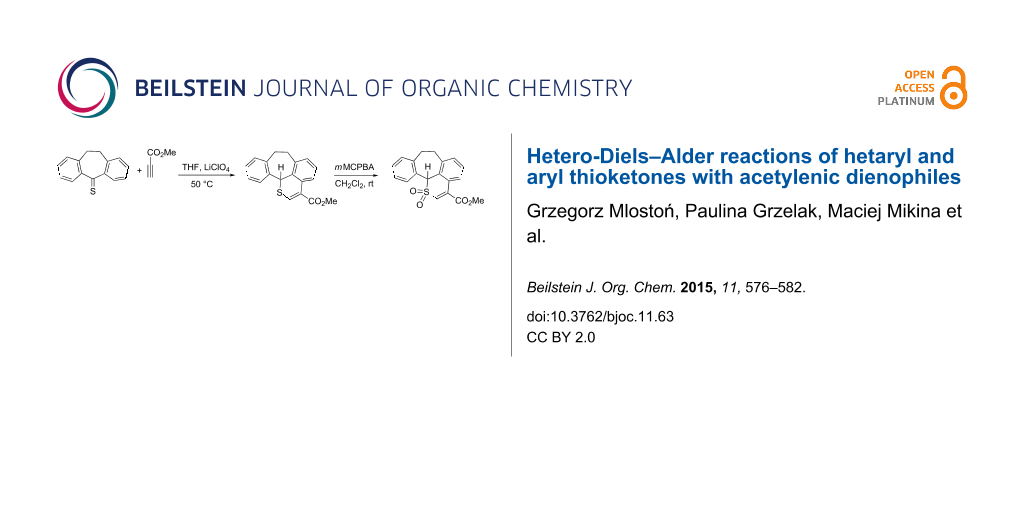
Abstract
Selected hetaryl and aryl thioketones react with acetylenecarboxylates under thermal conditions in the presence of LiClO4 or, alternatively, under high-pressure conditions (5 kbar) at room temperature yielding thiopyran derivatives. The hetero-Diels–Alder reaction occurs in a chemo- and regioselective manner. The initially formed [4 + 2] cycloadducts rearrange via a 1,3-hydrogen shift sequence to give the final products. The latter were smoothly oxidized by treatment with mCPBA to the corresponding sulfones.

Graphical Abstract
Introduction
A series of recent publications evidence that, in contrast to earlier opinions, thioketones are useful building blocks for the preparation of diverse sulfur heterocycles [1-3]. Studies performed by Huisgen and coworkers are of special importance and they resulted in the formulation of the name ‘superdipolarophiles’ for aromatic thioketones [4-6]. In addition, Sauer and coworkers called them ‘superdienophiles’ based on kinetic studies [7,8]. Moreover, thiobenzophenone (1a) was reported to react as a heterodiene smoothly with cyclooctyne, dicyanoacetylene, and dimethyl acetylenedicarboxylate (2a) to give [4 + 2] cycloadducts of type 3a, which spontaneously rearrange via a 1,3-hydrogen shift yielding rearomatized products of type 4a (Scheme 1) [9-11].
![[1860-5397-11-63-i1]](/bjoc/content/inline/1860-5397-11-63-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Hetero-Diels–Alder reaction of thiobenzophenone (1a) with dimethyl acetylenedicarboxylate (2a) [10].
Scheme 1: Hetero-Diels–Alder reaction of thiobenzophenone (1a) with dimethyl acetylenedicarboxylate (2a) [10].
The same transformation occurred faster under photolytic conditions [9,12]. The hetero-Diels–Alder reaction of 4-substituted analogues of 1a with in situ generated benzyne is also known [13]. Heteroaromatic thioketones are reported to undergo a hetero-Diels–Alder reaction with dienophiles such as maleic anhydride, acrylonitrile, styrene, and α-chloroacrylonitrile [14,15]. In the latter case, the stabilization of the initially formed cycloadduct occurred via HCl elimination, whereas, in the other cases, the 1,3-hydrogen shift led to rearomatized products.
In our ongoing studies on thioketones, we reported recently on selected reactions of new symmetrical and non-symmetrical hetaryl thioketones [16]. Among others, the reactions of 2a with phenyl thien-2-yl thioketone as well as with bis(thien-2-yl) thioketone were described. The goal of the present study is to examine the reactions of diverse hetaryl thioketones with both 2a and methyl propiolate (2b). Moreover, along with standard procedures, the high-pressure technique was applied. Finally, selected examples of the obtained polycyclic 2H-thiopyrans were oxidized to give the corresponding sulfones.
