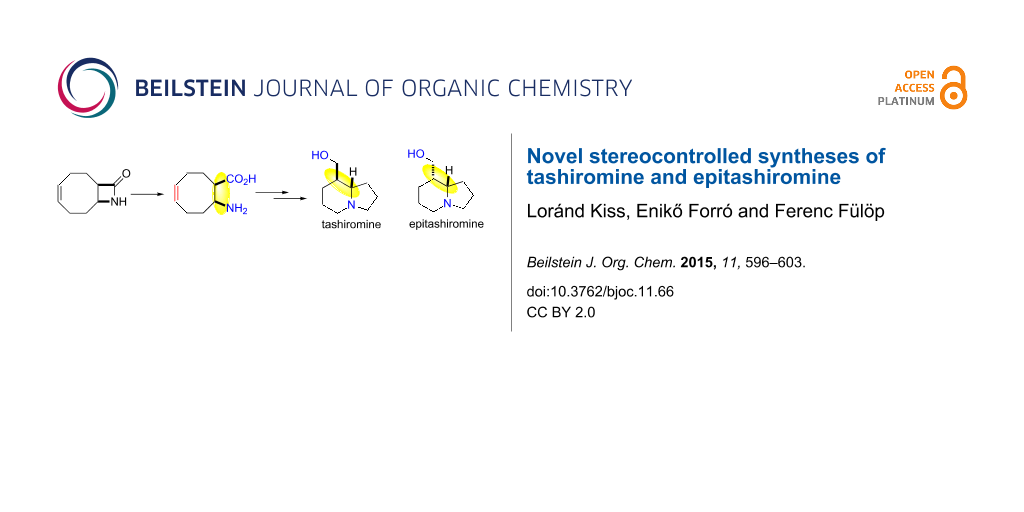
Abstract
A novel stereocontrolled approach has been developed for the syntheses of tashiromine and epitashiromine alkaloids from cyclooctene β-amino acids. The synthetic concept is based on the azetidinone opening of a bicyclic β-lactam, followed by oxidative ring opening through ring C–C double bond and reductive ring-closure reactions of the cis- or trans-cyclooctene β-amino acids.

Graphical Abstract
Introduction
Indolizidine alkaloids are an important class of naturally occurring compounds which have received considerable attention as a result of their valuable physiological properties. A number of representatives of this class exhibit glycosidase inhibitory activity or antimetastatic, anticancer, antitumour or anti-HIV properties [1-3]. A large number of natural products contain an indolizidine framework, among them (−)-δ-coniceine, (−)-swainsonine, indolizidine 167B [4-10], (+)-lentiginosine [11-15], (+)-slaframine [16], (−)-elaeokanine C [17], (+)-cyclizidine [18], lepadiformine [19], the highly oxygenated (+)-castanospermine [20,21], or pumiliotoxin [22]. Figure 1 illustrates the structures of several such compounds.
![[1860-5397-11-66-1]](/bjoc/content/figures/1860-5397-11-66-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Tashiromine is a natural indolizidine alkaloid isolated from Maackia tashiroi (1990). Strategies for the synthesis of indolizidine derivatives have received considerable interest from synthetic and medicinal chemists (Figure 2). A number of synthetic approaches have been described earlier for construction of the indolizidine framework; access to tashiromine in racemic form can be achieved through the alkylation of succinimide, followed by ring closure via acyliminium intermediates [23,24], the reduction of cyclized pyridinium salts [25], iminium cascade cyclization [26], alkyne-mediated hydroformylation–cyclization [27], or electrophilic pyrolidinone alkylation followed by ring closure [28,29]. Pyrrolidine alkylation and nucleophilic ring closure followed by C–C double bond hydroboration [30] leads to racemic epitashiromine, as does the N-alkylated succinimide transformation through the corresponding indolizidinone [31].
![[1860-5397-11-66-2]](/bjoc/content/figures/1860-5397-11-66-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Approaches to racemic tashiromine and epitashiromine.
Figure 2: Approaches to racemic tashiromine and epitashiromine.
Several synthetic procedures have also been developed for the preparation of tashiromine or epitashiromine enantiomers.
(+)-Tashiromine has been synthetized from a pyrrolidinone derivative through chiral Lewis acid-catalysed cyclization to substituted pyrrolidinones [17], by the intramolecular cyclization of a chiral alkenylated pyrrolidinone, followed by hydroxylation [32], or by the intramolecular ring closure of chiral pyrrolidine diesters followed by ester and oxo group reduction [33], while the syntheses of (+)-epitashiromine starts from a chiral morpholine derivative, with nitrone 1,3-dipolar cycloaddition and reduction [34] (Figure 3).
![[1860-5397-11-66-3]](/bjoc/content/figures/1860-5397-11-66-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Synthetic routes to (+)-tashiromine and (+)-epitashiromine.
Figure 3: Synthetic routes to (+)-tashiromine and (+)-epitashiromine.
(−)-Tashiromine has been accessed through the ring closure of difunctionalized acyclic chiral sulfonamide-based β-amino acids [35], the cyclization of pyrrole derivatives with a chiral side-chain [36], or the enantioselective arylation of pyrrole, followed by saturation [37]. The transformation of chiral functionalized pyrrole or pyrrolidine derivatives has served as the basis of the construction of (−)-epitashiromine [38,39] (Figure 4).
![[1860-5397-11-66-4]](/bjoc/content/figures/1860-5397-11-66-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Synthetic routes to (−)-tashiromine and (−)-epitashiromine.
Figure 4: Synthetic routes to (−)-tashiromine and (−)-epitashiromine.
