Hopf, H.; Narayanan, S. V.; Jones, P. G. Beilstein J. Org. Chem. 2014, 10, 2021–2026. doi:10.3762/bjoc.10.210
-
Aly, A. A.; Brown, A. B. Tetrahedron 2009, 65, 8055–8089. doi:10.1016/j.tet.2009.06.034
Return to citation in text: [1] -
David, O. R. P. Tetrahedron 2012, 68, 8977–8993. doi:10.1016/j.tet.2012.08.009
Return to citation in text: [1] -
Brown, C. J.; Farthing, A. C. Nature 1949, 164, 915–916. doi:10.1038/164915b0
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Xie, P.; Zhang, L.; Xu, W.; Chang, Y.; Abboud, K. A. J. Org. Chem. 2008, 73, 2469–2472. doi:10.1021/jo7026849
Return to citation in text: [1] -
Chow, S. W.; Pilato, L. A.; Wheelwright, W. L. J. Org. Chem. 1970, 35, 20–22. doi:10.1021/jo00826a005
Return to citation in text: [1] -
Grechkina, E. V.; Sochilin, V. A.; Pebalk, A. V.; Kardash, I. E. Russ. J. Org. Chem. 1993, 29, 1663–1665.
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Asghar, M. A.; Pan, H.-Q.; Celewicz, L. J. Org. Chem. 1993, 58, 1827–1830. doi:10.1021/jo00059a038
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Rong, X. X.; Yuelian, X.; Beach, W. F. J. Org. Chem. 1997, 62, 7500–7502. doi:10.1021/jo970927q
Return to citation in text: [1] -
Filler, R.; Miller, F. N. Chem. Ind. 1965, 767–768.
Return to citation in text: [1] -
Filler, R.; Cantrell, G. L.; Wolanin, D.; Naqvi, S. M. J. Fluorine Chem. 1986, 30, 399–414. doi:10.1016/S0022-1139(00)85095-2
Return to citation in text: [1] -
Nagel, M.; Allmann, R.; Eltamany, S.; Hopf, H. Chem. Ber. 1982, 115, 3203–3207. doi:10.1002/cber.19821150923
Return to citation in text: [1] -
Longone, D. T.; Simanyi, L. H. J. Org. Chem. 1964, 29, 3245–3249. doi:10.1021/jo01034a028
Return to citation in text: [1] -
Bondarenko, L.; Dix, I.; Hinrichs, H.; Hopf, H. Synthesis 2004, 2751–2759. doi:10.1055/s-2004-834872
Return to citation in text: [1] -
Morisaki, Y.; Chujo, Y. Angew. Chem., Int. Ed. 2006, 45, 6430–6437. doi:10.1002/anie.200600752
Return to citation in text: [1] -
Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969
Return to citation in text: [1] -
Broggi, A.; Tomasi, I.; Bianchi, L.; Marrocchi, A.; Vaccaro, L. ChemPlusChem 2014, 79, 486–507. doi:10.1002/cplu.201400001
Return to citation in text: [1] -
Cram, D. J.; Allinger, N. L. J. Am. Chem. Soc. 1955, 77, 6289–6294. doi:10.1021/ja01628a067
Return to citation in text: [1] -
El Shaieb, K.; Narayanan, V.; Hopf, H.; Dix, I.; Fischer, A.; Jones, P. G.; Ernst, L.; Ibrom, K. Eur. J. Org. Chem. 2003, 567–577. doi:10.1002/ejoc.200390095
Return to citation in text: [1] [2] [3] [4] [5] -
Zitt, H.; Dix, I.; Hopf, H.; Jones, P. G. Eur. J. Org. Chem. 2002, 2298–2307. doi:10.1002/1099-0690(200207)2002:14<2298::AID-EJOC2298>3.0.CO;2-E
Return to citation in text: [1] -
Hopf, H.; Lenich, F. T. Chem. Ber. 1974, 107, 1891–1902. doi:10.1002/cber.19741070613
Return to citation in text: [1] -
Hopf, H.; Popova, E. L.; unpublished results from this laboratory. The “crossed” tetraacid was prepared from the corresponding tetrabromide by lithiation followed by a carbon dioxide quench.