Results and Discussion
Under standard conditions (benzene, rt), the reaction of 1a with 2a is slow, and it requires several days to be complete [9]. For that reason, we modified the procedure by using THF as a solvent and LiClO4 as a known, efficient catalyst applied frequently in diverse Diels–Alder reactions [17]. Heating the mixture in a closed tube to 50 °C resulted in completion of the reaction after only 24 h, and after chromatographic work-up, the known product 4a was obtained in 84% yield. Another attempt to optimize the reaction conditions was based on the use of the high-pressure method. This approach offers some advantages, especially in the case of cycloaddition reactions [18]. To the best of our knowledge, reactions of thioketones under high-pressure conditions have never been studied. After a series of optimization experiments, a solution of 1a and 2a in a molar ratio of 1:2 in toluene was placed in a high-pressure vessel at 5 kbar, and after 24 h at room temperature, the product 4a was isolated in 70% yield.
The reaction conditions with THF and LiClO4 were used for further reactions of aryl thioketones, i.e., thiodibenzosuberone 1b and thiodibenzosuberenone 1c, with 2a. The expected polycyclic thiopyran derivatives 4b and 4c, respectively, were obtained in 90 and 46% yield (Scheme 2). In the 1H NMR spectra of both compounds the low-field shifted CHS signal appeared at 5.82 and 4.49 ppm. Finally, the 2H-thiopyran structure of 4b was established by X-ray single crystal structure determination (Figure 1).
![[1860-5397-11-63-i2]](/bjoc/content/inline/1860-5397-11-63-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of polycyclic thiopyrans via the hetero-Diels–Alder reaction/1,3-hydrogen shift sequence.
Scheme 2: Synthesis of polycyclic thiopyrans via the hetero-Diels–Alder reaction/1,3-hydrogen shift sequence.
![[1860-5397-11-63-1]](/bjoc/content/figures/1860-5397-11-63-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP Plot [19] of the molecular structure of 4b, drawn using 50% probability displacement ellipsoids.
Figure 1: ORTEP Plot [19] of the molecular structure of 4b, drawn using 50% probability displacement ellipsoids.
The analogous reactions of 2a with bis(N-methylpyrrol-2-yl) thioketone (1d) afforded the pyrrolo[2,3-c]thiopyran derivative 4d in modest yield (35%) (Scheme 2). In the case of bis(thien-2-yl) thioketone (1e), the reaction with 2a was carried out in toluene at room temperature under a pressure of 5 kbar. After 72 h, the corresponding thieno[2,3-c]thiopyran 4e was obtained in 35% yield (Scheme 2).
In an extension of the study, reactions of methyl propiolate (2b) with selected aryl and hetaryl thioketones were performed under thermal and high-pressure conditions. The reaction of 1a with 2b in THF and a catalytic amount of LiClO4 led to the benzothiopyran 5a in 61% yield (Scheme 3, Table 1). The 1H NMR analysis of the crude mixture indicated the presence of a single regioisomer for which the structure 5a is proposed. In the alternative preparation of this product under high-pressure (toluene, 5 kbar), 66% of 5a were isolated. Both methods were applied for analogous reactions with the symmetrical bis-hetaryl thioketones 1e–g. Under thermal conditions, the products 5d–f were also formed regioselectively in 83, 5, and 6% yield, respectively. In these cases, the high-pressure experiments were also performed with 1e (16% yield of 5d) and 1f (33% yield of 5e), respectively; thioketone 1g was not tested under high pressure. However, attempted isolations of both 5e and 5f were unsuccessful as the products underwent decomposition upon chromatographic work-up. The experiment performed with the non-symmetrical phenyl thien-2-yl thioketone (1h) under thermal conditions resulted also in the formation of a single product 5g in a chemo- and regioselective manner in 74% yield.
![[1860-5397-11-63-i3]](/bjoc/content/inline/1860-5397-11-63-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of aryl/hetaryl thioketones with methyl propiolate (Table 1).
Scheme 3: Reactions of aryl/hetaryl thioketones with methyl propiolate (Table 1).
Table 1: Fused 2H-thiopyrans 5.
| 1 | Ph/Hetar | Hetar/Ph | 5 |
Method Aa
yield [%] |
Method Ba
yield [%] |
|---|---|---|---|---|---|
| a | Ph | Ph | a | 61 | 66 |
| b | dibenzosuberone | b | 94 | – | |
| c | dibenzosuberenone | c | 61 | – | |
| e | thien-2-yl | thien-2-yl | d | 83 | 16 |
| f | selenophen-2-yl | selenophen-2-yl | e | 5b | 33c |
| g | furan-2-yl | furan-2-yl | f | 6b | – |
| h | Ph | thien-2-yl | g | 74 | 17 |
aMethods A and B see Experimental; bnot isolated; cdecomposes during attempted chromatographic separation.