The oxidative functionalization of cyclic β-amino acid derivatives has been reported to be a convenient route for the preparation of N-heterocyclic β-amino acid derivatives [40,41] or for the stereocontrolled synthesis of functionalized cispentacins [42] and their acyclic counterparts [43,44] (Figure 5). The oxidative ring cleavage of various vicinal diols and the transformation of the resulting dialdehyde intermediates has been efficiently applied in recent years for the synthesis of a series of valuable organic molecules [45-52]. In particular, Davies and co-workers have utilized the oxidative ring opening of cyclic vicinal diols followed by ring closure for access to pyrrolizidine alkaloids [45].
![[1860-5397-11-66-5]](/bjoc/content/figures/1860-5397-11-66-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Oxidative functionalizations of cyclic β-amino acids.
Figure 5: Oxidative functionalizations of cyclic β-amino acids.
Results and Discussion
We describe here a novel access route for the synthesis of tashiromine and epitashiromine by starting from an unsaturated bicyclic β-lactam. The retrosynthetic concept of the synthesis is represented on Scheme 1 and was based on the lactam ring opening, in continuation followed by oxidative ring opening of the formed β-amino esters and by reductive ring closure as key steps.
![[1860-5397-11-66-i1]](/bjoc/content/inline/1860-5397-11-66-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of tashiromine and epitashiromine.
Scheme 1: Retrosynthesis of tashiromine and epitashiromine.
Bicyclic β-lactam (±)-1 [53,54] was first transformed by azetidinone opening to the corresponding amino ester hydrochloride (±)-2 [53,54], N-protection of which with benzyl chloroformate (Z-Cl) afforded protected amino ester (±)-3 in 78% yield. In agreement with our earlier observations [40-42] C–C double bond functionalization of the cyclooctene β-amino ester via dihydroxylation with N-methyl morpholine N-oxide (NMO) in the presence of OsO4 afforded the corresponding all-cis dihydroxylated ethyl β-aminocyclooctanecarboxylate (±)-4 in 90% yield (for dihydroxylation, see also reference [54]) (Scheme 2). Amino ester (±)-4 was next subjected through its vicinal diol moiety to oxidative ring opening with NaIO4 in MeOH at 20 °C, which resulted (monitored by TLC) in the corresponding ring-opened unstable diformyl intermediate (I1), which after work-up was immediately used without further purification (for several similar types of acyclic diformyl intermediates, see references [40,41,43,44]). Thus, the crude material was submitted to catalytic hydrogenolysis and after N-deprotection underwent double cyclization–reduction to furnish indolizidine ester (±)-5 in 41% yield after purification by chromatography.
![[1860-5397-11-66-i2]](/bjoc/content/inline/1860-5397-11-66-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of (±)-tashiromine ((±)-6).
Scheme 2: Synthesis of (±)-tashiromine ((±)-6).
Reduction of the ester group of (±)-5 with an excess of LiAlH4 in THF at 20 °C gave the corresponding tashiromine (±)-6 [35-37], which was isolated in 48% yield after purification by column chromatography (Scheme 2). The stereochemistry of (±)-6 was unequivocally assigned by NMR data, which were consistent with those reported [35-37].
A similar strategy was applied for the synthesis of epitashiromine. On reaction with NaOEt in EtOH at 20 °C, ethyl cis-β-aminocyclooctenecarboxylate (±)-3 underwent epimerization at C-1, leading after 18 h to an equilibrium mixture of cis and trans amino esters (1:1 ratio determined by 1H NMR on the crude mixture), the required trans isomer (±)-7 being separated from the cis counterpart and isolated in a yield of 48% by means of column chromatography. Dihydroxylation of (±)-7 with NMO/OsO4 next afforded an oily mixture of cis and trans dihydroxylated cyclooctane β-amino esters (diastereomeric mixture of (±)-8) in 77% overall yield after column chromatography. Our attempts to separate this nearly 1:1 mixture of the two dihydroxylated stereoisomers (determined on the basis of 1H NMR data) failed, but the mixture could be applied in the next ring-opening oxidation step, since it gave only one open-chain diformyl intermediate I2.
Similarly to the cis isomer, this unstable dialdehyde intermediate was subjected without isolation to catalytic hydrogenolysis, followed by reductive cyclization, to give the corresponding indolizidine ester (±)-9 in 40% yield. Finally, ester reduction with LiAlH4 in THF resulted in epitashiromine (±)-10 [32,34,39] in 53% yield after isolation by chromatography (Scheme 3). The stereochemistry of (±)-epitashiromine was assigned by NMR data, which were in agreement with those reported [32,34,39].
![[1860-5397-11-66-i3]](/bjoc/content/inline/1860-5397-11-66-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of (±)-epitashiromine ((±)-10).
Scheme 3: Synthesis of (±)-epitashiromine ((±)-10).
Conclusion
In summary, a novel stereocontrolled efficient method has been presented for the synthesis of tashiromine and epitashiromine alkaloids in six or seven steps, based on the preparation of cis or trans cyclooctene β-amino esters, followed by their oxidative ring cleavage and double reductive ring-closure reactions.
Experimental
General procedure for the Z-protection of amino esters
To a solution of amino ester hydrochloride ((±)-2 or (−)-2) [29] (17.8 mmol) in THF (40 mL), Et3N (9 mL) was added at 0 °C, followed by 7.8 mL (1 equivalent) of Z-Cl (a 50% solution in toluene). The mixture was stirred for 14 h at 20 °C, and then was diluted with EtOAc (120 mL). The organic layer was washed with H2O (3 × 60 mL), dried (Na2SO4), and concentrated under reduced pressure. The crude material was purified by column chromatography on silica gel (n-hexane/EtOAc 4:1), affording the amino ester.
General procedure for the dihydroxylation of amino esters
To a solution of cis or trans Z-protected amino ester ((±)-3, (−)-3 or (±)-7) (2.9 mmol) in acetone (30 mL) and H2O (1 mL), NMO (1.5 equivalents) and 2% OsO4 in t-BuOH (0.7 mL) were added and the mixture was stirred at 20 °C for 4 h. A saturated aqueous solution of Na2SO3 (40 mL) was then added, the mixture was extracted with CH2Cl2 (3 × 30 mL), and the organic layer was dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (n-hexane/EtOAc 1:4) to give the dihydroxylated amino ester.
