
Abstract
A series of pyrroles functionalized in the 3-position with p-dimethoxybenzene via various linkers (CH2, CH2CH2, CH=CH, C≡C) has been synthesized. Their electronic properties have been deduced from 1H NMR, 13C NMR, and UV–vis spectra to detect possible interactions between the two aromatic subunits. The extent of conjugation between the subunits is largely controlled by the nature of the linker, with the largest conjugation found with the trans-ethene linker and the weakest with the aliphatic linkers. DFT calculations revealed substantial changes in the HOMO–LUMO gap that correlated with the extent of conjugation found experimentally. The results of this work are expected to open up for use of the investigated compounds as components of redox-active materials in sustainable, organic electrical energy storage devices.
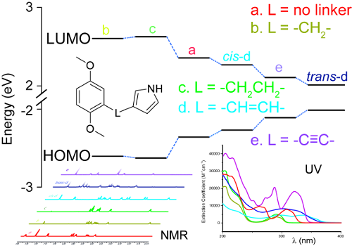
Graphical Abstract
Introduction
Quinone–pyrrole dyads have attracted interest in various applications due to the possibility of modulating the electronic interaction between the two subunits, with porphyrin–quinone dyads being well-known examples [1-3]. Recently, we have shown the suitability of the quinone–hydroquinone redox couple (Figure 1) as the redox active and capacity carrying component in conducting redox polymers (CRPs) [4,5]. To further investigate the interaction between the molecular components in these systems, a series of compounds with different linkers between the pyrrole and hydroquinone subunits was designed [6]. Although it is known that N-protected pyrrole (Py) can be selectively functionalized at the β-position [7], we found the published procedures to be unsuitable for our purpose (vide infra). Therefore, we have developed improved procedures for the synthesis of quinone–pyrrole dyads with a variety of linkers between the two subunits.
![[1860-5397-12-10-1]](/bjoc/content/figures/1860-5397-12-10-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of pyrrole/hydroquinone derivatives 3-(2,5-dimethoxyphenyl)-1H-pyrrole (1) and 3-(1,4-dihydroxyphenyl)-1H-pyrrole (2) [5].
Figure 1: Structure of pyrrole/hydroquinone derivatives 3-(2,5-dimethoxyphenyl)-1H-pyrrole (1) and 3-(1,4-dih...
The target compounds thus consist of a pyrrole unit as well as a hydroquinone group (protected as dimethyl ether, i.e., dimethoxybenzene, DMB), connected by different linkers, resulting in varying distance and degree of conjugation between the two subunits (Figure 2). DMB was chosen due to its superior stability compared to the hydroquinone counterpart during the synthesis. The corresponding hydroquinone analogue can be achieved by straightforward demethylation with BBr3 [8-10]. The electronic properties of the monomers were investigated by NMR and UV–vis spectroscopy as well as density functional theory (DFT) calculations.
![[1860-5397-12-10-2]](/bjoc/content/figures/1860-5397-12-10-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Hydroquinone dimethyl ether functionalized pyrroles with linkers L discussed in this study.
Figure 2: Hydroquinone dimethyl ether functionalized pyrroles with linkers L discussed in this study.
Results and Discussion
Synthesis
Methyl linker: Synthesis of 3-(2,5-dimethoxybenzyl)-1H-pyrrole (3a)
For our target compound 3-(2,5-dimethoxybenzyl)-1H-pyrrole (3a), we initially considered some published procedures, but none of them were deemed adequate for our requirements. For example, Foos et al. have devised a synthetic route using alkylation of pyrrylmagnesium bromide with the benzyl bromide 5 [10], but yielding only 3.1% after a tedious work-up. Aquino-Binag et al. achieved a total yield of 70% [11], however, their procedure relies on a Wolff–Kishner reduction of the corresponding acylpyrrole with hydrazine hydrate, making it impractical in countries where the use of hydrazine has been restricted by law [12]. As an alternative, the Suzuki–Miyaura reaction can be utilized for C–C cross coupling of benzyl halides with heteroarylboronic acids. However, in contrast to its use for the synthesis of thiophene and furane derivatives, it has rarely been employed for the coupling of pyrrolylboronic acids with benzyl halides [13-17]. We applied the Suzuki–Miyaura cross-coupling as shown in Scheme 1, with a total yield of nearly 50% (from 2,5-dimethoxybenzyl bromide (5)).
