Abstract
Two pairs of divalent and tetravalent porphyrin building blocks carrying the complementary supramolecular crown ether/secondary ammonium ion binding motif have been synthesized and their derived pseudorotaxanes have been studied by a combination of NMR spectroscopy in solution and ESI mass spectrometry in the gas phase. By simple mixing of the components the formation of discrete dimeric and trimeric (metallo)porphyrin complexes predominates, in accordance to binding stoichiometry, while the amount of alternative structures can be neglected. Our results illustrate the power of multivalency to program the multicomponent self-assembly of specific entities into discrete functional nanostructures.
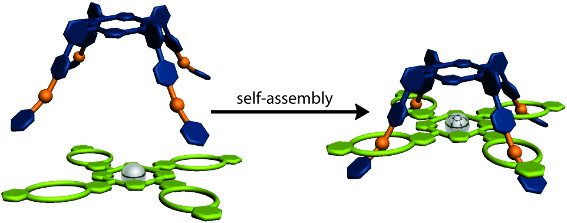
Graphical Abstract
Introduction
Supramolecular chemistry [1], the chemistry “beyond the molecule“ [2], has immensely reshaped the concepts of chemistry by putting the intermolecular interaction into the focus. Different fields of chemistry, from materials [3-6] and analytical sciences [7-12] to life science [13-17] have benefited from the development of the basic concepts of molecular recognition, templation [18], self-assembly [19], or self-sorting [20,21], just to name a few. More recently, multivalent binding [22-24] and cooperativity [25,26] have attracted significant attention mediated in particular by the desire to understand biological phenomena, such as virus docking to cells [27], toxin inhibition [28], or leucocyte recruitment in inflammation processes of the endothelium [29]. Multivalency has also inspired synthetic supramolecular architecture as it not only contributes to binding enhancement, but also helps to exert control over complex formation. For example, “molecular elevators” have been constructed by Stoddart et al. [30,31] and giant porphyrin wheels were prepared by Anderson and co-workers [32,33], both using a multivalent template strategy.
The crown ether/secondary ammonium ion binding motif [34] is a powerful tool to create well-defined pseudorotaxane structures [35-39], which have also served as precursors in rotaxane syntheses [40-42] thus providing access to interlocked, mechanically bound molecules. Based on these structures, functional supramolecular architectures such as molecular switches and motors [43-45] as well as artificial muscles [46-50], have been synthesized.
Due to their four-fold symmetry, porphyrins are excellent candidates to extend these concepts to tetravalent supramolecules. Beyond being a mere spacer and scaffold connecting the binding sites, porphyrins also offer interesting physical and optical properties [51,52]. Therefore, they have played a pivotal role in supramolecular chemistry [53-66], for example as potential candidates for artificial light-harvesting systems [67-73].
Here, we report the synthesis of two new porphyrin-based di- and tetravalent ammonium guest molecules A2 and A4 and their complementary porphyrin-based di- and tetravalent crown ether hosts C2 and C4 (Figure 1). The selection of these building blocks is based on force-field calculations, which suggest a good geometric fit between the crown ether hosts and the ammonium ion guests. The two monovalent building blocks A1 and C1 serve as control compounds. Based on this “toolbox”, which can be expanded in the future with other functional building blocks, the formation of specific multiply threaded pseudorotaxanes was achieved, thereby demonstrating the ability to program complex multicomponent self-assembly [74,75].
![[1860-5397-11-85-1]](/bjoc/content/figures/1860-5397-11-85-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Mono-, di-, and tetravalent axles A1, A2 and A4 and mono-, di-, and tetravalent hosts C1, C2 and C4. Numbers and letters are assigned to specific H atoms as discussed later in the main text.
Figure 1: Mono-, di-, and tetravalent axles A1, A2 and A4 and mono-, di-, and tetravalent hosts C1, C2 and C4...
Results and Discussion
Synthesis
The synthesis of the two ammonium-substituted porphyrins A2 and A4 was performed convergent by first preparing two different (zinc)porphyrin cores 1 and 2 (Scheme 1), which are equipped with two and four bromine atoms in the m-position of the meso-phenyl substituents, respectively, for further functionalization. Zinc porphyrins 1 and 2 have been synthesized following standard protocols for symmetrical [76] A4 and trans-disubstituted [77] A2B2 meso-functionalized porphyrins. The tetrabrominated core 1 was synthesized from aldehyde 3 and pyrrole (4) to form the free base porphyrin 5, which is subsequently converted into its zinc complex 1. On the other hand the difunctional core 2 was obtained through the condensation of aldehyde 3 with mesityldipyrromethane (6) followed by metalation of the intermediately formed free base porphyrin 7 to give its respective zinc complex 2. In the next step, axle precursor 8 was synthesized by reductive amination of 4-bromobenzaldehyde (9) and benzylamine yielding amine 10, which was subsequently Boc-protected, then reacted with trimethylsilylacetylene in a Sonogashira cross-coupling followed by desilylation. Finally, the porphyrin cores 1 and 2 were combined with axle precursor 8 in another two and four-fold Sonogashira cross-coupling reaction. After deprotection of the termini of the attached axles with trifluoroacetic acid (TFA), protonation of the free amines with HCl, and anion exchange with sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (NaBArF), the target compounds A2 and A4 were obtained. The weakly coordinating BArF counter-ion has been used to overcome solubility problems in organic solvents. It should be noted that the porphyrin is demetalated to yield the free base porphyrin during the deprotection of the Boc group. Furthermore, NMR integration of signals corresponding to the BArF protons relative to those corresponding to the macrocycle indicates that the porphyrin core is protonated (three BArF anions per divalent guest A2; five BArF anions per tetravalent guest A4). Based on the assumption that protonation of the porphyrin core, which is rather remote to the primary binding sites, does not influence the association strongly, no selective deprotonation of the porphyrin core has been attempted.
![[1860-5397-11-85-i1]](/bjoc/content/inline/1860-5397-11-85-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Overview of the synthesis of the guests A2 and A4. a) Pyrrole (4), BF3·Et2O, DDQ, CHCl3, rt; b) Zn(OAc)2, CHCl3/MeOH, rt; c) dipyrromethane 6, BF3·Et2O, DDQ, CHCl3, rt; d) Zn(OAc)2, CHCl3/MeOH, rt; e) 1. benzylamine, trimethyl orthoformate, rt, 2. NaBH4, THF/MeOH, rt; f) Boc2O, triethylamine, CH2Cl2, rt; g) 1. ethynyltrimethylsilane, CuI, PPh3, Pd(PPh3)4, TEA, toluene, 80 °C, 2. KOH, THF, rt; h) precursor 8, CuI, PPh3, Pd(PPh3)4, TEA, toluene, 80 °C; i) TFA, CH2Cl2, rt; j) 1. HCl, MeOH/CHCl3, rt, 2. NaBArF, MeOH.
Scheme 1: Overview of the synthesis of the guests A2 and A4. a) Pyrrole (4), BF3·Et2O, DDQ, CHCl3, rt; b) Zn(...
The preparation of the corresponding crown ether hosts (Scheme 2) involved an initial Williamson ether synthesis in which catechol (17) was first extended with 2-[2-(2-chloroethoxy)ethoxy]ethanol to diol 18, which was then converted in dibromide 19 by an Appel reaction. Macrocyclization of 19 with 3,4-dihydroxybenzaldehyde under “pseudo high-dilution” conditions, i.e., slow addition of the two reactants into a solution of Cs2CO3 in DMF at 100 °C provides the corresponding crown ether aldehyde 20. Porphyrin synthesis using 20 and pyrrole (4) following the Lindsey protocol [77] for A4 porphyrins gives the desired tetravalent porphyrin host as the free base 21, which is subsequently converted into the desired product C4 by metalation using zinc(II) acetate. Host C2 was synthesized according to the above-mentioned standard procedure [76] for trans-A2B2-porphyrins from 20 and mesityldipyrromethane 6 to form the divalent free base porphyrin 22. Final zinc insertion provides the desired host C2.