Return to citation in text: [1] -
Allgeier, H.; Siegel, M. G.; Helgeson, R. C.; Schmidt, E.; Cram, D. J. J. Am. Chem. Soc. 1975, 97, 3782–3789. doi:10.1021/ja00846a038
Return to citation in text: [1] [2] -
Rozenberg, V. I.; Popova, E. L.; Hopf, H. Helv. Chim. Acta 2002, 85, 431–441. doi:10.1002/1522-2675(200202)85:2<431::AID-HLCA431>3.0.CO;2-H
Return to citation in text: [1] [2] -
Antonov, D. Yu.; Sergeeva, E. V.; Vorontsov, E. V.; Rozenberg, V. I. Russ. Chem. Bull. 1977, 46, 1897–1900. doi:10.1007/BF02503781
Return to citation in text: [1] [2] -
Meissner, R.; Garcias, X.; Mecozzi, S.; Rebek, J., Jr. J. Am. Chem. Soc. 1997, 119, 77–85. doi:10.1021/ja962991f
Return to citation in text: [1] [2] -
Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3517–3526. doi:10.1021/ja01041a016
Return to citation in text: [1] -
Helgeson, R. C.; Tarnowski, T. L.; Timko, J. M.; Cram, D. J. J. Am. Chem. Soc. 1977, 99, 6411–6418. doi:10.1021/ja00461a038
The authors describe crownophanes derived from di-and tetrahydroxy[2.2]paracyclophanes.
Return to citation in text: [1] [2] -
Meyer-Eppler, G.; Sure, R.; Schneider, A.; Schnakenburg, G.; Grimme, S.; Lützen, A. J. Org. Chem. 2014, 79, 6679–6687. doi:10.1021/jo501212t
The authors have recently described effectives routes to various pseudo-meta-substituted [2.2]paracyclophanes, among them the pseudo-meta-isomer of 8.
Return to citation in text: [1] [2] -
Groom, C. R.; Allen, F. H. Angew. Chem., Int. Ed. 2014, 53, 662–671. doi:10.1002/anie.201306438
Return to citation in text: [1]
| 3. | Aly, A. A.; Brown, A. B. Tetrahedron 2009, 65, 8055–8089. doi:10.1016/j.tet.2009.06.034 |
| 4. | David, O. R. P. Tetrahedron 2012, 68, 8977–8993. doi:10.1016/j.tet.2012.08.009 |
| 11. | Filler, R.; Miller, F. N. Chem. Ind. 1965, 767–768. |
| 12. | Filler, R.; Cantrell, G. L.; Wolanin, D.; Naqvi, S. M. J. Fluorine Chem. 1986, 30, 399–414. doi:10.1016/S0022-1139(00)85095-2 |
| 27. | Meissner, R.; Garcias, X.; Mecozzi, S.; Rebek, J., Jr. J. Am. Chem. Soc. 1997, 119, 77–85. doi:10.1021/ja962991f |
| 7. | Chow, S. W.; Pilato, L. A.; Wheelwright, W. L. J. Org. Chem. 1970, 35, 20–22. doi:10.1021/jo00826a005 |
| 8. | Grechkina, E. V.; Sochilin, V. A.; Pebalk, A. V.; Kardash, I. E. Russ. J. Org. Chem. 1993, 29, 1663–1665. |
| 9. | Dolbier, W. R., Jr.; Asghar, M. A.; Pan, H.-Q.; Celewicz, L. J. Org. Chem. 1993, 58, 1827–1830. doi:10.1021/jo00059a038 |
| 10. | Dolbier, W. R., Jr.; Rong, X. X.; Yuelian, X.; Beach, W. F. J. Org. Chem. 1997, 62, 7500–7502. doi:10.1021/jo970927q |
| 20. | El Shaieb, K.; Narayanan, V.; Hopf, H.; Dix, I.; Fischer, A.; Jones, P. G.; Ernst, L.; Ibrom, K. Eur. J. Org. Chem. 2003, 567–577. doi:10.1002/ejoc.200390095 |
| 6. | Dolbier, W. R., Jr.; Xie, P.; Zhang, L.; Xu, W.; Chang, Y.; Abboud, K. A. J. Org. Chem. 2008, 73, 2469–2472. doi:10.1021/jo7026849 |
| 24. | Allgeier, H.; Siegel, M. G.; Helgeson, R. C.; Schmidt, E.; Cram, D. J. J. Am. Chem. Soc. 1975, 97, 3782–3789. doi:10.1021/ja00846a038 |
| 25. | Rozenberg, V. I.; Popova, E. L.; Hopf, H. Helv. Chim. Acta 2002, 85, 431–441. doi:10.1002/1522-2675(200202)85:2<431::AID-HLCA431>3.0.CO;2-H |
| 26. | Antonov, D. Yu.; Sergeeva, E. V.; Vorontsov, E. V.; Rozenberg, V. I. Russ. Chem. Bull. 1977, 46, 1897–1900. doi:10.1007/BF02503781 |
| 27. | Meissner, R.; Garcias, X.; Mecozzi, S.; Rebek, J., Jr. J. Am. Chem. Soc. 1997, 119, 77–85. doi:10.1021/ja962991f |
| 25. | Rozenberg, V. I.; Popova, E. L.; Hopf, H. Helv. Chim. Acta 2002, 85, 431–441. doi:10.1002/1522-2675(200202)85:2<431::AID-HLCA431>3.0.CO;2-H |