![[1860-5397-12-10-i1]](/bjoc/content/inline/1860-5397-12-10-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route for 3-(2,5-dimethoxybenzyl)-1H-pyrrole (3a). Conditions: i) Pd(PPh3)4, Na2CO3 (2 M aq), MeOH/toluene, microwave heating, 110 °C, 4 h, 56%; ii) TBAF, THF, rt, 0.5 h, 88%.
Scheme 1: Synthetic route for 3-(2,5-dimethoxybenzyl)-1H-pyrrole (3a). Conditions: i) Pd(PPh3)4, Na2CO3 (2 M ...
Starting material 5 was prepared from 2,5-dimethoxybenzylalcohol by a reported procedure [18] in 95% yield. Using typical conditions, the yield of the coupling reaction was 56%, which was considered satisfactory because of the ready availability of starting materials and the modest reaction time of 4 hours. Since the starting materials in the Suzuki–Miyaura cross-coupling tolerate a wide variety of functional groups, facile and versatile combination of different dihydroxybenzyl halide derivatives and pyrrolylboronic acids should be possible.
Ethylene linker: Synthesis of 3-(2,5-dimethoxystyryl)-1H-pyrrole (3c)
Our initial approach of synthesizing vinyl linker dyad 3c was to use a Heck reaction, coupling 3-iodo-1-(triisopropylsilyl)-1H-pyrrole and dimethoxystyrene. A variation of reaction parameters was investigated, involving the selection of base and solvent. However, none of the attempted conditions gave the desired product. Instead, desilylation of 3-iodo-1-(triisopropylsilyl)-1H-pyrrole was observed during the reaction, and some of the dimethoxystyrene could be recovered (for details, see Supporting Information File 2).
According to a study by Liu et al. [19], the protecting group for 3-iodo pyrrole is essential in the Heck reaction involving pyrrole. Based on this suggestion, 3-iodo-1-tosyl-1H-pyrrole was also tested but did not result in any product. It is worth to mention that in their study, ten equivalents of styrene were used to obtain 28% yield, indicating that the reaction conditions were not optimal, and therefore cannot be applied to other vinyl substrates such as dimethoxystyrene directly. A synthetic route via hydrogenation of the ethynyl linker compound 4d was also investigated (vide infra), but did not produce satisfactory results.
Settambolo et al. [20] reported a synthetic route starting from an acylation of pyrrole by phenylacetyl chloride via a Friedel–Crafts reaction, followed by reduction by NaBH4, and elimination to give the vinylpyrrole product. When this strategy was applied to the reaction of (2,5-dimethoxyphenyl)acetyl chloride with 1-(triisopropylsilyl)-1H-pyrrole, nearly 50% yield was achieved in the first step, but requiring a reaction time of more than one day. Therefore, considering the total reaction time and availability of starting material, a Wittig reaction was chosen as an alternative.
The Wittig reaction approach is shown in Scheme 2. In this reaction, phosphonium salt 7 was prepared in quantitative yield from 5 and triphenylphosphine by reflux in toluene, according to a procedure reported for dimethoxybenzyl chloride [21]. The work-up involved simple filtration and washing with toluene. Use of t-BuOK as a base to deprotonate the phosphonium salt resulted in a dark red phosphorous ylide, to which 1-(triisopropylsilyl)-1H-pyrrole-3-carbaldehyde was added, followed by heating at 80 °C. The desired product 4c was obtained in 40% yield after 3 h. When the reaction was performed at room temperature, only 20% yield was obtained after 48 h. Close to equal quantities of cis-4c and trans-4c were formed in this reaction, and were separated chromatographically. Having access to both isomers was desirable, since the stereoisomers are expected to have different electronic properties. After desilylation, compound 3c was obtained in a combined total yield of 35% (sum of cis-3c and trans-3c) from the pyrrole-3-carbaldehyde. To obtain trans-4c selectively, a Wittig–Horner reaction was investigated, with dimethoxybenzyl bromide as starting material for the corresponding phosphonate. However, neither n-BuLi nor t-BuOK used as base succeeded to give the desired product.
![[1860-5397-12-10-i2]](/bjoc/content/inline/1860-5397-12-10-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic route for 3-(2,5-dimethoxystyryl)-1H-pyrrole (3c); cis-4c and trans-4c were separated chromatographically to achieve the synthesis of both isomers of 3c separately. Conditions: i) PPh3, toluene, reflux, overnight, quantitative; ii) t-BuOK, THF, 0.5 h, not isolated; iii) 1-(triisopropylsilyl)-1H-pyrrole-3-carbaldehyde, 80 °C, 3 h, 40%; iv) TBAF, THF, rt, 0.5 h, 86%.