General procedure for epimerization of the cis-amino ester
To a solution of cis N-protected amino ester ((±)-3 or (−)-3) (3.3 mmol) in EtOH (30 mL), EtONa (1.5 equivalents) was added at 0 °C and the mixture was stirred at 20 °C for 18 h. H2O (70 mL) was then added, the mixture was extracted with CH2Cl2 (3 × 30 mL), and the organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by column chromatography on silica gel (n-hexane/EtOAc 9:1) to give the trans isomer as a colourless oil.
General procedure for the oxidative ring opening/reductive ring closure of dihydroxylated amino esters
To a solution of dihydroxylated amino ester ((±)-4, (±)-8 or (−)-8) (2.46 mmol) in MeOH (25 mL), NaIO4 (2 equivalents) was added and the mixture was stirred at 20 °C for 45 min. It was then diluted with H2O (50 mL) and extracted with CH2Cl2 (3 × 20 mL). The organic layer was dried (Na2SO4) and concentrated under reduced pressure. The crude mixture was dissolved in EtOAc (30 mL), Pd/C (150 mg) was added and the mixture was stirred at 20 °C for 16 h. The catalyst was next filtered off through Celite. The crude mixture was then purified by column chromatography on silica gel (CH2Cl2/MeOH 95:5 or CH2Cl2/MeOH 9:1) to give the indolizidine derivative.
General procedure for reduction of the ester
To a solution of indolizidine carboxylate ((±)-5, (±)-9 or (−)-9) (1 mmol) in dry THF (15 mL), LiAlH4 (5 equivalents) was added at 0 °C and the mixture was stirred at 20 °C for 4 h. It was then cooled to 0 °C, H2O (2 mL) was added dropwise and the solid formed was filtered off through Celite. The filtrate was extracted with CH2Cl2 (3 × 15 mL), and the combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude oil was purified by column chromatography on silica gel (CH2Cl2/MeOH/NH4OH 90:8:2 or CH2Cl2/MeOH/NH4OH 90:5:5) to give the alkaloid.
Ethyl (1R*,2S*)-2-(benzyloxycarbonylamino)cyclooct-5-enecarboxylate ((±)-3)
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-66-i4.svg?max-width=637&scale=1.18182)
A colourless oil (Rf 0.6, n-hexane/EtOAc 4:1); yield: 78%; 1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.1 Hz, 3H, CH3), 1.76–1.87 (m, 2H, CH2), 1.88–1.97 (m, 1H, CH2), 1.98–2.07 (m, 1H, CH2), 2.09–2.18 (m, 2H, CH2), 2.22–2.31 (m, 1H, CH2), 2.43–2.52 (m, 1H, CH2), 2.82–2.89 (m, 1H, H-1), 4.12–4.21 (m, 2H, OCH2), 4.22–4.30 (m, 1H, H-2), 5.08 (s, 2H, OCH2), 5.30 (brs, 1H, N-H), 5.60–5.74 (m, 2H, H-5 and H-6), 7.33–7.48 (m, 5H, Ar-H); 13C NMR (100 MHz, DMSO) δ 14.9, 24.4, 25.1, 26.6, 32.3, 46.4, 52.1, 60.6, 66.1, 128.5, 128.6, 129.2, 129.5, 130.7, 138.0, 156.3, 174.5; anal. calcd for C19H25NO4: C, 68.86; H, 7.60; N, 4.23; found: C, 68.59; H, 7.31; N, 3.93.
Ethyl (1R*,2S*,5R*,6S*)-2-(benzyloxycarbonylamino)-5,6-dihydroxycyclooctanecarboxylate ((±)-4)
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-66-i5.svg?max-width=637&scale=1.18182)
A colourless oil (Rf 0.4, n-hexane/EtOAc 1:4); yield: 90%; 1H NMR (400 MHz, CDCl3) δ 1.26 (t, J = 7.15 Hz, 3H, CH3), 1.62–1.94 (m, 4H, CH2), 1.97–2.28 (m, 4H, CH2), 2.77–2.82 (m, 1H, H-1), 3.83–3.89 (m, 2H, H-5 and H-6), 4.02–4.10 (m, 1H, H-2), 4.11–4.19 (m, 2H, OCH2), 5.09 (m, 2H, OCH2), 5.49 (brs, 1H, N-H), 7.36–7.48 (m, 5H, Ar-H); 13C NMR (100 MHz, DMSO) δ 14.8, 21.2, 26.9, 28.5, 29.0, 45.4, 51.6, 60.7, 65.9, 72.3, 72.4, 128.4, 128.5, 129.1, 138.1, 156.3, 174.6; anal. calcd for C19H27NO6: C, 62.45; H, 7.45; N, 3.83; found: C, 62.19; H, 7.10; N, 4.13.
Ethyl (8R*,8aS*)-octahydroindolizine-8-carboxylate ((±)-5)
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-66-i6.svg?max-width=637&scale=1.18182)
A yellow oil (Rf 0.55, CH2Cl2/MeOH 95:5); yield: 41%; 1H NMR (400 MHz, CDCl3) δ 1.27 (t, J = 7.15 Hz, 3H, CH3), 1.46–1.53 (m, 2H, CH2), 1.57–1.92 (m, 5H, CH2), 1.99–2.10 (m, 3H, CH2), 2.13–2.20 (m, 1H, CH2), 2.25–2.31 (m, 1H, H-8), 3.03–3.10 (m, 2H, CH2 and H-8a); 13C NMR (100 MHz, CDCl3) δ 14.6, 20.9, 25.1, 29.5, 30.1, 48.4, 52.6, 54.4, 60.6, 65.6, 174.6; MS (ESI) m/z: 198.5 [M + 1]; anal. calcd for C11H19NO2: C, 66.97; H, 9.71; N, 7.10; found: C, 66.60; H, 10.02; N, 7.39.