The experiment under high-pressure conditions was much less successful and gave 5g in only 17% yield. The structures of the products were determined based on the spectroscopic data. Thus, similar to hetero-Diels–Alder reactions with maleic anhydride [14], the C=C bond of the thiophene ring of 1h is part of the reactive heterodiene system.
The reactions of 2b with thiobenzosuberone 1b and thiobenzosuberenone 1c were performed under thermal conditions, and in both cases, new polycyclic thiopyran derivatives 5b and 5c, respectively, were formed regioselectively and obtained in good yields (94 and 61%, respectively).
In order to test the scope and limitations of the hetero-Diels–Alder reaction with thioketones and acetylenic dienophiles, other easily available acetylenes were used in the reaction with 1a. In all reactions attempted with phenylacetylene, (pyridin-2-yl)acetylene, and (tert-butyl)acetylene, no conversion of 1a was observed as evidenced by the blue color of the reaction mixture even after 24 h. Unfortunately, the experiments performed under the described conditions (THF, LiClO4, temp.) with (trifluoromethyl)acetylene and (diethoxyphosphoryl)acetylene, activated by the presence of strongly electron-withdrawing substituents, were also unsuccessful.
Some of the thiopyran derivatives 4 and 5 obtained in the present study were used for oxidation reactions aimed at the preparation of the corresponding sulfoxides and sulfones. As demonstrated in a recent publication [20], polycyclic sulfones are attractive substrates for the synthesis of polycyclic hydrocarbons via thermal SO2 extrusion. In our experiments, thiopyrans 4a,b and 5a,b were oxidized in CH2Cl2 solution at room temperature using 3.0 equivalents of mCPBA. In the case of 4a, the progress of the reaction was monitored by TLC and 1H NMR spectroscopy. The spectrum recorded after 3 h showed that along with starting materials, two new products in a ratio of ca. 4:6 are present in the mixture. For that reason, the reaction time was extended to 3 days. Then, only one of these products was present, with characteristic signals of 2 MeO groups at 3.93 and 4.05 ppm. The signal of the CHS group was shifted downfield and appeared at 5.48 ppm. The ESI mass spectra as well as the elemental analysis confirmed the molecular formula C19H16SO6, which corresponds with the structure of the sulfone 6a obtained in 90% yield (Scheme 4). Based on this result, the intermediate product observed in the mixture after 3 h can be proposed as the corresponding sulfoxide. No attempts were made to isolate this product.
![[1860-5397-11-63-i4]](/bjoc/content/inline/1860-5397-11-63-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Oxidation of selected thiopyrans 4 and 5 to give the corresponding sulfones.
Scheme 4: Oxidation of selected thiopyrans 4 and 5 to give the corresponding sulfones.
The same oxidation protocol was applied to convert thiopyranes 5a, 4b, and 5b into the corresponding sulfones 6b–6d in 94, 70, and 80% yield, respectively. The structure of 6d was established by X-ray single crystal structure determination (Figure 2). The course of the oxidation reactions for these thiopyrans differs from a similar process reported for Se-containing systems. In these cases, ring contraction and elimination of an aryl group, but no formation of an oxidized product, was observed [21]. The same report describes the appearance of rearranged products (Pummerer-type rearrangement) upon treatment of 1H-2-benzothiopyran-3,4-dicarboxylates of type 4 with an equimolar amount of mCPBA.
![[1860-5397-11-63-2]](/bjoc/content/figures/1860-5397-11-63-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP Plot [19] of the molecular structure of one of the symmetry-independent molecules of 6d, drawn using 50% probability displacement ellipsoids.
Figure 2: ORTEP Plot [19] of the molecular structure of one of the symmetry-independent molecules of 6d, drawn us...
Conclusion
The present study shows that hetaryl thioketones react with activated acetylenecarboxylates in a hetero-Diels–Alder reaction followed by the 1,3-hydrogen shift to give fused thiopyran derivatives in a chemo- and regioselective manner. The thermal reaction can be catalyzed by LiClO4. Comparable reaction times were observed when the reaction was performed under high-pressure conditions at room temperature. The thiopyran derivatives can be oxidized by treatment with mCPBA at room temperature to give the corresponding sulfones. In this reaction, even when using equimolar amounts of mCPBA, selective formation of the sulfoxide could not be achieved. The obtained sulfones are promising substrates for thermal SO2 extrusion reactions aimed at the contraction of the ring. The importance of the presented study is amplified by the fact that benzothiopyran and related systems containing a thiopyran moiety, and especially their carboxylic derivatives, are known as important pharmacophores, with key importance for the biological activity of diverse sulfur polyheterocycles [22-24].