![[1860-5397-11-85-i2]](/bjoc/content/inline/1860-5397-11-85-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of crown ether hosts C4 and C2: a) K2CO3, LiBr, 17, 2-[2-(2-chloroethoxy)ethoxy]ethanol, DMF, 100 °C; b) CBr4, PPh3, CH2Cl2, rt; c) Cs2CO3, 3,4-dihydroxybenzaldehyde, DMF, 85 °C; d) 1. pyrrole (4), propionic acid, 140 °C, 2. Zn(OAc)2, MeOH/CHCl3, rt; e) 1. dipyrromethane (6), BF3·Et2O, DDQ, CHCl3, rt, 2. Zn(OAC)2, MeOH/CHCl3, rt.
Scheme 2: Synthesis of crown ether hosts C4 and C2: a) K2CO3, LiBr, 17, 2-[2-(2-chloroethoxy)ethoxy]ethanol, ...
For further detailed synthetic procedures and characterization data the reader is referred to Supporting Information File 1.
Formation and characterization of complexes
NMR spectroscopy of simple pseudorotaxanes prepared from crown ether wheels and secondary ammonium axles provides complexation-induced shift data, which can be easily interpreted and yield insight into complexation. Earlier experiences with divalent crown/ammonium pseudorotaxanes however also demonstrated that the NMR spectroscopic approach is often rather limited for more complex structures [78], as very complicated spectra are obtained with typically overlapping signals that prevent further (straightforward) analysis. Another complication, which makes the NMR analysis difficult, is the fact that the di- and tetravalent crown ethers C2 and C4 are achiral themselves, but become chiral, when complexed to axle components A2 and A4. Consequently, the signals for all methylene protons of the crown ethers split into two diastereotopic ones not only producing another set of signals, but also more complicated splitting patterns. Furthermore, the crown ethers are connected to the porphyrin core by single bonds, around which they can easily rotate in the non-complexed state. This rotation is, however, fixed upon complexation and two possible orientations of each of the crown ethers on its corresponding axle are possible. One can therefore expect a mixture of stereoisomers to form. In the simplest case, A2@C2, two enantiomers and one meso-form are expected to exist, which should result in two overlapping sets of signals. For the other three complexes, the situation is even more complicated. Therefore, a straightforward and easy analysis of the NMR spectra will likely be impossible.
In our earlier studies [37,78,79], however, electrospray ionization (ESI) mass spectrometry (MS) turned out to be a perfectly suited method to characterize the complexes present in solution. The formation of unspecific complexes as well as fragmentation upon ionization have been found to be quite limited so that the picture obtained from the mass spectra can be expected to provide realistic insights into the composition of the complexes present in solution. As all stereoisomers have the same elemental composition, their presence as a mixture does not obscure the mass spectrometric results. For these reasons, we describe our NMR spectroscopic data, but focus on ESI–MS of the complexes under study starting with the four possible combinations of A2 and A4 with monovalent dibenzo[24]crown-8 C1 as well as of C2 and C4 with monovalent dibenzylammonium A1 (Figure 2, top), followed by the results obtained for the multivalent 1:1 and 2:1 complexes A2@C2, A22@C4, A4@C22 and A4@C4 (Figure 2, bottom).
![[1860-5397-11-85-2]](/bjoc/content/figures/1860-5397-11-85-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Schematic representation of the host–guests complexes. Top: complexes A2@C12, A4@C14, A12@C2 and A14@C4, which are built from one multi- and several monovalent building blocks. Bottom: complexes A2@C2, A22@C4, A4@C22 and A4@C4, which are built from di- or tetravalent building blocks.
Figure 2: Schematic representation of the host–guests complexes. Top: complexes A2@C12, A4@C14, A12@C2 and A14...
[3]- and [5]pseudorotaxanes from monovalent building blocks
First the association of A2 and A4 with monovalent C1 as well as C2 and C4 with monovalent A1 was investigated and it can be seen that in all four cases successful complexation with the expected stoichiometry was achieved. For instance, upon addition of C1 to a 3 mM solution of A2 (Figure 3a) a continuous complexation, indicated by the appearance of a new set of signals due to slow exchange rates on the NMR-time scale, could be observed. Upon association the benzyl signals Hb/c shift downfield by approximately +0.3 ppm and split into two separate pair of signals, which is typical for a complexation of C1 with a dibenzylammonium moiety [36]. The aromatic signals of C1 H1/2 shift slightly upfield by −0.1 ppm and split as well. The signals of the crown ether region shift upfield by −0.05, −0.14, and −0.38 ppm due to complexation. An overstoichiometric addition of C1 results in no further association (see Figure 3a, inset), clearly proving the desired host–guest ratio in the supramolecular structure. Similar results are obtained for the other [3]- and [5]pseudorotaxanes (Figure 3b and Figure 4a,b). However, it should be noted that despite extensive titration experiments (see Supporting Information File 1 for details) a detailed analysis of the binding constants of these systems cannot be obtained as the binding constants are too high for a NMR-based method.
![[1860-5397-11-85-3]](/bjoc/content/figures/1860-5397-11-85-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR (500 MHz, 298 K, CD2Cl2, 3 mM) of a) C1 (top), A2@C12 (middle) and A2 (bottom); b) C1 (top), A4@C14 (middle) and A4 (bottom) showing clear evidence of the complexation. The red lines indicate the shift of the proton signals upon addition. The inserts show the titration curve of each complexation with the expected ratio of the complex formed.
Figure 3: 1H NMR (500 MHz, 298 K, CD2Cl2, 3 mM) of a) C1 (top), A2@C12 (middle) and A2 (bottom); b) C1 (top), ...
![[1860-5397-11-85-4]](/bjoc/content/figures/1860-5397-11-85-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR (500 MHz, 298 K, CD2Cl2, 3 mM) of a) C2 (top), A12@C2 (middle) and A1 (bottom) and b) C4 (top), A14@C4 (middle) and A1 (bottom) showing clear evidence of the complexation. The red lines indicate the shift of the proton signals upon addition. The inserts show the titration curve of each complexation with the expected ratio of the complex formed.
Figure 4: 1H NMR (500 MHz, 298 K, CD2Cl2, 3 mM) of a) C2 (top), A12@C2 (middle) and A1 (bottom) and b) C4 (to...
Guests A2 and A4 as well as the hosts C2 and C4 show typical absorption behavior for porphyrin-based molecules. All four have pronounced absorption maxima at around 420 nm (Soret band) and less intense absorption bands between 500 and 600 nm (Q-bands). However, A4 shows rather strong aggregation even in the µM concentration regime likely caused by electrostatic interactions mediated by the closely associated BArF counter-ions that are expected to be significant as rather non-polar solvents are being used. This aggregation results in a broad red-shifted absorption band. Upon complexation this aggregate is broken, resulting in the recovery of a typical sharp Soret band at 420 nm. Note that UV–vis titration shows no significant batho- or hypsochromic shift upon association (Figure 5) of neither di- and tetravalent guests A2 and A4 with monovalent host C1 nor of monovalent guest A1 to the di- and tetravalent hosts C2 and C4. The lack of such optical signature of the complexation event in the characteristic porphyrin absorption can be explained by the fact that the binding sites are electronically decoupled from the porphyrin core.