((8R*,8aS*)-Octahydroindolizin-8-yl)methanol; ((±)-tashiromine (±)-6) [35-37]
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-66-i7.svg?max-width=637&scale=1.18182)
A yellow oil (Rf 0.45, CH2Cl2/MeOH/NH4OH 90:8:2); yield: 48%; 1H NMR (400 MHz, CDCl3) δ 1.00–1.11 (m, 1H, CH2), 1.42–1.53 (m, 2H, CH2), 1.58–1.83 (m, 5H, CH2), 1.84–1.99 (m, 3H, CH2), 2.04–2.11 (m, 1H, CH2), 3.03–3.10 (m, 2H, N-CH), 3.41–3.46 (m, 1H, OCH2), 3.59–3.64 (m, 1H, OCH2); 13C NMR (100 MHz, CDCl3) δ 21.1, 25.5, 28.0, 29.4, 44.9, 53.1, 54.5, 66.0, 66.9; MS (ESI) m/z: 156.6 [M + 1]; anal. calcd for C9H17NO: C, 69.63; H, 11.04; N, 9.02; found: C, 69.28; H, 10.70; N, 8.76.
Ethyl (1S*,2S*)-2-(benzyloxycarbonylamino)cyclooct-5-enecarboxylate ((±)-7)
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-66-i8.svg?max-width=637&scale=1.18182)
A colourless oil (Rf 0.55, n-hexane/EtOAc 4:1); yield: 48%; 1H NMR (400 MHz, DMSO) δ 1.18 (t, J = 7.10 Hz, 3H, CH3), 1.55–1.64 (m, 2H, CH2), 1.73–1.97 (m, 2H, CH2), 2.04–2.19 (m, 2H, CH2), 2.33–2.47 (m, 2H, CH2), 2.68–2.75 (m, 1H, H-1), 3.88–4.02 (m, 3H, OCH2 and H-2), 4.96–5.02 (m, 2H, OCH2), 5.53–5.60 (m, 2H, H-5 and H-6), 7.30–7.44 (m, 5H, Ar-H); 13C NMR (100 MHz, DMSO) δ 14.8, 24.5, 25.1, 29.2, 33.8, 49.2, 52.1, 60.4, 65.9, 128.0, 128.4, 129.0, 129.1, 130.7, 138.2, 156.9, 174.5; anal. calcd for C19H25NO4: C, 68.86; H, 7.60; N, 4.23; found: C, 68.57; H, 7.28; N, 3.97.
Ethyl (8S*,8aS*)-octahydroindolizine-8-carboxylate ((±)-9)
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-66-i9.svg?max-width=637&scale=1.18182)
A yellow oil (Rf 0.45, CH2Cl2/MeOH 4:1); yield: 40%; 1H NMR (400 MHz, CDCl3) δ 1.28 (t, J = 7.15 Hz, 3H, CH3), 1.28–1.33 (m, 1H, CH2), 1.42–1.55 (m, 2H, CH2), 1.58–1.64 (m, 1H, CH2), 1.68–1.86 (m, 3H, CH2), 2.02–2.18 (m, 3H, CH2), 2.23–2.27 (m, 1H, CH2), 2.78–2.81 (m, 1H, H-8), 3.04–3.10 (m, 2H, CH2 and H-8a), 4.09–4.17 (m, 2H, OCH2); 13C NMR (100 MHz, DMSO) δ 15.1, 21.2, 22.6, 26.8, 27.3, 42.0, 53.4, 55.0, 60.5, 64.9, 170.4; MS (ESI) m/z: 198.7 [M + 1]; anal. calcd for C11H19NO2: C, 66.97; H, 9.71; N, 7.10; found: C, 67.28; H, 9.40; N, 6.78.
((8S*,8aS*)-Octahydroindolizin-8-yl)methanol; ((±)-epitashiromine, (±)-10) [32,34,39]
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-66-i10.svg?max-width=637&scale=1.18182)
A yellow oil (Rf 0.45, CH2Cl2/MeOH/NH4OH 88:8:4); yield: 53%; 1H NMR (400 MHz, CDCl3) δ 1.56–1.62 (m, 2H, CH2), 1.65–1.74 (m, 4H, CH2), 1.97–2.12 (m, 5H, CH2), 2.20–2.28 (m, 1H, H-8), 2.94–3.00 (m, 1H, CH2), 3.08–3.14 (m, 1H, N-CH), 3.70–3.75 (m, 1H, OCH2), 4.13–4.19 (m, 1H, OCH2); 13C NMR (100 MHz, CDCl3) δ 21.2, 23.7, 26.3, 30.1, 35.7, 54.0, 54.9, 66.0, 66.8; MS (ESI) m/z: 156.4 [M + 1]; anal. calcd for C9H17NO: C, 69.63; H, 11.04; N 9.02; found: C, 69.30; H, 10.71; N, 8.79.