It is worth of mentioning, that the described hetero-Diels–Alder reactions with aromatic thioketones as heterodienes, display certain similarities to the reported organometallic pathways observed in their reactions with triiron dodecacarbonyl Fe3(CO)12 [25,26].
Experimental
General information: Melting points were determined in capillaries using a MEL-TEMP II apparatus (Aldrich) and are uncorrected. IR spectra were recorded with a FTIR NEXUS spectrophotometer as KBr pellets; absorptions (ν) in cm−1. 1H and 13C NMR spectra were measured on a Bruker Avance III (1H at 600 and 13C at 150 MHz) instrument in CDCl3; chemical shifts (δ) are given in ppm, coupling constants (J) in Hz. The multiplicity of the 13C signals was deduced from DEPT, supported by 1H,13C HMQC spectra. 1H NMR data are presented as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant, integration. The mass spectra were recorded on a Finnigan MAT-95 (ESI), Bruker maxis (HRESI), or SYNAPT G2-S HMDS (HR MALDI–TOF) instrument. Elemental analyses were performed in the Microanalytical Laboraory of the Chemistry Faculty in Łódź. Reactions under high pressure were performed in a High Pressure Autoclave LC10T in the Laboratory of the Polish Academy of Sciences (CBMiM) in Łódź. Applied reagents such as DMAD, methyl propiolate, inorganic reagents, and solvents are commercially available (Aldrich) and were used as received.
Reaction of thioketones 1a–h with acetylenic dienophiles 2a or 2b – General procedures: Method A: A solution of 1 mmol of the corresponding thioketone, 2 mmol of the corresponding acetylenic dienophiles and 10 mol % of LiClO4 in 1 mL of dry THF was placed in a thick-wall glass tube, which then was closed with a screw cap. The mixture was heated at 50 °C for 24 h. The solvent was evaporated in vacuo. Subsequent separation on preparative plates coated with silica gel (eluent: CH2Cl2) gave pure products. Method B: The corresponding thioketone (1 mmol) and acetylenic dienophile (2 mmol) were placed in a 4 mL teflon vial that was then filled with dry toluene. The vial was closed and kept at 5 kbar at room temperature for 24 h. After depressurizing, the solvent was removed in vacuo. Subsequent separation on preparative plates coated with silica gel (eluent: CH2Cl2) gave pure products.
Dimethyl 11,12-dihydro-4bH-benzo[4,5]cyclohepta[1,2,3-ij]isothiochromene-6,7-dicarboxylate (4b): Yield: 328.5 mg (90%). Colorless crystals; mp 147.5–148.0 °C (MeOH); IR (KBr) ν: 3061 (w), 2948 (w), 1732 (s), 1725 (s), 1597 (w), 1562 (w), 1435 (m), 1270 (s), 1232 (s), 1030 (w), 751(m) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.61 (d, J = 12 Hz, 1Harom), 7.33–7.30 (m, 1Harom), 7.24–7.15 (m, 4Harom), 7.03 (d, J = 6 Hz, 1Harom), 5.82 (br s, S-CH), 3.92, 3.94 (2s, 6H, 2 OCH3), 3.55 (br s, 1CH), 3.37 (br s, 1CH), 3.12 (br s, 1CH), 3.01–2.97 (m, 1CH) ppm; 13C NMR (150 MHz, CDCl3) δ 168.1, 164.3 (2 C=O), 138.1, 138.0, 131.8, 130.6 (7 C(sp2)), 136.3, 128.0, 127.5, 126.4, 125.3 (7 CHarom), 53.1, 52.8 (2 OCH3), 33.0 (S-CH), 32.0 (broad, 2 CH2) ppm; HRMS (ESI): [M + Na]+ calcd for C21H18NaO4S, 389.08180; found, 389.08178; anal. calcd for C21H18O4S: C, 68.82; H, 4.95; S, 8.75; found: C, 68.71; H, 5.06; S, 8.82.