Scheme 2: Synthetic route for 3-(2,5-dimethoxystyryl)-1H-pyrrole (3c); cis-4c and trans-4c were separated chr...
Acetylene linker: Synthesis of 3-((2,5-dimethoxyphenyl)ethynyl)-1H-pyrrole (3d)
The synthesis strategy for the ethynyl-linked compound 3d was straightforward, using a Sonogashira coupling reaction (Scheme 3). Thus, 2,5-dimethoxyphenyl bromide (8) was converted to ((2,5-dimethoxyphenyl)ethynyl)trimethylsilane (9), which was desilylated by addition of NaOH (aq) to give 2-ethynyl-1,4-dimethoxybenzene (10). Sonogashira conditions were as described by Erdélyi et al. using microwave heating [22], allowing the reaction to be completed in half an hour with 95% yield. A second Sonogashira reaction of 10 with 3-iodo-1-(triisopropylsilyl)-1H-pyrrole afforded the protected pyrrole derivative 4d. For this second Sonogashira reaction, these conditions, when attempting to couple 10 to 3-bromo-1-(triisopropylsilyl)-1H-pyrrole resulted in low yield (20%) and also homocoupling. Considering that desilylation of triisopropylsilyl protected pyrrole might occur at high temperature in DMF, an overnight reaction at room temperature was performed, resulting in a yield for 4d of 50%. In summary, after a four step synthesis route, the total yield of 3d from 8 was 36%. Attempts were made to convert alkyne 4d into the corresponding alkene 4c. However, partial hydrogenations with a Lindlar catalyst [23] or with trimethylsilane using a palladium catalyst [24] were either unsuccessful or produced low yields (for details, see Supporting Information File 2).
![[1860-5397-12-10-i3]](/bjoc/content/inline/1860-5397-12-10-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 3-((2,5-dimethoxyphenyl)ethynyl)-1H-pyrrole (3d). Conditions: i) Ethynyltrimethylsilane, Pd(PPh3)2Cl2, CuI, PPh3, diethylamine, DMF, microwave heating, 120 °C, 30 min, 95%; ii) NaOH (1 M, aq), MeOH/CHCl3, rt, 0.5 h, 90%; iii) Pd(PPh3)2Cl2, CuI, triethylamine, 3-iodo-1-(triisopropylsilyl)-1H-pyrrole, THF, rt, overnight, 50%; iv) TBAF, THF, rt, 0.5 h, 85%.
Scheme 3: Synthesis of 3-((2,5-dimethoxyphenyl)ethynyl)-1H-pyrrole (3d). Conditions: i) Ethynyltrimethylsilan...
Ethyl linker: Synthesis of 3-(2,5-dimethoxyphenethyl)-1H-pyrrole (3b)
Compound 3b was prepared by reduction of 4d with H2 over Pd/C catalyst (Scheme 4). The N-protected 4d was used instead of 3d to minimize the risk of decomposition during the hydrogenation reaction, since unprotected pyrrole is known to be far less stable [25]. After desilylation by TBAF, 3-(2,5-dimethoxyphenethyl)-1H-pyrrole (3b) was obtained in a total yield of 36% over five steps from 8.
![[1860-5397-12-10-i4]](/bjoc/content/inline/1860-5397-12-10-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 3-(2,5-dimethoxyphenethyl)-1H-pyrrole (3b). Conditions: i) Pd/C, MeOH/acetone, rt, 1.5 h, 98%; ii) TBAF, THF, rt, 0.5 h, 85%.
Scheme 4: Synthesis of 3-(2,5-dimethoxyphenethyl)-1H-pyrrole (3b). Conditions: i) Pd/C, MeOH/acetone, rt, 1.5...