![[1860-5397-11-85-5]](/bjoc/content/figures/1860-5397-11-85-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Normalized UV–vis absorption spectra (CH2Cl2, 3 μM) of A2, A4, C2 and C4 and their complexes formed with the monovalent building blocks A1 and C1 showing no significant batho- or hypsochromic shift. Absorption spectrum of A4 was normalized to 0.5 because of the strong self-aggregation and the resulting broad Soret band.
Figure 5: Normalized UV–vis absorption spectra (CH2Cl2, 3 μM) of A2, A4, C2 and C4 and their complexes formed...
The [3]- and [5]pseudorotaxanes with the monovalent building blocks were further investigated by ESI-Q-TOF mass spectrometry. Separate solutions of hosts and guests were prepared (A1/C1: 4 mM, A2/C2: 2 mM, A4/C4: 1 mM all in CH2Cl2), and the same aliquots of the individual solutions were combined to obtain equal concentrations of ammonium ion functions and crown ether moieties in each solution. The solutions of the pseudorotaxanes were allowed to equilibrate for 24 hours at room temperature and diluted to 0.2 µM prior to analysis. The respective [3]- or [5]pseudorotaxanes could be detected for all mixtures (Figure 6). In the cases of the 1:2 and 1:4 mixtures of A2 and A4 with C1, respectively, the respective pseudorotaxanes A2@C12 and A4@C14 give rise to the second and third most abundant species (Figure 6a,b). One signal represents the desired doubly, respectively quadruply charged pseudorotaxane ([A2@C12]2+ at m/z 1094 and [A4@C14]4+ at m/z = 898). In addition, a second set of signals for the triply, respectively five-fold charged species ([A2@C12 + H]3+ at m/z = 729 and [A4@C14 + H]5+ at m/z = 719) could be observed. The most abundant species – most probably due to its high ESI response factor – is the one sodium ion containing molecular ion of C1 ([Na@C1]+ at m/z 471, see Supporting Information File 1). The spectra of the di- and tetravalent hosts C2 and C4 and the monovalent guest A1 show a more complex signal pattern (Figure 6c,d). In the mixture of divalent crown ether C2 with A1 three different species in a statistical distribution of 1:2:1 were detected: the host with two axles [A12@C2]2+ (m/z = 948), the host with one axle and one sodium ion [NaA1@C2]2+ (m/z = 861) and the host loaded with two sodium ions ([Na2@C2]2+ m/z = 773). This can be easily explained with the nature of the ESI spray process, which is known to cause the dissociation in multiply charged non-covalently bound complexes. The results of the NMR titrations, however, clearly indicate the doubly bound pseudorotaxane A12@C2 to be the most prominent species in solution (Figure 4a). The fact that the desired pseudorotaxane A12@C2 can be detected by mass spectrometry despite the likely dissociation of the multiply charged complex in the ion source shows that this technique gives reasonable results for determining the species present in solution. The 4:1 mixture of A1 and C4 gives rise to an even more complex signal pattern (Figure 6d). Due to the four binding sites of C4, there are numerous possibilities of A1 and sodium cations to bind. There are species with three or four guest ions detected with an approximately statistic distribution: [Na(4−x)A1x@C4]4+ (x = 1, 2, 3, 4) and [Na(3−y)A1y@C4]3+ (y = 1, 2, 3). The desired [5]pseudorotaxane is not very stable at the ionization conditions, but is nevertheless detected ([A14@C4]4+ at m/z = 737). As explained above, this shows that mass spectrometry gives a reasonable image of the species present in solution, because we already know from NMR titration studies that the [5]pseudorotaxane A14@C4 is the predominant species in solution (Figure 4b).
![[1860-5397-11-85-6]](/bjoc/content/figures/1860-5397-11-85-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: ESI-Q-TOF-MS spectra (CH2Cl2, 0.2 µM; left hand side) and respective experimental and calculated isotopic patterns of the desired [3]- or [5]pseudorotaxanes (right hand side): a) 1:2 mixture of A2 and C1, b) 1:4 mixture of A4 and C1, c) 2:1 mixture of A1 and C2, d) 4:1 mixture of A1 and C4. For reasons of clarity not all of the peaks are assigned (see Supporting Information File 1 for details).
Figure 6: ESI-Q-TOF-MS spectra (CH2Cl2, 0.2 µM; left hand side) and respective experimental and calculated is...
To summarize, all four desired [3]- or [5]pseudorotaxanes could be detected by mass spectrometry despite the likeliness of A12@C2 and A14@C4 to dissociate upon electrospray ionization. These results show that mass spectrometry should be a well suited method for the investigation of the multivalent pseudorotaxanes under study. These usually show much higher binding constants than the monovalent analogue and should therefore very likely survive the ionization process.
[2]- and [3]pseudorotaxanes from di- and tetravalent building blocks
Subsequently, we investigated the di- and tetravalent pseudorotaxanes formed between A2, A4, C2, and C4. As already mentioned above, NMR spectroscopy is limited for the given systems because of the numerous isomers that can be formed. However, some general conclusion can be made. In all four cases one can observe a shift of the benzylic protons Hb/c down field by approximately 0.5 ppm, which is typical for the threading in a crown ether/secondary ammonium ion binding motif. Furthermore, the signals for the crown ether region broaden significantly, which is in agreement with the assumption that upon complexation the number of signals increases because the methylene protons become diasterotopic and different supramolecular stereoisomers can form. However, based on the present NMR spectroscopy data (Figure 7 and Figure 8) one cannot exclude the formation of polymeric aggregates or only partially threaded structures. For this reason the formed complexes were analyzed in detail using mass spectrometry.
![[1860-5397-11-85-7]](/bjoc/content/figures/1860-5397-11-85-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: 1H NMR (500 MHz, 298 K, CD2Cl2, 1 mM) of a) C4 (top), A22@C4 (middle) and A2 (bottom); b) C2 (top), A2@C2 (middle) and A2 (bottom). Disappearance and shift of the signals (red lines) suggest complexation. Due to the presence of a complex stereoisomeric mixture only qualitative information of the complexation is possible.
Figure 7: 1H NMR (500 MHz, 298 K, CD2Cl2, 1 mM) of a) C4 (top), A22@C4 (middle) and A2 (bottom); b) C2 (top), ...
![[1860-5397-11-85-8]](/bjoc/content/figures/1860-5397-11-85-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: 1H NMR (500 MHz, 298 K, CD2Cl2, 1 mM) of a) C4 (top), A4@C4 (middle) and A4 (bottom) and b) C2 (top), A4@C22 (middle) and A4 (bottom). Disappearance and shift of the signals (red lines) suggest complexation. Due to the presence of a complex stereoisomeric mixture only qualitative information of the complexation is possible.
Figure 8: 1H NMR (500 MHz, 298 K, CD2Cl2, 1 mM) of a) C4 (top), A4@C4 (middle) and A4 (bottom) and b) C2 (top...
Comparing the absorption of the complexes (Figure 9), one can see that the tetravalent A4@C4 complex shows the strongest blue shift while the divalent A4@C22 shows almost no change in the spectrum (except breaking the A4 aggregate). The hypsochromic shift indicates a parallel alignment of the porphyrin moieties, which is in good agreement with the hypothesized structure. However, since the observed shifts are rather small the interactions, i.e., exciton coupling, between the two porphyrin chromophores seems to be rather weak.
![[1860-5397-11-85-9]](/bjoc/content/figures/1860-5397-11-85-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Normalized UV–vis absorption spectra (CH2Cl2, 2 μM) of the guests A2 and A4 (black), the hosts C2 and C4 (blue) and of the mixtures (red), showing a slight hypsochromic shift of the absorption maxima upon complexation.