Dimethyl 4bH-benzo[4,5]cyclohepta[1,2,3-ij]isothiochromene-6,7-dicarboxylate (4c): Yield: 83 mg (46%). Orange solid; mp 139.7–140.0 °C (purified chromatographically); IR (KBr) ν: 3018 (w), 2947 (w), 1728 (s), 1725 (s), 1596 (w), 1563 (w), 1433 (w), 1264 (s),1229 (s), 1097 (w), 771 (w) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.77 (d, J = 6 Hz, 1Harom), 7.49–7.45 (m, 2Harom), 7.42 (d, J = 6 Hz, 1Harom), 7.36 (d, J = 6 Hz, 1Harom), 7.30, 7.27 (2d, J = 6 Hz, 2 olefinic HC=), 7.23 (d, J = 6 Hz, 2Harom), 4.49 (s, S-CH), 3.92, 4.00 (2 s, 6H, 2 OCH3) ppm; 13C NMR (150 MHz, CDCl3) δ 167.8, 164.2 (2 C=O), 139.9, 135.8, 134.6, 132.5, 130.3, 128.8, 124.2 (7 C(sp2)), 132.2, 130.1, 129.3, 127.8, 127.6, 126.7, 126.7, 126.6, 125.2 (7 CHarom + 2 CHolefin), 53.2, 52.8 (2 OCH3), 41.8 (S-CH) ppm; HRMS (MALDI–TOF): [M + Na]+ calcd for C21H16NaO4S, 387.0668; found, 387.0667.
Methyl 11,12-dihydro-4bH-benzo[4,5]cyclohepta[1,2,3-ij]isothiochromene-7-carboxylate (5b): Yield: 288.5 mg (94%). Yellow solid; mp 116.0–116.5 °C (chromatographic purification); IR (KBr) ν: 3059 (w), 2946 (w), 1702 (s), 1591 (w), 1431 (w), 1221 (s), 1068 (m), 756 (m) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.93–7.87 (m, 1Harom), 7.84–7.80 (m, 1Harom), 7.61 (br s, S-CH=), 7.28–7.23 (m, 2Harom), 7.21–7.17 (m, 2Harom), 7.03 (br s, 1Harom), 5.79 (br s, S-CH), 3.87 (s, 3H, OCH3), 3.55 (br s, 1CH), 3.35 (br s, 1CH), 3.15 (br s, 1CH), 3.03–2.95 (m, 1CH) ppm; 13C NMR (150 MHz, CDCl3) δ 165.3 (C=O), 142.0, 138.7, 137.6, 135.2, 125.7, 125.4 (6 C(sp2)), 132.4, 130.6, 129.3, 128.0, 127.8, 127.1, 126.6, 126.1 (7 CHarom + S-CH=), 51.9 (OCH3), 35.0 (S-CH), 32.2 (broad, 2 CH2) ppm; HRMS (MALDI–TOF): [M + Na]+ calcd for C19H16NaO2S, 331.0761; found, 331.0769.
Methyl 4bH-benzo[4,5]cyclohepta[1,2,3-ij]isothiochromene-7-carboxylate (5c): Yield: 85 mg (61%). Yellow solid; mp 77.5–78.0 °C (chromatographic purification); IR (KBr) ν: 3057 (w), 2948 (w), 1709 (s), 1644 (m), 1589 (w), 1432 (w), 1235 (s), 1066 (m), 726 (m) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.98 (m, 1Harom), 7.95 (d, J = 6 Hz, 1Harom), 7.75 (d, J = 12 Hz, 1Harom), 7.43–7.41 (m, 1Harom), 7.37–7.34 (m, 2Harom), 7.27 (s, 1Harom), 7.28–7.24 (m, 1Harom), 7.22, 7.18 (2 d, J = 12 Hz, 2 olefinic HC=), 4.45 (s, S-CH), 3.86 (s, 3H, OCH3) ppm; 13C NMR (150 MHz, CDCl3) δ 165.2 (C=O), 139.7, 134.9, 134.7, 131.6, 131.1, 125.0 (6 C(sp2)), 138.7, 131.9, 131.6, 130.7, 130.2, 128.8, 127.9, 127.8, 126.5, 126.1 (7 CHarom + 2 CHolefin + S-CH=), 51.9 (OCH3), 41.3 (S-CH) ppm; MS (ESI) m/z (%): 207 (100, [M − 74]+), 245 (55, [M − 36]+), 297 (45, [M − 16]+); anal. calcd for C19H14O2S: C, 74.48; H, 4.61; S, 10.46; found: C, 74.53; H, 4.99; S, 10.64.
General procedure for the oxidation of thiopyran derivatives 4a,b and 5a,b: A solution of 1 mmol of the corresponding thiopyran and 3 mmol of mCPBA (70% purity) in dichloromethane was stirred at room temperature for 3 days. Then the reaction mixture was extracted with aqueous saturated NaHCO3 (3 × 10 mL) and distilled water (1 × 10 mL). The organic phase was dried over anhydrous MgSO4 and concentrated in vacuo.