Electronic properties: Evidence from NMR spectroscopy, UV–vis spectroscopy and DFT calculations
In the NMR spectra, conjugation between the DMB and the pyrrole units is indicated by increased chemical shifts for DMB-H-6 (7.13–7.03 for 1, 3c and 3d) as compared to the non-conjugated compounds (6.83–6.76 for 3a and 3b) (Figure 3). In particular, the chemical shift of the pyrrole H-2 (Pyr-2) is lower than that of the pyrrole H-5 (Pyr-5) in the non-conjugated compounds 3a and 3b, whereas pyrrole H-2 has considerably higher chemical shift than H-5 in the conjugated compounds 1, 3c and 3d. This proton might therefore be used as an indicator of conjugation. Also, in the 13C NMR spectra, DMB C-1 (Ph-1) has a lower chemical shift in the conjugated compounds (128.7–113.9 for 1, 3c and 3d) as compared to the non-conjugated ones (132.3 for 3a and 3b) (Figure 4).
![[1860-5397-12-10-3]](/bjoc/content/figures/1860-5397-12-10-3.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra (400 MHz, CDCl3 solution) of the DMB-pyrrole dyads (aliphatic signals not shown).
Figure 3: 1H NMR spectra (400 MHz, CDCl3 solution) of the DMB-pyrrole dyads (aliphatic signals not shown).
![[1860-5397-12-10-4]](/bjoc/content/figures/1860-5397-12-10-4.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 4: 13C NMR spectra (100.6 MHz, CDCl3 solution) of the DMB-pyrrole dyads (aliphatic signals not shown).
Figure 4: 13C NMR spectra (100.6 MHz, CDCl3 solution) of the DMB-pyrrole dyads (aliphatic signals not shown).
To rationalize these chemical shift patterns, DFT calculations were performed. The observed 1H and 13C NMR chemical shifts are closely matched by those derived from the DFT calculations (see Supporting Information File 2 for details) [26,27], with average errors of 0.13 ppm for 1H (RMS error = 0.17), and 1.9 ppm for 13C (RMS error = 2.6). The above mentioned pattern of δpyrrole H-2 > δpyrrole H-5 for the conjugated compounds, with reversal for the non-conjugated compounds, was also present in the calculated chemical shifts, except for 3a and trans-3c. We notice that in these two compounds, the deviation between observed and calculated chemical shift is untypically high at 0.23 ppm for both Py-5 in 3a and Py-2 in trans-3c. This illustrates the well-known caveat for chemical shift predictions [26].
To assess the electronic properties as indicated in UV–vis spectra, reference spectra of pyrrole, DMB, 1,4-dimethoxy-2-vinylbenzene (DMB-VI), and 1,4-dimethoxy-2-ethynylbenzene (DMB-EN) were recorded. The UV–vis spectra of non-conjugated 3a and 3b closely resemble the sum of absorptions from the individual DMB and pyrrole spectra, indicating the absence of substantial electronic effects from the substituent. On the other hand, for the conjugated 3c, 3d and 1, a red shift is observed when compared to the individual reference compound spectra (Figure 5).
![[1860-5397-12-10-5]](/bjoc/content/figures/1860-5397-12-10-5.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 5: UV–vis absorption spectra of 1, 3a–d, (full lines) and the reference compounds DMB, DMB-VI, DMB-EN (dotted lines). The calculated wavelengths for absorption maxima from DFT calculations for each linker compound are indicated by vertical red lines.
Figure 5: UV–vis absorption spectra of 1, 3a–d, (full lines) and the reference compounds DMB, DMB-VI, DMB-EN ...
According to DFT calculations (see Supporting Information File 2 for details), the HOMO–LUMO transition is allowed for all compounds and has π–π* character. The HOMO of DMB couples strongly to the pyrrole HOMO, resulting in increased HOMO energy in 1, cis-3c, trans-3c, and 3d. Additionally, the corresponding coupling between the DMB and pyrrole LUMOs results in a lowering of the LUMO level in the combined molecular systems, since they are anti-bonding. The perturbation of the HOMOs and LUMOs depends on the degree of interaction between the two subunits, and is therefore a measure of the charge delocalization in the molecule. This effect can be viewed as an increased delocalized system that results in a lowering of the HOMO–LUMO gap, which can be traced by the red shifts of the highest wavelength absorption in UV–vis spectra (Table 1).
The extent of conjugation among the series of linker compounds can be assessed by the HOMO–LUMO transition (Table 1). Since there is no conjugation between the two subunits in 3a and 3b, the HOMO and LUMO levels do not shift with respect to the reference compound DMB. This is also revealed by the electron distribution on both of the rings at the HOMO level. For both 3a and 3b, the HOMO is entirely localized at the DMB unit (Figure 6a), whereas for conjugated compounds such as 3d, the HOMO is distributed over both rings (Figure 6b).