References
-
Michael, J. P. Nat. Prod. Rep. 2008, 25, 139–165. doi:10.1039/B612166G
Return to citation in text: [1] -
Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem. – Eur. J. 2009, 15, 7808. doi:10.1002/chem.200900707
Return to citation in text: [1] -
Macchi, B.; Minutolo, A.; Grelli, S.; Cardona, F.; Cordero, F. M.; Mastino, A.; Brandi, A. Glycobiology 2010, 20, 500–506. doi:10.1093/glycob/cwp202
Return to citation in text: [1] -
Felpin, F. X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193
Return to citation in text: [1] -
Hu, X.-G.; Hunter, L. Beilstein J. Org. Chem. 2013, 9, 2696–2708. doi:10.3762/bjoc.9.306
Return to citation in text: [1] -
Faria, A. R.; Salvador, E. L.; Correia, C. R. D. J. Org. Chem. 2002, 67, 3651–3661. doi:10.1021/jo016189u
Return to citation in text: [1] -
Jiang, X.-P.; Cheng, Y.; Shi, G.-F.; Kang, Z.-M. J. Org. Chem. 2007, 72, 2212–2215. doi:10.1021/jo0624290
Return to citation in text: [1] -
Trajkovic, M.; Balanac, V.; Ferjancic, Z.; Saicic, R. N. RSC Adv. 2014, 4, 53722–53724. doi:10.1039/C4RA11978A
Return to citation in text: [1] -
Davies, S. G.; Fletcher, A. M.; Foster, E. M.; Houlsby, I. T. T.; Roberts, P. M.; Schofield, T. M.; Thomson, J. E. Org. Biomol. Chem. 2014, 12, 9223–9235. doi:10.1039/C4OB01737D
Return to citation in text: [1] -
Rao, N. N.; Parida, B. B.; Cha, J. K. Org. Lett. 2014, 16, 6208–6211. doi:10.1021/ol503136s
Return to citation in text: [1] -
Kim, G.-W.; Jin, T.; Kim, J.-S.; Park, S.-H.; Lee, K.-H.; Kim, S.-S.; Myeong, I.-S.; Ham, W.-H. Tetrahedron: Asymmetry 2014, 25, 87–91. doi:10.1016/j.tetasy.2013.11.009
Return to citation in text: [1] -
Du-a-man, S.; Soorukram, D.; Kuhakarn, C.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. Eur. J. Org. Chem. 2014, 1708–1715. doi:10.1002/ejoc.201301671
Return to citation in text: [1] -
Yoon, H.; Cho, K. S.; Sim, T. Tetrahedron: Asymmetry 2014, 25, 497–502. doi:10.1016/j.tetasy.2014.02.009
Return to citation in text: [1] -
Kauloorkar, S. V.; Jha, V.; Jogdand, G.; Kumar, P. Org. Biomol. Chem. 2014, 12, 4454–4460. doi:10.1039/c4ob00461b
Return to citation in text: [1] -
Lingamurthy, M.; Rajander, A.; Rao, B. V. Tetrahedron: Asymmetry 2014, 25, 860–863. doi:10.1016/j.tetasy.2014.04.011
Return to citation in text: [1] -
Ortiz, G. X., Jr.; Kang, B.; Wang, Q. J. Org. Chem. 2014, 79, 571–581. doi:10.1021/jo4022666
Return to citation in text: [1] -
Suga, H.; Hashimoto, Y.; Yasumura, S.; Takezawa, R.; Itoh, K.; Kakehi, A. J. Org. Chem. 2013, 78, 10840–10852. doi:10.1021/jo401837d
Return to citation in text: [1] [2] -
Hanessian, S.; Soma, U.; Dorich, S.; Deschênes-Simard, B. Org. Lett. 2011, 13, 1048–1051. doi:10.1021/ol103094j
Return to citation in text: [1] -
Han, M.-Y.; Jia, J.-Y.; Wang, W. Tetrahedron Lett. 2014, 55, 784–794. doi:10.1016/j.tetlet.2013.11.048
Return to citation in text: [1] -
Singh, P.; Manna, S. K.; Panda, G. Tetrahedron 2014, 70, 1363–1374. doi:10.1016/j.tet.2013.11.074
Return to citation in text: [1] -
Tiwari, D. K.; Bharadwaj, K. C.; Puranik, V. G.; Tiwari, D. K. Org. Biomol. Chem. 2014, 12, 7389–7396. doi:10.1039/C4OB00948G
Return to citation in text: [1] -
Jouanno, L.-A.; Di Mascio, V.; Tognetti, V.; Joubert, L.; Sabot, C.; Renard, P.-Y. J. Org. Chem. 2014, 79, 1303. doi:10.1021/jo402729a
Return to citation in text: [1] -
McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072
Return to citation in text: [1] -
Marsden, S. P.; McElhinney, A. D. Beilstein J. Org. Chem. 2008, 4, No. 8. doi:10.1186/1860-5397-4-8
Return to citation in text: [1] -
Bates, R. W.; Boonsombat, J. J. Chem. Soc., Perkin Trans. 1 2001, 654–656. doi:10.1039/b100407g
Return to citation in text: [1] -
Amorde, S. M.; Jewett, I. T.; Martin, S. F. Tetrahedron 2009, 65, 3222–3231. doi:10.1016/j.tet.2008.10.074
Return to citation in text: [1] -
Chiou, W.-H.; Lin, Y.-H.; Chen, G.-T.; Gao, Y.-K.; Tseng, Y.-C.; Kao, C.-L.; Tsai, J.-C. Chem. Commun. 2011, 47, 3562–3564. doi:10.1039/c0cc05646d
Return to citation in text: [1] -
Pohmakotr, M.; Prateeptongkum, S.; Chooprayoon, S.; Tuchinda, P.; Reutrakul, V. Tetrahedron 2008, 64, 2339–2347. doi:10.1016/j.tet.2008.01.008
Return to citation in text: [1] -
Bélanger, G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. J. Org. Chem. 2006, 71, 704–712. doi:10.1021/jo052141v
Return to citation in text: [1] [2] -
Dieter, R. K.; Watson, R. Tetrahedron Lett. 2002, 43, 7725–7728. doi:10.1016/S0040-4039(02)01835-X
Return to citation in text: [1] -
Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383
Return to citation in text: [1] -
Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094
Return to citation in text: [1] [2] [3] [4] -
David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.-C.; Haviari, G.; Célérier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. J. Org. Chem. 1999, 64, 3122–3131. doi:10.1021/jo982169p
Return to citation in text: [1] -
Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078
Return to citation in text: [1] [2] [3] [4] -
Cutter, A. C.; Miller, I. R.; Keily, J. F.; Bellingham, R. K.; Light, M. E.; Brown, R. C. D. Org. Lett. 2011, 13, 3988–3991. doi:10.1021/ol2015048
Return to citation in text: [1] [2] [3] [4] -
Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9
Return to citation in text: [1] [2] [3] [4] -
Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902
Return to citation in text: [1] [2] [3] [4] -
Reddy, K. K. S.; Rao, B. V.; Raju, S. S. Tetrahedron: Asymmetry 2011, 22, 662–668. doi:10.1016/j.tetasy.2011.03.009
Return to citation in text: [1] -
Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a
Return to citation in text: [1] [2] [3] [4] -
Kazi, B.; Kiss, L.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2010, 51, 82–85. doi:10.1016/j.tetlet.2009.10.072
Return to citation in text: [1] [2] [3] -
Kiss, L.; Kazi, B.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2008, 49, 339–342. doi:10.1016/j.tetlet.2007.11.043
Return to citation in text: [1] [2] [3] -
Kiss, L.; Cherepanova, M.; Forró, E.; Fülöp, F. Chem. – Eur. J. 2013, 19, 2102–2107. doi:10.1002/chem.201203183
Return to citation in text: [1] [2] -
Cherepanova, M.; Kiss, L.; Fülöp, F. Tetrahedron 2014, 70, 2515–2522. doi:10.1016/j.tet.2014.02.063
Return to citation in text: [1] [2] -
Cherepanova, M.; Kiss, L.; Forró, E.; Fülöp, F. Eur. J. Org. Chem. 2014, 403–409. doi:10.1002/ejoc.201301281
Return to citation in text: [1] [2] -
Brambilla, M.; Davies, S. G.; Fletcher, A. M.; Roberts, P. M.; Thomson, J. E. Tetrahedron 2014, 70, 204–211. doi:10.1016/j.tet.2013.11.094
Return to citation in text: [1] [2] -
Lee, M.-r.; Stahl, S. S.; Gellman, S. H. Org. Lett. 2008, 10, 5317–5319. doi:10.1021/ol802274x
Return to citation in text: [1] -
Fricke, Y.; Kopp, N.; Wünsch, B. Synthesis 2010, 791–796. doi:10.1055/s-0029-1218622
Return to citation in text: [1] -
Malik, G.; Guinchard, X.; Crich, D. Org. Lett. 2012, 14, 596–599. doi:10.1021/ol203213f
Return to citation in text: [1] -
Sousa, C. A. D.; Rizzo-Aguiar, F.; Vale, M. L. C.; García-Mera, X.; Caamaño, O.; Rodríguez-Borges, J. E. Tetrahedron Lett. 2012, 53, 1029–1032. doi:10.1016/j.tetlet.2011.12.037
Return to citation in text: [1] -
Pérez-Bautista, J. A.; Sosa-Rivadeneyra, M.; Quintero, L.; Höpfl, H.; Tejeda-Dominguez, F. A.; Sartillo-Piscil, F. Tetrahedron Lett. 2009, 50, 5572–5574. doi:10.1016/j.tetlet.2009.07.070
Return to citation in text: [1] -
Caputo, F.; Cattaneo, C.; Clerici, F.; Gelmi, M. L.; Pellegrino, S. J. Org. Chem. 2006, 71, 8467–8472. doi:10.1021/jo061391o
Return to citation in text: [1] -
Robinson, A.; Thomas, G. L.; Spandl, R. J.; Welch, M.; Spring, D. R. Org. Biomol. Chem. 2008, 6, 2978–2981. doi:10.1039/b809038f
Return to citation in text: [1] -
Forró, E.; Árva, J.; Fülöp, F. Tetrahedron: Asymmetry 2001, 12, 643–649. doi:10.1016/S0957-4166(01)00100-8
Return to citation in text: [1] [2] -
Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R. Tetrahedron: Asymmetry 2010, 21, 957–961. doi:10.1016/j.tetasy.2010.05.003
Return to citation in text: [1] [2] [3]
| 45. | Brambilla, M.; Davies, S. G.; Fletcher, A. M.; Roberts, P. M.; Thomson, J. E. Tetrahedron 2014, 70, 204–211. doi:10.1016/j.tet.2013.11.094 |
| 46. | Lee, M.-r.; Stahl, S. S.; Gellman, S. H. Org. Lett. 2008, 10, 5317–5319. doi:10.1021/ol802274x |
| 47. | Fricke, Y.; Kopp, N.; Wünsch, B. Synthesis 2010, 791–796. doi:10.1055/s-0029-1218622 |
| 48. | Malik, G.; Guinchard, X.; Crich, D. Org. Lett. 2012, 14, 596–599. doi:10.1021/ol203213f |
| 49. | Sousa, C. A. D.; Rizzo-Aguiar, F.; Vale, M. L. C.; García-Mera, X.; Caamaño, O.; Rodríguez-Borges, J. E. Tetrahedron Lett. 2012, 53, 1029–1032. doi:10.1016/j.tetlet.2011.12.037 |
| 50. | Pérez-Bautista, J. A.; Sosa-Rivadeneyra, M.; Quintero, L.; Höpfl, H.; Tejeda-Dominguez, F. A.; Sartillo-Piscil, F. Tetrahedron Lett. 2009, 50, 5572–5574. doi:10.1016/j.tetlet.2009.07.070 |
| 51. | Caputo, F.; Cattaneo, C.; Clerici, F.; Gelmi, M. L.; Pellegrino, S. J. Org. Chem. 2006, 71, 8467–8472. doi:10.1021/jo061391o |