Methyl 11,12-dihydro-4bH-benzo[4,5]cyclohepta[1,2,3-ij]isothiochromene-7-carboxylate 5,5-dioxide (6d): Yield: 88 mg (80%). Colorless crystals; mp 187.5–188.0 °C (MeOH); IR (KBr) ν: 3041 (w), 2950 (w), 1717 (s), 1600 (w), 1433 (w), 1307 (s), 1262 (s), 1130 (s), 785 (m) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.60–7.48 (m, 1Harom), 7.36 (br s, 1Harom + S-CH=), 7.32–7.28 (m, 2Harom), 7.27–7.20 (m, 3Harom), 5.72 (br s S-CH), 3.96 (s, 3H, OCH3), 3.58 (br s, 1H), 3.10 (br s, 2H), 2.91 (br s, 1H) ppm; 13C NMR (150 MHz, CDCl3) δ 164.9 (C=O), 134.5, 134.3, 132.2, 131.0, 130.8, 129.2 (6 C(sp2)), 134.1, 133.3, 130.0, 129.7, 129.1, 128.0, 127.5, 126.1 (7 CHarom + S-CH=), 62.5 (OCH3), 53.4 (S-CH), 37.3, 34.7 (2 broad signals, 2 CH2) ppm; HRMS (ESI): [M + Na]+ calcd for C19H16NaO4S, 363.06615; found, 363.06614.
Supporting Information
| Supporting Information File 1: Experimental data for selected compounds 4–6, details of the crystal structure determination, and the original 1H and 13C NMR spectra for all products. CCDC-1038599 and 1038600 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Mlostoń, G.; Heimgartner, H. In Targets in Heterocyclic Systems; Chemistry and Properties; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, 2005; Vol. 9, pp 141–157.
Return to citation in text: [1] -
Mlostoń, G.; Heimgartner, H. In Targets in Heterocyclic Systems; Chemistry and Properties; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, 2006; Vol. 10, pp 266–300.
Return to citation in text: [1] -
Okuma, K. Sulfur Rep. 2002, 23, 209–241. doi:10.1080/01961770208047971
Return to citation in text: [1] -
Huisgen, R.; Fisera, L.; Giera, H.; Sustmann, R. J. Am. Chem. Soc. 1995, 117, 9671–9678. doi:10.1021/ja00143a008
Return to citation in text: [1] -
Huisgen, R.; Langhals, E. Heteroat. Chem. 2006, 17, 433–442. doi:10.1002/hc.20262
Return to citation in text: [1] -
Huisgen, R.; Li, X.; Giera, H.; Langhals, E. Helv. Chim. Acta 2001, 84, 981–999. doi:10.1002/1522-2675(20010516)84:5<981::AID-HLCA981>3.0.CO;2-O
Return to citation in text: [1] -
Schatz, J.; Sauer, J. Tetrahedron Lett. 1994, 35, 4767–4770. doi:10.1016/S0040-4039(00)76962-0
Return to citation in text: [1] -
Rohr, U.; Schatz, J.; Sauer, J. Eur. J. Org. Chem. 1998, 2875–2883. doi:10.1002/(SICI)1099-0690(199812)1998:12<2875::AID-EJOC2875>3.0.CO;2-N
Return to citation in text: [1] -
Gotthardt, H.; Nieberl, S. Liebigs Ann. Chem. 1980, 867–872. doi:10.1002/jlac.198019800607
Return to citation in text: [1] [2] [3] -
Rapp, J.; Huisgen, R. Tetrahedron 1997, 53, 961–970. doi:10.1016/S0040-4020(96)01069-1
Return to citation in text: [1] [2] -
Huisgen, R.; Rapp, J. Heterocycles 1997, 45, 507–525. doi:10.3987/COM-96-7706
Return to citation in text: [1] -
Ohno, A.; Koizumi, T.; Ohnishi, Y. Bull. Chem. Soc. Jpn. 1971, 44, 2511–2515. doi:10.1246/bcsj.44.2511
Return to citation in text: [1] -
Okuma, K.; Yamamoto, T.; Shirokawa, T.; Kitamura, T.; Fujiwara, Y. Tetrahedron Lett. 1996, 37, 8883–8886. doi:10.1016/S0040-4039(96)02074-6
Return to citation in text: [1] -
Ohmura, H.; Motoki, S. Bull. Chem. Soc. Jpn. 1984, 57, 1131–1137. doi:10.1246/bcsj.57.1131
Return to citation in text: [1] [2] -
Saito, T.; Shizuta, T.; Kikuchi, H.; Nakagawa, J.; Hirotsu, K.; Ohmura, H.; Motoki, S. Synthesis 1994, 727–732. doi:10.1055/s-1994-25558
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Gębicki, K.; Grzelak, P.; Heimgartner, H. Heteroat. Chem. 2014, 25, 548–555. doi:10.1002/hc.21191
Return to citation in text: [1] -
Fringuelli, F.; Taticchi, A., Eds. The Diels-Alder Reaction; Selected Practical Methods; J. Wiley & Sons Ltd.: Chichester, U.K., 2002.