![[1860-5397-12-10-6]](/bjoc/content/figures/1860-5397-12-10-6.png?scale=1.68&max-width=1024&background=FFFFFF)
Figure 6: Calculated HOMO for 3a (a) and 3d (b).
Figure 6: Calculated HOMO for 3a (a) and 3d (b).
Among the conjugated compounds, 1 has the largest HOMO–LUMO gap. This may be explained by the smaller overall conjugated π system as compared to cis-3c, 3d and trans-3c. The vinyl-linker structures (cis- and trans-3c) have more extended π systems, which yield lower energy HOMO–LUMO transitions. However, a significant difference is observed between trans-3c and cis-3c, which arises from the difference in their geometry: Whereas trans-3c has a planar structure, for cis-3c the pyrrole and DMB substituents on the vinyl linker were found to be twisted due to steric hindrance between them, which might also explain the low chemical shift of the pyrrole-H-4 in cis-3c as the result of being exposed to an anisotropy effect of the DMB ring.
Conclusion
In summary, synthesis protocols for a series of methyl-protected hydroquinone-pyrrole dyads with good overall yields have been devised. DFT calculations allow insight into the electronic properties and conjugation within these compounds, with good agreement between calculations and experimental spectroscopic data. The extent of conjugation was found to vary with the linker, with trans-3c (trans-3-(2,5-dimethoxystyryl)-1H-pyrrole) having the strongest conjugation, as judged by the HOMO–LUMO gap, while 3a (3-(2,5-dimethoxybenzyl)-1H-pyrrole) and 3b (3-(2,5-dimethoxyphenethyl)-1H-pyrrole) have no conjugation, with HOMO–LUMO gaps similar to DMB (p-dimethoxybenzene). These compounds should be interesting as components of redox active materials, as we have recently shown in the synthesis of conducting redox polymers for use as the active material in organic electrical energy storage [6].
Acknowledgements
The Swedish Foundation for Strategic Research (SSF), The Swedish Energy Agency (Project SweGRIDS), the Carl Trygger Foundation as well as the Olle Byggmästare Foundation are acknowledged for their financial support of this work. We thank Bo Ek for HRMS measurements as well as Julien Andrès for invaluable discussions and proofreading of the manuscript.
References
-
Tsue, H.; Imahori, H.; Kaneda, T.; Tanaka, Y.; Okada, T.; Tamaki, K.; Sakata, Y. J. Am. Chem. Soc. 2000, 122, 2279–2288. doi:10.1021/ja9900454
Return to citation in text: [1] -
Kacprzak, S.; Kaupp, M. J. Phys. Chem. B 2006, 110, 8158–8165. doi:10.1021/jp061105c
Return to citation in text: [1] -
Karr, P. A.; Zandler, M. E.; Beck, M.; Jaeger, J. D.; McCarty, A. L.; Smith, P. M.; D'Souza, F. J. Mol. Struct.: THEOCHEM 2006, 765, 91–103. doi:10.1016/j.theochem.2006.03.012
Return to citation in text: [1] -
Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. J. Phys. Chem. C 2013, 117, 23558–23567. doi:10.1021/jp408567h
Return to citation in text: [1] -
Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. J. Phys. Chem. C 2014, 118, 23499–23508. doi:10.1021/jp506821z
Return to citation in text: [1] [2] -
Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. RSC Adv. 2015, 5, 11309–11316. doi:10.1039/C4RA15708G
Return to citation in text: [1] [2] -
Bray, B. L.; Mathies, P. H.; Naef, R.; Solas, D. R.; Tidwell, T. T.; Artis, D. R.; Muchowski, J. M. J. Org. Chem. 1990, 55, 6317–6328. doi:10.1021/jo00313a019
Return to citation in text: [1] -
Kon, A. B.; Foos, J. S.; Rose, T. L. Chem. Mater. 1992, 4, 416–424. doi:10.1021/cm00020a034
Return to citation in text: [1] -
Weissman, S. A.; Zewge, D. Tetrahedron 2005, 61, 7833–7863. doi:10.1016/j.tet.2005.05.041
Return to citation in text: [1] -
Foos, J. S.; Degnan, S. M.; Glennon, D. G.; Beebe, X. J. Electrochem. Soc. 1990, 137, 2530–2533. doi:10.1149/1.2086982
Return to citation in text: [1] [2] -
Aquino-Binag, C.; Kumar, N.; Pigram, P. Org. Prep. Proced. Int. 1995, 27, 700–703. doi:10.1080/00304949509458537
Return to citation in text: [1] -
Swedish Regulation, "Förordning (2008:245) om kemiska produkter och biotekniska organismer".