Figure 9: Normalized UV–vis absorption spectra (CH2Cl2, 2 μM) of the guests A2 and A4 (black), the hosts C2 a...
For mass spectrometric analysis (ESI-Q-TOF MS) of the desired pseudorotaxanes separate solutions of hosts and guests were prepared (CH2Cl2, A2/C2: 0.6 mM, A4/C4: 0.3 mM). They were mixed in the respective 1:1, 1:2, and 2:1 molar ratios and allowed to equilibrate for 14 hours at 6 °C, after which no further changes in the mass spectra were observed and thus equilibrium was reached. The pseudorotaxane solutions were diluted to 0.2 µM prior to analysis. The respective mass spectra are shown in Figure 10. Guest A2 was combined with host C2 as well as C4 in 1:1 and 2:1 ratios, respectively. The expected pseudorotaxanes [A2@C2]2+ (m/z = 1396) and [A22@C4]4+ (m/z = 1185) are detected as the major species (Figure 10a,b). A species with only one guest A2 in host C4 [Na2A2@C4]4+ (m/z = 873) could also be detected but with very low intensity. This partly bound species A2@C4 could in principle allow formation of small oligomers, if present in solution. The fact that no oligomers could be detected and the very small abundance of the signal of the partly bound state [Na2A2@C4]4+ (m/z = 873) leads to the conclusion, that this partly bound pseudorotaxane is most probably a product of the electrospray ionization process.
![[1860-5397-11-85-10]](/bjoc/content/figures/1860-5397-11-85-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: ESI-Q-TOF-MS spectra (CH2Cl2, 0.2 µM; left hand side) and respective experimental and calculated isotopic patterns of the desired [2]- or [3]pseudorotaxanes (right hand side): a) 1:1 mixture of A2 and C2, b) 2:1 mixture of A2 and C4, c) 1:1 mixture of A4 and C4, d) 1:2 mixture of A4 and C2.
Figure 10: ESI-Q-TOF-MS spectra (CH2Cl2, 0.2 µM; left hand side) and respective experimental and calculated is...
In cases of the 1:1 mixture of A4 and C4 and the 1:2 mixture of A4 and C2 the desired pseudorotaxanes [A4@C4]4+ (m/z = 989) and [A4@C22]4+ (m/z = 1200) are the most abundant species and there are again only traces of the possible 1:1 pseudorotaxane [A4@C2]4+ (m/z = 825) detected (Figure 10c,d). As mentioned above, this is most probably a product of the ionization process. The free hosts C4 and C2 are detected in only small amounts or traces. Again, in both cases no oligomers are observed.
In summary, the formation of all desired multivalent pseudorotaxanes of building blocks A2, A4, C2, and C4 could be verified by mass spectrometry. The defined stoichiometry for the observed pseudorotaxanes in the gas phase ([A2@C2]2+, [A22@C4]4+, [A4@C4]4+, [A4@C22]4+), the only slight abundance of partly bound pseudorotaxanes ([Na2A2@C4]4+, [A4@C2]4+) and the absence of any oligomeric species gives clear evidence, that this specific binding situation is also present in solution.
Conclusion
The successful synthesis of di- and tetravalent porphyrin-based guests A2 and A4 as well as their complementary di- and tetravalent hosts C2 and C4 could be achieved. All four molecules show strong binding even to simple monovalent building blocks A1 and C1, respectively, which could be shown by NMR-titration experiments as well as mass spectrometry. Furthermore, the formation of the di- and tetravalent pseudorotaxanes A2@C2, A22@C4, A4@C22, and A4@C4 could be demonstrated qualitatively by NMR spectroscopy and was investigated in detail by mass spectrometry. Since the association constants in the monovalent cases are already too high to be determined by NMR-titration experiments, currently ongoing work is dealing with the daunting task to quantify the binding constants for the di- and tetravalent multiporphyrin complexes for example using isothermal calorimetry (ITC), in order to analyze the thermodynamics and kinetics of multivalent binding in these architectures in detail. In the future, we will continue to exploit the concept of complementary multivalent binding to program the increasingly complex self-assembly of multiple different chromophore components into functional supramolecular architectures.
Supporting Information
| Supporting Information File 1: Detailed synthetic procedures. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Steed, J. W.; Atwood, J. L. Supramolecular Chemistry; Wiley: Chichester, UK, 2007.
Return to citation in text: [1] -
Lehn, J.-M. Angew. Chem., Int. Ed. 1988, 27, 89–112. doi:10.1002/anie.198800891
Return to citation in text: [1] -
Desiraju, G. R. Angew. Chem., Int. Ed. 2007, 46, 8342–8356. doi:10.1002/anie.200700534
Return to citation in text: [1] -
Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022
Return to citation in text: [1] -
Bhosale, R.; Mišek, J.; Sakai, N.; Matile, S. Chem. Soc. Rev. 2010, 39, 138–149. doi:10.1039/B906115K
Return to citation in text: [1] -
Zhao, Y.; Sakai, F.; Su, L.; Liu, Y.; Wei, K.; Chen, G.; Jiang, M. Adv. Mater. 2013, 25, 5215–5256. doi:10.1002/adma.201302215
Return to citation in text: [1] -
Cametti, M.; Crousse, B.; Metrangolo, P.; Milani, R.; Resnati, G. Chem. Soc. Rev. 2012, 41, 31–42. doi:10.1039/C1CS15084G
Return to citation in text: [1] -
Valeur, B.; Leray, I. Coord. Chem. Rev. 2000, 205, 3–40. doi:10.1016/S0010-8545(00)00246-0
Return to citation in text: [1] -
Flink, S.; van Veggel, F. C. J. M.; Reinhoudt, D. N. J. Phys. Chem. B 1999, 103, 6515–6520. doi:10.1021/jp990014v
Return to citation in text: [1] -
Beer, P. D.; Gale, P. A. Angew. Chem., Int. Ed. 2001, 40, 486–516. doi:10.1002/1521-3773(20010202)40:3<486::AID-ANIE486>3.0.CO;2-P
Return to citation in text: [1] -
Martínez-Máñez, R.; Sancenón, F. Chem. Rev. 2003, 103, 4419–4476. doi:10.1021/cr010421e
Return to citation in text: [1] -
Llinares, J. M.; Powell, D.; Bowman-James, K. Coord. Chem. Rev. 2003, 240, 57–75. doi:10.1016/S0010-8545(03)00019-5
Return to citation in text: [1] -
Chen, Y.; Liu, Y. Chem. Soc. Rev. 2010, 39, 495–505. doi:10.1039/B816354P
Return to citation in text: [1] -
Feiters, M. C.; Rowan, A. E.; Nolte, R. J. M. Chem. Soc. Rev. 2000, 29, 375–384. doi:10.1039/a804252g
Return to citation in text: [1] -
Wang, L.; Li, L.-L.; Ma, H. L.; Wang, H. Chin. Chem. Lett. 2013, 24, 351–358. doi:10.1016/j.cclet.2013.03.018
Return to citation in text: [1] -
Huskens, J. Curr. Opin. Chem. Biol. 2006, 10, 537–543. doi:10.1016/j.cbpa.2006.09.007
Return to citation in text: [1] -
Vigneron, J.-P. Molecules 1999, 4, 180–203. doi:10.3390/40700180
Return to citation in text: [1] -
Diederich, F.; Stang, P. J. Templated Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2000.