Return to citation in text: [1] -
Matsumoto, M.; Hamana, H.; Iida, H. Helv. Chim. Acta 2005, 88, 2033–2234. doi:10.1002/hlca.200590156
Return to citation in text: [1] -
Johnson, C. K. ORTEP II, Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, Tennessee, 1976.
Return to citation in text: [1] [2] -
Aitken, R. A.; Hauduc, C.; Hossain, M. S.; McHale, E.; Schwan, A. L.; Slawin, A. M. Z.; Stewart, C. A. Aust. J. Chem. 2014, 67, 1288–1295. doi:10.1071/CH14155
Return to citation in text: [1] -
Okuma, K.; Kojima, K.; Koga, Y.; Shioji, K. Heterocycles 2000, 52, 1021–1024. doi:10.3987/COM-99-S118
Return to citation in text: [1] -
Kaminskyy, D.; Kryshchyshyn, A.; Nektegayev, I.; Vasylenko, O.; Grellier, P.; Lesyk, R. Eur. J. Med. Chem. 2014, 75, 57–66. doi:10.1016/j.ejmech.2014.01.028
Return to citation in text: [1] -
Tavakolinia, F.; Baghipour, T.; Hossaini, Z.; Zareyee, D.; Khalilzadeh, M. A.; Rajabi, M. Nucleic Acid Ther. 2012, 22, 265–270. doi:10.1089/nat.2012.0346
Return to citation in text: [1] -
Rajabi, M.; Khalilzadeh, M. A.; Mehrzad, J. DNA Cell Biol. 2012, 31, 128–134. doi:10.1089/dna.2011.1291
Return to citation in text: [1] -
Daraosheh, A. Q.; Görls, H.; El-khateeb, M.; Mlostoń, G.; Weigand, W. Eur. J. Inorg. Chem. 2011, 349–355. doi:10.1002/ejic.201000770
Return to citation in text: [1] -
Daraosheh, A. Q.; Apfel, U.-P.; Görls, H.; Friebe, C.; Schubert, U.-S.; El-khateeb, M.; Mlostoń, G.; Weigand, W. Eur. J. Inorg. Chem. 2012, 318–326. doi:10.1002/ejic.201101032
Return to citation in text: [1]
| 25. | Daraosheh, A. Q.; Görls, H.; El-khateeb, M.; Mlostoń, G.; Weigand, W. Eur. J. Inorg. Chem. 2011, 349–355. doi:10.1002/ejic.201000770 |
| 26. | Daraosheh, A. Q.; Apfel, U.-P.; Görls, H.; Friebe, C.; Schubert, U.-S.; El-khateeb, M.; Mlostoń, G.; Weigand, W. Eur. J. Inorg. Chem. 2012, 318–326. doi:10.1002/ejic.201101032 |
| 19. | Johnson, C. K. ORTEP II, Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, Tennessee, 1976. |
| 22. | Kaminskyy, D.; Kryshchyshyn, A.; Nektegayev, I.; Vasylenko, O.; Grellier, P.; Lesyk, R. Eur. J. Med. Chem. 2014, 75, 57–66. doi:10.1016/j.ejmech.2014.01.028 |
| 23. | Tavakolinia, F.; Baghipour, T.; Hossaini, Z.; Zareyee, D.; Khalilzadeh, M. A.; Rajabi, M. Nucleic Acid Ther. 2012, 22, 265–270. doi:10.1089/nat.2012.0346 |
| 24. | Rajabi, M.; Khalilzadeh, M. A.; Mehrzad, J. DNA Cell Biol. 2012, 31, 128–134. doi:10.1089/dna.2011.1291 |
| 1. | Mlostoń, G.; Heimgartner, H. In Targets in Heterocyclic Systems; Chemistry and Properties; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, 2005; Vol. 9, pp 141–157. |
| 2. | Mlostoń, G.; Heimgartner, H. In Targets in Heterocyclic Systems; Chemistry and Properties; Attanasi, O. A.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, 2006; Vol. 10, pp 266–300. |