| 52. | Robinson, A.; Thomas, G. L.; Spandl, R. J.; Welch, M.; Spring, D. R. Org. Biomol. Chem. 2008, 6, 2978–2981. doi:10.1039/b809038f |
| 45. | Brambilla, M.; Davies, S. G.; Fletcher, A. M.; Roberts, P. M.; Thomson, J. E. Tetrahedron 2014, 70, 204–211. doi:10.1016/j.tet.2013.11.094 |
| 53. | Forró, E.; Árva, J.; Fülöp, F. Tetrahedron: Asymmetry 2001, 12, 643–649. doi:10.1016/S0957-4166(01)00100-8 |
| 54. | Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R. Tetrahedron: Asymmetry 2010, 21, 957–961. doi:10.1016/j.tetasy.2010.05.003 |
| 1. | Michael, J. P. Nat. Prod. Rep. 2008, 25, 139–165. doi:10.1039/B612166G |
| 2. | Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F. M.; Goti, A. Chem. – Eur. J. 2009, 15, 7808. doi:10.1002/chem.200900707 |
| 3. | Macchi, B.; Minutolo, A.; Grelli, S.; Cardona, F.; Cordero, F. M.; Mastino, A.; Brandi, A. Glycobiology 2010, 20, 500–506. doi:10.1093/glycob/cwp202 |
| 17. | Suga, H.; Hashimoto, Y.; Yasumura, S.; Takezawa, R.; Itoh, K.; Kakehi, A. J. Org. Chem. 2013, 78, 10840–10852. doi:10.1021/jo401837d |
| 30. | Dieter, R. K.; Watson, R. Tetrahedron Lett. 2002, 43, 7725–7728. doi:10.1016/S0040-4039(02)01835-X |
| 32. | Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094 |
| 34. | Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078 |
| 39. | Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a |
| 16. | Ortiz, G. X., Jr.; Kang, B.; Wang, Q. J. Org. Chem. 2014, 79, 571–581. doi:10.1021/jo4022666 |
| 31. | Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383 |
| 32. | Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094 |
| 34. | Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078 |
| 39. | Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a |
| 11. | Kim, G.-W.; Jin, T.; Kim, J.-S.; Park, S.-H.; Lee, K.-H.; Kim, S.-S.; Myeong, I.-S.; Ham, W.-H. Tetrahedron: Asymmetry 2014, 25, 87–91. doi:10.1016/j.tetasy.2013.11.009 |
| 12. | Du-a-man, S.; Soorukram, D.; Kuhakarn, C.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. Eur. J. Org. Chem. 2014, 1708–1715. doi:10.1002/ejoc.201301671 |
| 13. | Yoon, H.; Cho, K. S.; Sim, T. Tetrahedron: Asymmetry 2014, 25, 497–502. doi:10.1016/j.tetasy.2014.02.009 |
| 14. | Kauloorkar, S. V.; Jha, V.; Jogdand, G.; Kumar, P. Org. Biomol. Chem. 2014, 12, 4454–4460. doi:10.1039/c4ob00461b |
| 15. | Lingamurthy, M.; Rajander, A.; Rao, B. V. Tetrahedron: Asymmetry 2014, 25, 860–863. doi:10.1016/j.tetasy.2014.04.011 |
| 27. | Chiou, W.-H.; Lin, Y.-H.; Chen, G.-T.; Gao, Y.-K.; Tseng, Y.-C.; Kao, C.-L.; Tsai, J.-C. Chem. Commun. 2011, 47, 3562–3564. doi:10.1039/c0cc05646d |
| 35. | Cutter, A. C.; Miller, I. R.; Keily, J. F.; Bellingham, R. K.; Light, M. E.; Brown, R. C. D. Org. Lett. 2011, 13, 3988–3991. doi:10.1021/ol2015048 |
| 36. | Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9 |
| 37. | Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902 |
| 4. | Felpin, F. X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193 |
| 5. | Hu, X.-G.; Hunter, L. Beilstein J. Org. Chem. 2013, 9, 2696–2708. doi:10.3762/bjoc.9.306 |
| 6. | Faria, A. R.; Salvador, E. L.; Correia, C. R. D. J. Org. Chem. 2002, 67, 3651–3661. doi:10.1021/jo016189u |
| 7. | Jiang, X.-P.; Cheng, Y.; Shi, G.-F.; Kang, Z.-M. J. Org. Chem. 2007, 72, 2212–2215. doi:10.1021/jo0624290 |
| 8. | Trajkovic, M.; Balanac, V.; Ferjancic, Z.; Saicic, R. N. RSC Adv. 2014, 4, 53722–53724. doi:10.1039/C4RA11978A |
| 9. | Davies, S. G.; Fletcher, A. M.; Foster, E. M.; Houlsby, I. T. T.; Roberts, P. M.; Schofield, T. M.; Thomson, J. E. Org. Biomol. Chem. 2014, 12, 9223–9235. doi:10.1039/C4OB01737D |
| 10. | Rao, N. N.; Parida, B. B.; Cha, J. K. Org. Lett. 2014, 16, 6208–6211. doi:10.1021/ol503136s |
| 28. | Pohmakotr, M.; Prateeptongkum, S.; Chooprayoon, S.; Tuchinda, P.; Reutrakul, V. Tetrahedron 2008, 64, 2339–2347. doi:10.1016/j.tet.2008.01.008 |
| 29. | Bélanger, G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. J. Org. Chem. 2006, 71, 704–712. doi:10.1021/jo052141v |
| 35. | Cutter, A. C.; Miller, I. R.; Keily, J. F.; Bellingham, R. K.; Light, M. E.; Brown, R. C. D. Org. Lett. 2011, 13, 3988–3991. doi:10.1021/ol2015048 |
| 36. | Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9 |
| 37. | Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902 |
| 22. | Jouanno, L.-A.; Di Mascio, V.; Tognetti, V.; Joubert, L.; Sabot, C.; Renard, P.-Y. J. Org. Chem. 2014, 79, 1303. doi:10.1021/jo402729a |