Return to citation in text: [1] -
Whitesides, G. M.; Mathias, J. P.; Seto, C. T. Science 1991, 254, 1312–1319. doi:10.1126/science.1962191
Return to citation in text: [1] -
Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784–5814. doi:10.1021/cr100357h
Return to citation in text: [1] -
He, Z.; Jiang, W.; Schalley, C. A. Chem. Soc. Rev. 2015, 44, 779–789. doi:10.1039/C4CS00305E
Return to citation in text: [1] -
Mammen, M.; Choi, S.-K.; Whitesides, G. M. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. doi:10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3
Return to citation in text: [1] -
Mulder, A.; Huskens, J.; Reinhoudt, D. N. Org. Biomol. Chem. 2004, 2, 3409–3424. doi:10.1039/b413971b
Return to citation in text: [1] -
Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Angew. Chem., Int. Ed. 2012, 51, 10472–10498. doi:10.1002/anie.201201114
Return to citation in text: [1] -
Hunter, C. A.; Anderson, H. L. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. doi:10.1002/anie.200902490
Return to citation in text: [1] -
Ercolani, G.; Schiaffino, L. Angew. Chem., Int. Ed. 2011, 50, 1762–1768. doi:10.1002/anie.201004201
Return to citation in text: [1] -
Papp, I.; Sieben, C.; Sisson, A. L.; Kostka, J.; Böttcher, C.; Ludwig, K.; Herrmann, A.; Haag, R. ChemBioChem 2011, 12, 887–895. doi:10.1002/cbic.201000776
Return to citation in text: [1] -
Kitov, P. I.; Sadowska, J. M.; Mulvey, G.; Armstrong, G. D.; Ling, H.; Pannu, N. S.; Read, R. J.; Bundle, D. R. Nature 2000, 403, 669–672. doi:10.1038/35001095
Return to citation in text: [1] -
Simanek, E. E.; McGarvey, G. J.; Jablonowski, J. A.; Wong, C.-H. Chem. Rev. 1998, 98, 833–862. doi:10.1021/cr940226i
Return to citation in text: [1] -
Badjić, J. D.; Balzani, V.; Credi, A.; Lowe, J. N.; Silvi, S.; Stoddart, J. F. Chem. – Eur. J. 2004, 10, 1926–1935. doi:10.1002/chem.200305687
Return to citation in text: [1] -
Balzani, V.; Clemente-León, M.; Credi, A.; Lowe, J. N.; Badjić, J. D.; Stoddart, J. F.; Williams, D. J. Chem. – Eur. J. 2003, 9, 5348–5360. doi:10.1002/chem.200304979
Return to citation in text: [1] -
O’Sullivan, M. C.; Sprafke, J. K.; Kondratuk, D. V.; Rinfray, C.; Claridge, T. D. W.; Saywell, A.; Blunt, M. O.; O’Shea, J. N.; Beton, P. H.; Malfois, M.; Anderson, H. L. Nature 2011, 469, 72–75. doi:10.1038/nature09683
Return to citation in text: [1] -
Kondratuk, D. V.; Perdigao, L. M. A.; O'Sullivan, M. C.; Svatek, S.; Smith, G.; O'Shea, J. N.; Beton, P. H.; Anderson, H. L. Angew. Chem., Int. Ed. 2012, 51, 6696–6699. doi:10.1002/anie.201202870
Return to citation in text: [1] -
Timko, J. M.; Moore, S. S.; Walba, D. M.; Hiberty, P. C.; Cram, D. J. J. Am. Chem. Soc. 1977, 99, 4207–4219. doi:10.1021/ja00455a001
Return to citation in text: [1] -
Ashton, P. R.; Campbell, P. J.; Glink, P. T.; Philp, D.; Spencer, N.; Stoddart, J. F.; Chrystal, E. J. T.; Menzer, S.; Williams, D. J.; Tasker, P. A. Angew. Chem., Int. Ed. Engl. 1995, 34, 1865–1869. doi:10.1002/anie.199518651
Return to citation in text: [1] -
Ashton, P. R.; Chrystal, E. J. T.; Glink, P. T.; Menzer, S.; Schiavo, C.; Spencer, N.; Stoddart, J. F.; Tasker, P. A.; White, A. J. P.; Williams, D. J. Chem. – Eur. J. 1996, 2, 709–728. doi:10.1002/chem.19960020616
Return to citation in text: [1] [2] -
Jiang, W.; Schalley, C. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10425–10429. doi:10.1073/pnas.0809512106
Return to citation in text: [1] [2] -
Clifford, T.; Abushamleh, A.; Busch, D. H. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 4830–4836. doi:10.1073/pnas.062639799
Return to citation in text: [1] -
Dong, S.; Zheng, B.; Wang, F.; Huang, F. Acc. Chem. Res. 2014, 47, 1982–1994. doi:10.1021/ar5000456
Return to citation in text: [1] -
Amabilino, D. B.; Stoddart, J. F. Chem. Rev. 1995, 95, 2725–2828. doi:10.1021/cr00040a005
Return to citation in text: [1] -
Yamaguchi, N.; Gibson, W. H. Chem. Commun. 1999, 789–790. doi:10.1039/a901044k
Return to citation in text: [1] -
Jiang, W.; Sattler, D.; Rissanen, K.; Schalley, C. A. Org. Lett. 2011, 13, 4502–4505. doi:10.1021/ol201618f
Return to citation in text: [1] -
Balzani, V.; Credi, A.; Raymo, F. M.; Stoddart, J. F. Angew. Chem., Int. Ed. 2000, 39, 3348–3391. doi:10.1002/1521-3773(20001002)39:19<3348::AID-ANIE3348>3.0.CO;2-X
Return to citation in text: [1] -
Kay, E. R.; Leigh, D. A.; Zerbetto, F. Angew. Chem., Int. Ed. 2007, 46, 72–191. doi:10.1002/anie.200504313
Return to citation in text: [1] -
Vukotic, V. N.; Loeb, S. J. Chem. Soc. Rev. 2012, 41, 5896–5906. doi:10.1039/c2cs35141b
Return to citation in text: [1] -
Liu, Y.; Flood, A. H.; Bonvallet, P. A.; Vignon, S. A.; Northrop, B. H.; Tseng, H.-R.; Jeppesen, J. O.; Huang, T. J.; Brough, B.; Baller, M.; Magonov, S.; Solares, S. D.; Goddard, W. A.; Ho, C.-M.; Stoddart, J. F. J. Am. Chem. Soc. 2005, 127, 9745–9759. doi:10.1021/ja051088p
Return to citation in text: [1] -
Coskun, A.; Banaszak, M.; Astumian, R. D.; Stoddart, J. F.; Grzybowski, B. A. Chem. Soc. Rev. 2012, 41, 19–30. doi:10.1039/C1CS15262A
Return to citation in text: [1] -
Bruns, C. J.; Stoddart, J. F. Acc. Chem. Res. 2014, 47, 2186–2199. doi:10.1021/ar500138u
Return to citation in text: [1] -
Romuald, C.; Busseron, E.; Coutrot, F. J. Org. Chem. 2010, 75, 6516–6531. doi:10.1021/jo101234u
Return to citation in text: [1] -
Clark, P. G.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2009, 131, 13631–13633. doi:10.1021/ja905924u
Return to citation in text: [1] -
Milgrom, L. R. The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds; Oxford University Press: Oxford, UK, 2001.