| 3. | Okuma, K. Sulfur Rep. 2002, 23, 209–241. doi:10.1080/01961770208047971 |
| 10. | Rapp, J.; Huisgen, R. Tetrahedron 1997, 53, 961–970. doi:10.1016/S0040-4020(96)01069-1 |
| 20. | Aitken, R. A.; Hauduc, C.; Hossain, M. S.; McHale, E.; Schwan, A. L.; Slawin, A. M. Z.; Stewart, C. A. Aust. J. Chem. 2014, 67, 1288–1295. doi:10.1071/CH14155 |
| 9. | Gotthardt, H.; Nieberl, S. Liebigs Ann. Chem. 1980, 867–872. doi:10.1002/jlac.198019800607 |
| 10. | Rapp, J.; Huisgen, R. Tetrahedron 1997, 53, 961–970. doi:10.1016/S0040-4020(96)01069-1 |
| 11. | Huisgen, R.; Rapp, J. Heterocycles 1997, 45, 507–525. doi:10.3987/COM-96-7706 |
| 21. | Okuma, K.; Kojima, K.; Koga, Y.; Shioji, K. Heterocycles 2000, 52, 1021–1024. doi:10.3987/COM-99-S118 |
| 7. | Schatz, J.; Sauer, J. Tetrahedron Lett. 1994, 35, 4767–4770. doi:10.1016/S0040-4039(00)76962-0 |
| 8. | Rohr, U.; Schatz, J.; Sauer, J. Eur. J. Org. Chem. 1998, 2875–2883. doi:10.1002/(SICI)1099-0690(199812)1998:12<2875::AID-EJOC2875>3.0.CO;2-N |
| 19. | Johnson, C. K. ORTEP II, Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, Tennessee, 1976. |
| 4. | Huisgen, R.; Fisera, L.; Giera, H.; Sustmann, R. J. Am. Chem. Soc. 1995, 117, 9671–9678. doi:10.1021/ja00143a008 |
| 5. | Huisgen, R.; Langhals, E. Heteroat. Chem. 2006, 17, 433–442. doi:10.1002/hc.20262 |
| 6. | Huisgen, R.; Li, X.; Giera, H.; Langhals, E. Helv. Chim. Acta 2001, 84, 981–999. doi:10.1002/1522-2675(20010516)84:5<981::AID-HLCA981>3.0.CO;2-O |
| 14. | Ohmura, H.; Motoki, S. Bull. Chem. Soc. Jpn. 1984, 57, 1131–1137. doi:10.1246/bcsj.57.1131 |
| 16. | Mlostoń, G.; Urbaniak, K.; Gębicki, K.; Grzelak, P.; Heimgartner, H. Heteroat. Chem. 2014, 25, 548–555. doi:10.1002/hc.21191 |
| 17. | Fringuelli, F.; Taticchi, A., Eds. The Diels-Alder Reaction; Selected Practical Methods; J. Wiley & Sons Ltd.: Chichester, U.K., 2002. |
| 14. | Ohmura, H.; Motoki, S. Bull. Chem. Soc. Jpn. 1984, 57, 1131–1137. doi:10.1246/bcsj.57.1131 |
| 15. | Saito, T.; Shizuta, T.; Kikuchi, H.; Nakagawa, J.; Hirotsu, K.; Ohmura, H.; Motoki, S. Synthesis 1994, 727–732. doi:10.1055/s-1994-25558 |
| 18. | Matsumoto, M.; Hamana, H.; Iida, H. Helv. Chim. Acta 2005, 88, 2033–2234. doi:10.1002/hlca.200590156 |
| 13. | Okuma, K.; Yamamoto, T.; Shirokawa, T.; Kitamura, T.; Fujiwara, Y. Tetrahedron Lett. 1996, 37, 8883–8886. doi:10.1016/S0040-4039(96)02074-6 |
| 9. | Gotthardt, H.; Nieberl, S. Liebigs Ann. Chem. 1980, 867–872. doi:10.1002/jlac.198019800607 |
| 12. | Ohno, A.; Koizumi, T.; Ohnishi, Y. Bull. Chem. Soc. Jpn. 1971, 44, 2511–2515. doi:10.1246/bcsj.44.2511 |
| 9. | Gotthardt, H.; Nieberl, S. Liebigs Ann. Chem. 1980, 867–872. doi:10.1002/jlac.198019800607 |
© 2015 Mlostoń et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)