| 25. | Bates, R. W.; Boonsombat, J. J. Chem. Soc., Perkin Trans. 1 2001, 654–656. doi:10.1039/b100407g |
| 54. | Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R. Tetrahedron: Asymmetry 2010, 21, 957–961. doi:10.1016/j.tetasy.2010.05.003 |
| 20. | Singh, P.; Manna, S. K.; Panda, G. Tetrahedron 2014, 70, 1363–1374. doi:10.1016/j.tet.2013.11.074 |
| 21. | Tiwari, D. K.; Bharadwaj, K. C.; Puranik, V. G.; Tiwari, D. K. Org. Biomol. Chem. 2014, 12, 7389–7396. doi:10.1039/C4OB00948G |
| 26. | Amorde, S. M.; Jewett, I. T.; Martin, S. F. Tetrahedron 2009, 65, 3222–3231. doi:10.1016/j.tet.2008.10.074 |
| 40. | Kazi, B.; Kiss, L.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2010, 51, 82–85. doi:10.1016/j.tetlet.2009.10.072 |
| 41. | Kiss, L.; Kazi, B.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2008, 49, 339–342. doi:10.1016/j.tetlet.2007.11.043 |
| 43. | Cherepanova, M.; Kiss, L.; Fülöp, F. Tetrahedron 2014, 70, 2515–2522. doi:10.1016/j.tet.2014.02.063 |
| 44. | Cherepanova, M.; Kiss, L.; Forró, E.; Fülöp, F. Eur. J. Org. Chem. 2014, 403–409. doi:10.1002/ejoc.201301281 |
| 19. | Han, M.-Y.; Jia, J.-Y.; Wang, W. Tetrahedron Lett. 2014, 55, 784–794. doi:10.1016/j.tetlet.2013.11.048 |
| 53. | Forró, E.; Árva, J.; Fülöp, F. Tetrahedron: Asymmetry 2001, 12, 643–649. doi:10.1016/S0957-4166(01)00100-8 |
| 54. | Palkó, M.; Benedek, G.; Forró, E.; Wéber, E.; Hänninen, M.; Sillanpää, R. Tetrahedron: Asymmetry 2010, 21, 957–961. doi:10.1016/j.tetasy.2010.05.003 |
| 18. | Hanessian, S.; Soma, U.; Dorich, S.; Deschênes-Simard, B. Org. Lett. 2011, 13, 1048–1051. doi:10.1021/ol103094j |
| 23. | McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072 |
| 24. | Marsden, S. P.; McElhinney, A. D. Beilstein J. Org. Chem. 2008, 4, No. 8. doi:10.1186/1860-5397-4-8 |
| 40. | Kazi, B.; Kiss, L.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2010, 51, 82–85. doi:10.1016/j.tetlet.2009.10.072 |
| 41. | Kiss, L.; Kazi, B.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2008, 49, 339–342. doi:10.1016/j.tetlet.2007.11.043 |
| 42. | Kiss, L.; Cherepanova, M.; Forró, E.; Fülöp, F. Chem. – Eur. J. 2013, 19, 2102–2107. doi:10.1002/chem.201203183 |
| 33. | David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.-C.; Haviari, G.; Célérier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. J. Org. Chem. 1999, 64, 3122–3131. doi:10.1021/jo982169p |
| 17. | Suga, H.; Hashimoto, Y.; Yasumura, S.; Takezawa, R.; Itoh, K.; Kakehi, A. J. Org. Chem. 2013, 78, 10840–10852. doi:10.1021/jo401837d |
| 29. | Bélanger, G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. J. Org. Chem. 2006, 71, 704–712. doi:10.1021/jo052141v |
| 32. | Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094 |
| 35. | Cutter, A. C.; Miller, I. R.; Keily, J. F.; Bellingham, R. K.; Light, M. E.; Brown, R. C. D. Org. Lett. 2011, 13, 3988–3991. doi:10.1021/ol2015048 |
| 36. | Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9 |
| 37. | Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902 |
| 32. | Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094 |
| 34. | Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078 |
| 39. | Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a |
| 42. | Kiss, L.; Cherepanova, M.; Forró, E.; Fülöp, F. Chem. – Eur. J. 2013, 19, 2102–2107. doi:10.1002/chem.201203183 |
| 43. | Cherepanova, M.; Kiss, L.; Fülöp, F. Tetrahedron 2014, 70, 2515–2522. doi:10.1016/j.tet.2014.02.063 |
| 44. | Cherepanova, M.; Kiss, L.; Forró, E.; Fülöp, F. Eur. J. Org. Chem. 2014, 403–409. doi:10.1002/ejoc.201301281 |
| 38. | Reddy, K. K. S.; Rao, B. V.; Raju, S. S. Tetrahedron: Asymmetry 2011, 22, 662–668. doi:10.1016/j.tetasy.2011.03.009 |
| 39. | Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a |
| 40. | Kazi, B.; Kiss, L.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2010, 51, 82–85. doi:10.1016/j.tetlet.2009.10.072 |
| 41. | Kiss, L.; Kazi, B.; Forró, E.; Fülöp, F. Tetrahedron Lett. 2008, 49, 339–342. doi:10.1016/j.tetlet.2007.11.043 |
| 36. | Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9 |
| 37. | Conrad, J. C.; Kong, J.; Laforteza, B. N.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11640–11641. doi:10.1021/ja9026902 |
| 34. | Thorat, R. G.; Pansare, S. V. Eur. J. Org. Chem. 2013, 7282–7285. doi:10.1002/ejoc.201301078 |
| 35. | Cutter, A. C.; Miller, I. R.; Keily, J. F.; Bellingham, R. K.; Light, M. E.; Brown, R. C. D. Org. Lett. 2011, 13, 3988–3991. doi:10.1021/ol2015048 |
© 2015 Kiss et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)