Return to citation in text: [1] -
Ishii, K.; Kobayashi, N. In The Porphyrin Handbook; Kadish, K.; Guilard, R.; Smith, K. M., Eds.; Academic Press: Amsterdam, 2003; pp 1–42. doi:10.1016/B978-0-08-092390-1.50007-2
Return to citation in text: [1] -
Durot, S.; Taesch, J.; Heitz, V. Chem. Rev. 2014, 114, 8542–8578. doi:10.1021/cr400673y
Return to citation in text: [1] -
Mandal, S.; Rahaman, M.; Sadhu, S.; Nayak, S. K.; Patra, A. J. Phys. Chem. C 2013, 117, 3069–3077. doi:10.1021/jp3100188
Return to citation in text: [1] -
Liu, Y.; Huang, Z.; Liu, K.; Kelgtermans, H.; Dehaen, W.; Wang, Z.; Zhang, X. Polym. Chem. 2014, 5, 53–56. doi:10.1039/C3PY01036H
Return to citation in text: [1] -
Fathalla, M.; Strutt, N. L.; Barnes, J. C.; Stern, C. L.; Ke, C.; Stoddart, J. F. Eur. J. Org. Chem. 2014, 2873–2877. doi:10.1002/ejoc.201402018
Return to citation in text: [1] -
Watanabe, K.; Kitagishi, H.; Kano, K. Angew. Chem., Int. Ed. 2013, 52, 6894–6897. doi:10.1002/anie.201302470
Return to citation in text: [1] -
Puglisi, A.; Purrello, R.; Rizzarelli, E.; Sortino, S.; Vecchio, G. New J. Chem. 2007, 31, 1499–1506. doi:10.1039/b703680a
Return to citation in text: [1] -
Králová, J.; Kejik, Z.; Břiza, T.; Poučková, P.; Král, A.; Martásek, P.; Král, V. J. Med. Chem. 2010, 53, 128–138. doi:10.1021/jm9007278
Return to citation in text: [1] -
Kiba, T.; Suzuki, H.; Hosokawa, K.; Kobayashi, H.; Baba, S.; Kakuchi, T.; Sato, S.-I. J. Phys. Chem. B 2009, 113, 11560–11563. doi:10.1021/jp905904h
Return to citation in text: [1] -
Kano, K.; Kitagishi, H.; Dagallier, C.; Kodera, M.; Matsuo, T.; Hayashi, T.; Hisaeda, Y.; Hirota, S. Inorg. Chem. 2006, 45, 4448–4460. doi:10.1021/ic060137b
Return to citation in text: [1] -
Kim, Y.-H.; Hong, J.-I. Chem. Commun. 2002, 512–513. doi:10.1039/b109596j
Return to citation in text: [1] -
D'Souza, F.; Chitta, R.; Gadde, S.; Zandler, M. E.; Sandanayaka, A. S. D.; Araki, Y.; Ito, O. Chem. Commun. 2005, 1279–1281. doi:10.1039/b416736h
Return to citation in text: [1] -
Even, P.; Boitrel, B. Coord. Chem. Rev. 2006, 250, 519–541. doi:10.1016/j.ccr.2005.09.003
Return to citation in text: [1] -
Zhang, H.; Liu, Q.; Li, J.; Qu, D.-H. Org. Lett. 2013, 15, 338–341. doi:10.1021/ol3032686
Return to citation in text: [1] -
Jahan, M.; Safari, N.; Khosravi, H.; Moghimi, A.; Notash, B. Polyhedron 2005, 24, 1682–1688. doi:10.1016/j.poly.2005.04.033
Return to citation in text: [1] -
Imahori, H.; Fukuzumi, S. Adv. Funct. Mater. 2004, 14, 525–536. doi:10.1002/adfm.200305172
Return to citation in text: [1] -
Imahori, H.; Sakata, Y. Eur. J. Org. Chem. 1999, 2445–2457. doi:10.1002/(SICI)1099-0690(199910)1999:10<2445::AID-EJOC2445>3.0.CO;2-G
Return to citation in text: [1] -
Yong, C.-K.; Parkinson, P.; Kondratuk, D. V.; Chen, W.-H.; Stannard, A.; Summerfield, A.; Sprafke, J. K.; O'Sullivan, M. C.; Beton, P. H.; Anderson, H. L.; Herz, L. M. Chem. Sci. 2015, 6, 181–189. doi:10.1039/C4SC02424A
Return to citation in text: [1] -
Parkinson, P.; Knappke, C. E. I.; Kamonsutthipaijit, N.; Sirithip, K.; Matichak, J. D.; Anderson, H. L.; Herz, L. M. J. Am. Chem. Soc. 2014, 136, 8217–8220. doi:10.1021/ja504730j
Return to citation in text: [1] -
Liu, J.-Y.; Huang, Y.; Menting, R.; Röder, B.; Ermilov, E. A.; Ng, D. K. P. Chem. Commun. 2013, 49, 2998–3000. doi:10.1039/c3cc00262d
Return to citation in text: [1] -
Menting, R.; Lau, J. T. F.; Xu, H.; Ng, D. K. P.; Röder, B.; Ermilov, E. A. Chem. Commun. 2012, 48, 4597–4599. doi:10.1039/c2cc30286a
Return to citation in text: [1] -
Burrell, A. K.; Officer, D. L.; Plieger, P. G.; Reid, D. C. W. Chem. Rev. 2001, 101, 2751–2796. doi:10.1021/cr0000426
Return to citation in text: [1] -
Yamada, Y.; Okamoto, M.; Furukawa, K.; Kato, T.; Tanaka, K. Angew. Chem., Int. Ed. 2012, 51, 709–713. doi:10.1002/anie.201107104
Return to citation in text: [1] -
van Nostrum, C. F.; Picken, S. J.; Schouten, A.-J.; Nolte, R. J. M. J. Am. Chem. Soc. 1995, 117, 9957–9965. doi:10.1021/ja00145a004
Return to citation in text: [1] -
Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836. doi:10.1021/jo00381a022
Return to citation in text: [1] [2] -
Geier, G. R., III; Lindsey, J. S. Tetrahedron 2004, 60, 11435–11444. doi:10.1016/j.tet.2004.09.081
Return to citation in text: [1] [2] -
Jiang, W.; Schäfer, A.; Mohr, P. C.; Schalley, C. A. J. Am. Chem. Soc. 2010, 132, 2309–2320. doi:10.1021/ja9101369
Return to citation in text: [1] [2] -
Jiang, W.; Schalley, C. A. J. Mass Spectrom. 2010, 45, 788–798. doi:10.1002/jms.1769
Return to citation in text: [1]
| 76. | Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836. doi:10.1021/jo00381a022 |
| 78. | Jiang, W.; Schäfer, A.; Mohr, P. C.; Schalley, C. A. J. Am. Chem. Soc. 2010, 132, 2309–2320. doi:10.1021/ja9101369 |
| 37. | Jiang, W.; Schalley, C. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10425–10429. doi:10.1073/pnas.0809512106 |
| 78. | Jiang, W.; Schäfer, A.; Mohr, P. C.; Schalley, C. A. J. Am. Chem. Soc. 2010, 132, 2309–2320. doi:10.1021/ja9101369 |
| 79. | Jiang, W.; Schalley, C. A. J. Mass Spectrom. 2010, 45, 788–798. doi:10.1002/jms.1769 |
| 1. | Steed, J. W.; Atwood, J. L. Supramolecular Chemistry; Wiley: Chichester, UK, 2007. |
| 13. | Chen, Y.; Liu, Y. Chem. Soc. Rev. 2010, 39, 495–505. doi:10.1039/B816354P |