| 26. | Antonov, D. Yu.; Sergeeva, E. V.; Vorontsov, E. V.; Rozenberg, V. I. Russ. Chem. Bull. 1977, 46, 1897–1900. doi:10.1007/BF02503781 |
| 19. | Cram, D. J.; Allinger, N. L. J. Am. Chem. Soc. 1955, 77, 6289–6294. doi:10.1021/ja01628a067 |
| 21. | Zitt, H.; Dix, I.; Hopf, H.; Jones, P. G. Eur. J. Org. Chem. 2002, 2298–2307. doi:10.1002/1099-0690(200207)2002:14<2298::AID-EJOC2298>3.0.CO;2-E |
| 16. | Morisaki, Y.; Chujo, Y. Angew. Chem., Int. Ed. 2006, 45, 6430–6437. doi:10.1002/anie.200600752 |
| 17. | Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969 |
| 18. | Broggi, A.; Tomasi, I.; Bianchi, L.; Marrocchi, A.; Vaccaro, L. ChemPlusChem 2014, 79, 486–507. doi:10.1002/cplu.201400001 |
| 22. | Hopf, H.; Lenich, F. T. Chem. Ber. 1974, 107, 1891–1902. doi:10.1002/cber.19741070613 |
| 23. | Hopf, H.; Popova, E. L.; unpublished results from this laboratory. The “crossed” tetraacid was prepared from the corresponding tetrabromide by lithiation followed by a carbon dioxide quench. |
| 15. | Bondarenko, L.; Dix, I.; Hinrichs, H.; Hopf, H. Synthesis 2004, 2751–2759. doi:10.1055/s-2004-834872 |
| 13. | Nagel, M.; Allmann, R.; Eltamany, S.; Hopf, H. Chem. Ber. 1982, 115, 3203–3207. doi:10.1002/cber.19821150923 |
| 14. | Longone, D. T.; Simanyi, L. H. J. Org. Chem. 1964, 29, 3245–3249. doi:10.1021/jo01034a028 |
| 20. | El Shaieb, K.; Narayanan, V.; Hopf, H.; Dix, I.; Fischer, A.; Jones, P. G.; Ernst, L.; Ibrom, K. Eur. J. Org. Chem. 2003, 567–577. doi:10.1002/ejoc.200390095 |
| 24. | Allgeier, H.; Siegel, M. G.; Helgeson, R. C.; Schmidt, E.; Cram, D. J. J. Am. Chem. Soc. 1975, 97, 3782–3789. doi:10.1021/ja00846a038 |
| 29. |
Helgeson, R. C.; Tarnowski, T. L.; Timko, J. M.; Cram, D. J. J. Am. Chem. Soc. 1977, 99, 6411–6418. doi:10.1021/ja00461a038
The authors describe crownophanes derived from di-and tetrahydroxy[2.2]paracyclophanes. |
| 28. | Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3517–3526. doi:10.1021/ja01041a016 |
| 29. |
Helgeson, R. C.; Tarnowski, T. L.; Timko, J. M.; Cram, D. J. J. Am. Chem. Soc. 1977, 99, 6411–6418. doi:10.1021/ja00461a038
The authors describe crownophanes derived from di-and tetrahydroxy[2.2]paracyclophanes. |
| 31. | Groom, C. R.; Allen, F. H. Angew. Chem., Int. Ed. 2014, 53, 662–671. doi:10.1002/anie.201306438 |
| 30. |
Meyer-Eppler, G.; Sure, R.; Schneider, A.; Schnakenburg, G.; Grimme, S.; Lützen, A. J. Org. Chem. 2014, 79, 6679–6687. doi:10.1021/jo501212t
The authors have recently described effectives routes to various pseudo-meta-substituted [2.2]paracyclophanes, among them the pseudo-meta-isomer of 8. |
| 30. |
Meyer-Eppler, G.; Sure, R.; Schneider, A.; Schnakenburg, G.; Grimme, S.; Lützen, A. J. Org. Chem. 2014, 79, 6679–6687. doi:10.1021/jo501212t
The authors have recently described effectives routes to various pseudo-meta-substituted [2.2]paracyclophanes, among them the pseudo-meta-isomer of 8. |
| 20. | El Shaieb, K.; Narayanan, V.; Hopf, H.; Dix, I.; Fischer, A.; Jones, P. G.; Ernst, L.; Ibrom, K. Eur. J. Org. Chem. 2003, 567–577. doi:10.1002/ejoc.200390095 |
| 20. | El Shaieb, K.; Narayanan, V.; Hopf, H.; Dix, I.; Fischer, A.; Jones, P. G.; Ernst, L.; Ibrom, K. Eur. J. Org. Chem. 2003, 567–577. doi:10.1002/ejoc.200390095 |
| 20. | El Shaieb, K.; Narayanan, V.; Hopf, H.; Dix, I.; Fischer, A.; Jones, P. G.; Ernst, L.; Ibrom, K. Eur. J. Org. Chem. 2003, 567–577. doi:10.1002/ejoc.200390095 |
© 2015 Hopf et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)