Return to citation in text: [1] -
Alvarez, A.; Guzman, A.; Ruiz, A.; Velarde, E.; Muchowski, J. M. J. Org. Chem. 1992, 57, 1653–1656. doi:10.1021/jo00032a011
Return to citation in text: [1] -
Molander, G. A.; Eliac, M. D. J. Org. Chem. 2006, 71, 9198–9202. doi:10.1021/jo061699f
Return to citation in text: [1] -
Burns, M. J.; Fairlamb, I. J. S.; Kapdi, A. R.; Sehnal, P.; Taylor, R. J. K. Org. Lett. 2007, 9, 5397–5400. doi:10.1021/ol702291r
Return to citation in text: [1] -
Molander, G. A.; Canturk, B.; Kennedy, L. E. J. Org. Chem. 2009, 74, 973–980. doi:10.1021/jo802590b
Return to citation in text: [1] -
Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. J. Org. Chem. 2014, 79, 230–239. doi:10.1021/jo4024123
Return to citation in text: [1] -
Kostikov, A. P.; Popik, V. V. J. Org. Chem. 2007, 72, 9190–9194. doi:10.1021/jo701426j
Return to citation in text: [1] -
Liu, J.-H.; Chan, H.-W.; Wong, H. N. C. J. Org. Chem. 2000, 65, 3274–3283. doi:10.1021/jo991531c
Return to citation in text: [1] -
Settambolo, R.; Lazzaroni, R.; Messeri, T.; Mazzetti, M.; Salvadori, P. J. Org. Chem. 1993, 58, 7899–7902. doi:10.1021/jo00079a040
Return to citation in text: [1] -
Rosowsky, A.; Papoulis, A. T.; Forsch, R. A.; Queener, S. F. J. Med. Chem. 1999, 42, 1007–1017. doi:10.1021/jm980572i
Return to citation in text: [1] -
Erdélyi, M.; Gogoll, A. J. Org. Chem. 2001, 66, 4165–4169. doi:10.1021/jo0057250
Return to citation in text: [1] -
Beccalli, E. M.; Marchesini, A.; Pilati, T. Tetrahedron 1994, 50, 12697–12712. doi:10.1016/S0040-4020(01)89402-3
Return to citation in text: [1] -
Luo, F.; Pan, C.; Wang, W.; Ye, Z.; Cheng, J. Tetrahedron 2010, 66, 1399–1403. doi:10.1016/j.tet.2009.11.098
Return to citation in text: [1] -
Jolicoeur, B.; Chapman, E. E.; Thompson, A.; Lubell, W. D. Tetrahedron 2006, 62, 11531–11563. doi:10.1016/j.tet.2006.08.071
Return to citation in text: [1] -
Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v
Return to citation in text: [1] [2] -
Willoughby, P. H.; Jansma, M. J.; Hoye, T. R. Nat. Protoc. 2014, 9, 643–660. doi:10.1038/nprot.2014.042
Return to citation in text: [1]
| 26. | Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v |
| 27. | Willoughby, P. H.; Jansma, M. J.; Hoye, T. R. Nat. Protoc. 2014, 9, 643–660. doi:10.1038/nprot.2014.042 |
| 24. | Luo, F.; Pan, C.; Wang, W.; Ye, Z.; Cheng, J. Tetrahedron 2010, 66, 1399–1403. doi:10.1016/j.tet.2009.11.098 |
| 25. | Jolicoeur, B.; Chapman, E. E.; Thompson, A.; Lubell, W. D. Tetrahedron 2006, 62, 11531–11563. doi:10.1016/j.tet.2006.08.071 |
| 1. | Tsue, H.; Imahori, H.; Kaneda, T.; Tanaka, Y.; Okada, T.; Tamaki, K.; Sakata, Y. J. Am. Chem. Soc. 2000, 122, 2279–2288. doi:10.1021/ja9900454 |
| 2. | Kacprzak, S.; Kaupp, M. J. Phys. Chem. B 2006, 110, 8158–8165. doi:10.1021/jp061105c |
| 3. | Karr, P. A.; Zandler, M. E.; Beck, M.; Jaeger, J. D.; McCarty, A. L.; Smith, P. M.; D'Souza, F. J. Mol. Struct.: THEOCHEM 2006, 765, 91–103. doi:10.1016/j.theochem.2006.03.012 |