| 14. | Feiters, M. C.; Rowan, A. E.; Nolte, R. J. M. Chem. Soc. Rev. 2000, 29, 375–384. doi:10.1039/a804252g |
| 15. | Wang, L.; Li, L.-L.; Ma, H. L.; Wang, H. Chin. Chem. Lett. 2013, 24, 351–358. doi:10.1016/j.cclet.2013.03.018 |
| 16. | Huskens, J. Curr. Opin. Chem. Biol. 2006, 10, 537–543. doi:10.1016/j.cbpa.2006.09.007 |
| 17. | Vigneron, J.-P. Molecules 1999, 4, 180–203. doi:10.3390/40700180 |
| 32. | O’Sullivan, M. C.; Sprafke, J. K.; Kondratuk, D. V.; Rinfray, C.; Claridge, T. D. W.; Saywell, A.; Blunt, M. O.; O’Shea, J. N.; Beton, P. H.; Malfois, M.; Anderson, H. L. Nature 2011, 469, 72–75. doi:10.1038/nature09683 |
| 33. | Kondratuk, D. V.; Perdigao, L. M. A.; O'Sullivan, M. C.; Svatek, S.; Smith, G.; O'Shea, J. N.; Beton, P. H.; Anderson, H. L. Angew. Chem., Int. Ed. 2012, 51, 6696–6699. doi:10.1002/anie.201202870 |
| 7. | Cametti, M.; Crousse, B.; Metrangolo, P.; Milani, R.; Resnati, G. Chem. Soc. Rev. 2012, 41, 31–42. doi:10.1039/C1CS15084G |
| 8. | Valeur, B.; Leray, I. Coord. Chem. Rev. 2000, 205, 3–40. doi:10.1016/S0010-8545(00)00246-0 |
| 9. | Flink, S.; van Veggel, F. C. J. M.; Reinhoudt, D. N. J. Phys. Chem. B 1999, 103, 6515–6520. doi:10.1021/jp990014v |
| 10. | Beer, P. D.; Gale, P. A. Angew. Chem., Int. Ed. 2001, 40, 486–516. doi:10.1002/1521-3773(20010202)40:3<486::AID-ANIE486>3.0.CO;2-P |
| 11. | Martínez-Máñez, R.; Sancenón, F. Chem. Rev. 2003, 103, 4419–4476. doi:10.1021/cr010421e |
| 12. | Llinares, J. M.; Powell, D.; Bowman-James, K. Coord. Chem. Rev. 2003, 240, 57–75. doi:10.1016/S0010-8545(03)00019-5 |
| 34. | Timko, J. M.; Moore, S. S.; Walba, D. M.; Hiberty, P. C.; Cram, D. J. J. Am. Chem. Soc. 1977, 99, 4207–4219. doi:10.1021/ja00455a001 |
| 3. | Desiraju, G. R. Angew. Chem., Int. Ed. 2007, 46, 8342–8356. doi:10.1002/anie.200700534 |
| 4. | Hirst, A. R.; Escuder, B.; Miravet, J. F.; Smith, D. K. Angew. Chem., Int. Ed. 2008, 47, 8002–8018. doi:10.1002/anie.200800022 |
| 5. | Bhosale, R.; Mišek, J.; Sakai, N.; Matile, S. Chem. Soc. Rev. 2010, 39, 138–149. doi:10.1039/B906115K |
| 6. | Zhao, Y.; Sakai, F.; Su, L.; Liu, Y.; Wei, K.; Chen, G.; Jiang, M. Adv. Mater. 2013, 25, 5215–5256. doi:10.1002/adma.201302215 |
| 29. | Simanek, E. E.; McGarvey, G. J.; Jablonowski, J. A.; Wong, C.-H. Chem. Rev. 1998, 98, 833–862. doi:10.1021/cr940226i |
| 2. | Lehn, J.-M. Angew. Chem., Int. Ed. 1988, 27, 89–112. doi:10.1002/anie.198800891 |
| 30. | Badjić, J. D.; Balzani, V.; Credi, A.; Lowe, J. N.; Silvi, S.; Stoddart, J. F. Chem. – Eur. J. 2004, 10, 1926–1935. doi:10.1002/chem.200305687 |
| 31. | Balzani, V.; Clemente-León, M.; Credi, A.; Lowe, J. N.; Badjić, J. D.; Stoddart, J. F.; Williams, D. J. Chem. – Eur. J. 2003, 9, 5348–5360. doi:10.1002/chem.200304979 |
| 22. | Mammen, M.; Choi, S.-K.; Whitesides, G. M. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. doi:10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3 |
| 23. | Mulder, A.; Huskens, J.; Reinhoudt, D. N. Org. Biomol. Chem. 2004, 2, 3409–3424. doi:10.1039/b413971b |
| 24. | Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Angew. Chem., Int. Ed. 2012, 51, 10472–10498. doi:10.1002/anie.201201114 |
| 27. | Papp, I.; Sieben, C.; Sisson, A. L.; Kostka, J.; Böttcher, C.; Ludwig, K.; Herrmann, A.; Haag, R. ChemBioChem 2011, 12, 887–895. doi:10.1002/cbic.201000776 |
| 20. | Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784–5814. doi:10.1021/cr100357h |
| 21. | He, Z.; Jiang, W.; Schalley, C. A. Chem. Soc. Rev. 2015, 44, 779–789. doi:10.1039/C4CS00305E |
| 28. | Kitov, P. I.; Sadowska, J. M.; Mulvey, G.; Armstrong, G. D.; Ling, H.; Pannu, N. S.; Read, R. J.; Bundle, D. R. Nature 2000, 403, 669–672. doi:10.1038/35001095 |
| 19. | Whitesides, G. M.; Mathias, J. P.; Seto, C. T. Science 1991, 254, 1312–1319. doi:10.1126/science.1962191 |
| 36. | Ashton, P. R.; Chrystal, E. J. T.; Glink, P. T.; Menzer, S.; Schiavo, C.; Spencer, N.; Stoddart, J. F.; Tasker, P. A.; White, A. J. P.; Williams, D. J. Chem. – Eur. J. 1996, 2, 709–728. doi:10.1002/chem.19960020616 |
| 18. | Diederich, F.; Stang, P. J. Templated Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2000. |
| 25. | Hunter, C. A.; Anderson, H. L. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. doi:10.1002/anie.200902490 |
| 26. | Ercolani, G.; Schiaffino, L. Angew. Chem., Int. Ed. 2011, 50, 1762–1768. doi:10.1002/anie.201004201 |
| 43. | Balzani, V.; Credi, A.; Raymo, F. M.; Stoddart, J. F. Angew. Chem., Int. Ed. 2000, 39, 3348–3391. doi:10.1002/1521-3773(20001002)39:19<3348::AID-ANIE3348>3.0.CO;2-X |
| 44. | Kay, E. R.; Leigh, D. A.; Zerbetto, F. Angew. Chem., Int. Ed. 2007, 46, 72–191. doi:10.1002/anie.200504313 |
| 45. | Vukotic, V. N.; Loeb, S. J. Chem. Soc. Rev. 2012, 41, 5896–5906. doi:10.1039/c2cs35141b |
| 35. | Ashton, P. R.; Campbell, P. J.; Glink, P. T.; Philp, D.; Spencer, N.; Stoddart, J. F.; Chrystal, E. J. T.; Menzer, S.; Williams, D. J.; Tasker, P. A. Angew. Chem., Int. Ed. Engl. 1995, 34, 1865–1869. doi:10.1002/anie.199518651 |
| 36. | Ashton, P. R.; Chrystal, E. J. T.; Glink, P. T.; Menzer, S.; Schiavo, C.; Spencer, N.; Stoddart, J. F.; Tasker, P. A.; White, A. J. P.; Williams, D. J. Chem. – Eur. J. 1996, 2, 709–728. doi:10.1002/chem.19960020616 |
| 37. | Jiang, W.; Schalley, C. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 10425–10429. doi:10.1073/pnas.0809512106 |
| 38. | Clifford, T.; Abushamleh, A.; Busch, D. H. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 4830–4836. doi:10.1073/pnas.062639799 |
| 39. | Dong, S.; Zheng, B.; Wang, F.; Huang, F. Acc. Chem. Res. 2014, 47, 1982–1994. doi:10.1021/ar5000456 |
| 40. | Amabilino, D. B.; Stoddart, J. F. Chem. Rev. 1995, 95, 2725–2828. doi:10.1021/cr00040a005 |
| 41. | Yamaguchi, N.; Gibson, W. H. Chem. Commun. 1999, 789–790. doi:10.1039/a901044k |
| 42. | Jiang, W.; Sattler, D.; Rissanen, K.; Schalley, C. A. Org. Lett. 2011, 13, 4502–4505. doi:10.1021/ol201618f |
| 77. | Geier, G. R., III; Lindsey, J. S. Tetrahedron 2004, 60, 11435–11444. doi:10.1016/j.tet.2004.09.081 |
| 77. | Geier, G. R., III; Lindsey, J. S. Tetrahedron 2004, 60, 11435–11444. doi:10.1016/j.tet.2004.09.081 |
| 74. | Yamada, Y.; Okamoto, M.; Furukawa, K.; Kato, T.; Tanaka, K. Angew. Chem., Int. Ed. 2012, 51, 709–713. doi:10.1002/anie.201107104 |
| 75. | van Nostrum, C. F.; Picken, S. J.; Schouten, A.-J.; Nolte, R. J. M. J. Am. Chem. Soc. 1995, 117, 9957–9965. doi:10.1021/ja00145a004 |
| 76. | Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836. doi:10.1021/jo00381a022 |
| 53. | Durot, S.; Taesch, J.; Heitz, V. Chem. Rev. 2014, 114, 8542–8578. doi:10.1021/cr400673y |
| 54. | Mandal, S.; Rahaman, M.; Sadhu, S.; Nayak, S. K.; Patra, A. J. Phys. Chem. C 2013, 117, 3069–3077. doi:10.1021/jp3100188 |
| 55. | Liu, Y.; Huang, Z.; Liu, K.; Kelgtermans, H.; Dehaen, W.; Wang, Z.; Zhang, X. Polym. Chem. 2014, 5, 53–56. doi:10.1039/C3PY01036H |
| 56. | Fathalla, M.; Strutt, N. L.; Barnes, J. C.; Stern, C. L.; Ke, C.; Stoddart, J. F. Eur. J. Org. Chem. 2014, 2873–2877. doi:10.1002/ejoc.201402018 |
| 57. | Watanabe, K.; Kitagishi, H.; Kano, K. Angew. Chem., Int. Ed. 2013, 52, 6894–6897. doi:10.1002/anie.201302470 |
| 58. | Puglisi, A.; Purrello, R.; Rizzarelli, E.; Sortino, S.; Vecchio, G. New J. Chem. 2007, 31, 1499–1506. doi:10.1039/b703680a |
| 59. | Králová, J.; Kejik, Z.; Břiza, T.; Poučková, P.; Král, A.; Martásek, P.; Král, V. J. Med. Chem. 2010, 53, 128–138. doi:10.1021/jm9007278 |
| 60. | Kiba, T.; Suzuki, H.; Hosokawa, K.; Kobayashi, H.; Baba, S.; Kakuchi, T.; Sato, S.-I. J. Phys. Chem. B 2009, 113, 11560–11563. doi:10.1021/jp905904h |
| 61. | Kano, K.; Kitagishi, H.; Dagallier, C.; Kodera, M.; Matsuo, T.; Hayashi, T.; Hisaeda, Y.; Hirota, S. Inorg. Chem. 2006, 45, 4448–4460. doi:10.1021/ic060137b |
| 62. | Kim, Y.-H.; Hong, J.-I. Chem. Commun. 2002, 512–513. doi:10.1039/b109596j |
| 63. | D'Souza, F.; Chitta, R.; Gadde, S.; Zandler, M. E.; Sandanayaka, A. S. D.; Araki, Y.; Ito, O. Chem. Commun. 2005, 1279–1281. doi:10.1039/b416736h |
| 64. | Even, P.; Boitrel, B. Coord. Chem. Rev. 2006, 250, 519–541. doi:10.1016/j.ccr.2005.09.003 |
| 65. | Zhang, H.; Liu, Q.; Li, J.; Qu, D.-H. Org. Lett. 2013, 15, 338–341. doi:10.1021/ol3032686 |
| 66. | Jahan, M.; Safari, N.; Khosravi, H.; Moghimi, A.; Notash, B. Polyhedron 2005, 24, 1682–1688. doi:10.1016/j.poly.2005.04.033 |
| 67. | Imahori, H.; Fukuzumi, S. Adv. Funct. Mater. 2004, 14, 525–536. doi:10.1002/adfm.200305172 |
| 68. | Imahori, H.; Sakata, Y. Eur. J. Org. Chem. 1999, 2445–2457. doi:10.1002/(SICI)1099-0690(199910)1999:10<2445::AID-EJOC2445>3.0.CO;2-G |
| 69. | Yong, C.-K.; Parkinson, P.; Kondratuk, D. V.; Chen, W.-H.; Stannard, A.; Summerfield, A.; Sprafke, J. K.; O'Sullivan, M. C.; Beton, P. H.; Anderson, H. L.; Herz, L. M. Chem. Sci. 2015, 6, 181–189. doi:10.1039/C4SC02424A |
| 70. | Parkinson, P.; Knappke, C. E. I.; Kamonsutthipaijit, N.; Sirithip, K.; Matichak, J. D.; Anderson, H. L.; Herz, L. M. J. Am. Chem. Soc. 2014, 136, 8217–8220. doi:10.1021/ja504730j |
| 71. | Liu, J.-Y.; Huang, Y.; Menting, R.; Röder, B.; Ermilov, E. A.; Ng, D. K. P. Chem. Commun. 2013, 49, 2998–3000. doi:10.1039/c3cc00262d |
| 72. | Menting, R.; Lau, J. T. F.; Xu, H.; Ng, D. K. P.; Röder, B.; Ermilov, E. A. Chem. Commun. 2012, 48, 4597–4599. doi:10.1039/c2cc30286a |
| 73. | Burrell, A. K.; Officer, D. L.; Plieger, P. G.; Reid, D. C. W. Chem. Rev. 2001, 101, 2751–2796. doi:10.1021/cr0000426 |
| 46. | Liu, Y.; Flood, A. H.; Bonvallet, P. A.; Vignon, S. A.; Northrop, B. H.; Tseng, H.-R.; Jeppesen, J. O.; Huang, T. J.; Brough, B.; Baller, M.; Magonov, S.; Solares, S. D.; Goddard, W. A.; Ho, C.-M.; Stoddart, J. F. J. Am. Chem. Soc. 2005, 127, 9745–9759. doi:10.1021/ja051088p |
| 47. | Coskun, A.; Banaszak, M.; Astumian, R. D.; Stoddart, J. F.; Grzybowski, B. A. Chem. Soc. Rev. 2012, 41, 19–30. doi:10.1039/C1CS15262A |
| 48. | Bruns, C. J.; Stoddart, J. F. Acc. Chem. Res. 2014, 47, 2186–2199. doi:10.1021/ar500138u |
| 49. | Romuald, C.; Busseron, E.; Coutrot, F. J. Org. Chem. 2010, 75, 6516–6531. doi:10.1021/jo101234u |
| 50. | Clark, P. G.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2009, 131, 13631–13633. doi:10.1021/ja905924u |
| 51. | Milgrom, L. R. The Colours of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds; Oxford University Press: Oxford, UK, 2001. |
| 52. | Ishii, K.; Kobayashi, N. In The Porphyrin Handbook; Kadish, K.; Guilard, R.; Smith, K. M., Eds.; Academic Press: Amsterdam, 2003; pp 1–42. doi:10.1016/B978-0-08-092390-1.50007-2 |
© 2015 Lohse et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)