| 5. | Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. J. Phys. Chem. C 2014, 118, 23499–23508. doi:10.1021/jp506821z |
| 22. | Erdélyi, M.; Gogoll, A. J. Org. Chem. 2001, 66, 4165–4169. doi:10.1021/jo0057250 |
| 7. | Bray, B. L.; Mathies, P. H.; Naef, R.; Solas, D. R.; Tidwell, T. T.; Artis, D. R.; Muchowski, J. M. J. Org. Chem. 1990, 55, 6317–6328. doi:10.1021/jo00313a019 |
| 23. | Beccalli, E. M.; Marchesini, A.; Pilati, T. Tetrahedron 1994, 50, 12697–12712. doi:10.1016/S0040-4020(01)89402-3 |
| 6. | Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. RSC Adv. 2015, 5, 11309–11316. doi:10.1039/C4RA15708G |
| 20. | Settambolo, R.; Lazzaroni, R.; Messeri, T.; Mazzetti, M.; Salvadori, P. J. Org. Chem. 1993, 58, 7899–7902. doi:10.1021/jo00079a040 |
| 4. | Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. J. Phys. Chem. C 2013, 117, 23558–23567. doi:10.1021/jp408567h |
| 5. | Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. J. Phys. Chem. C 2014, 118, 23499–23508. doi:10.1021/jp506821z |
| 21. | Rosowsky, A.; Papoulis, A. T.; Forsch, R. A.; Queener, S. F. J. Med. Chem. 1999, 42, 1007–1017. doi:10.1021/jm980572i |
| 12. | Swedish Regulation, "Förordning (2008:245) om kemiska produkter och biotekniska organismer". |
| 18. | Kostikov, A. P.; Popik, V. V. J. Org. Chem. 2007, 72, 9190–9194. doi:10.1021/jo701426j |
| 11. | Aquino-Binag, C.; Kumar, N.; Pigram, P. Org. Prep. Proced. Int. 1995, 27, 700–703. doi:10.1080/00304949509458537 |
| 19. | Liu, J.-H.; Chan, H.-W.; Wong, H. N. C. J. Org. Chem. 2000, 65, 3274–3283. doi:10.1021/jo991531c |
| 10. | Foos, J. S.; Degnan, S. M.; Glennon, D. G.; Beebe, X. J. Electrochem. Soc. 1990, 137, 2530–2533. doi:10.1149/1.2086982 |
| 26. | Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v |
| 8. | Kon, A. B.; Foos, J. S.; Rose, T. L. Chem. Mater. 1992, 4, 416–424. doi:10.1021/cm00020a034 |
| 9. | Weissman, S. A.; Zewge, D. Tetrahedron 2005, 61, 7833–7863. doi:10.1016/j.tet.2005.05.041 |
| 10. | Foos, J. S.; Degnan, S. M.; Glennon, D. G.; Beebe, X. J. Electrochem. Soc. 1990, 137, 2530–2533. doi:10.1149/1.2086982 |
| 13. | Alvarez, A.; Guzman, A.; Ruiz, A.; Velarde, E.; Muchowski, J. M. J. Org. Chem. 1992, 57, 1653–1656. doi:10.1021/jo00032a011 |
| 14. | Molander, G. A.; Eliac, M. D. J. Org. Chem. 2006, 71, 9198–9202. doi:10.1021/jo061699f |
| 15. | Burns, M. J.; Fairlamb, I. J. S.; Kapdi, A. R.; Sehnal, P.; Taylor, R. J. K. Org. Lett. 2007, 9, 5397–5400. doi:10.1021/ol702291r |
| 16. | Molander, G. A.; Canturk, B.; Kennedy, L. E. J. Org. Chem. 2009, 74, 973–980. doi:10.1021/jo802590b |
| 17. | Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. J. Org. Chem. 2014, 79, 230–239. doi:10.1021/jo4024123 |
| 6. | Karlsson, C.; Huang, H.; Strømme, M.; Gogoll, A.; Sjödin, M. RSC Adv. 2015, 5, 11309–11316. doi:10.1039/C4RA15708G |
© 2016 Huang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
