Abstract
The synthesis and optoelectronic properties of novel S,N-heterotetracenes consisting of fused heterocyclic thiophene and pyrrole rings are presented. Tetracyclic and benzannulated derivatives with a varying number and sequence of sulfur and nitrogen heteroatoms were synthesized in multistep synthetic routes. A Buchwald–Hartwig amination of brominated precursors, thermolysis of azide precursors, and a Cadogan reaction of nitro-substituted precursors were successfully applied to eventually build-up pyrrole rings to stable and soluble fused systems. The various obtained heteroatom sequences ‘SSNS’ (SN4), ‘SNNS’ (SN4’’), and ‘NSSN’ (SN4’) allowed for evaluation of structure–property relationships relative to the sulfur analogue tetrathienoacene (‘SSSS’). In line with the results for the whole series of S,N-heteroacenes, we find that replacement of sulfur by nitrogen atoms in the tetra- and hexacyclic systems leads to a red-shift in absorption, a decrease in oxidation potential and energy gap. On the other hand, the replacement of a thiophene ring by benzene leads to the opposite effects.

Graphical Abstract
Introduction
Thienoacenes and related S,N-heteroacenes have been developed to important π-conjugated systems mainly for application as p-type semiconductor in organic electronic devices with excellent results [1-3]. The series of thienoacenes consisting of only fused thiophene rings has been synthesized up to an octamer, whereby the longer members in the series quickly lose solubility [4-6]. The implementation of pyrrole rings into the fused ladder-type structure leads to the class of S,N-heteroacenes, whereby the incorporation of the electron-rich pyrrole rings not only modifies the electronic properties compared to thienoacenes, but also allows for the attachment of side chains at the pyrrole nitrogen as a useful synthetic tool to enhance solubility [7]. The smaller members of the series of S,N-heteroacenes, dimeric thieno[3,2-b]pyrrole [8] and trimeric dithieno[3,2-b:2’,3’-d]pyrrole (DTP) [9], are long known but still frequently used as building block for organic electronic materials [10]. By application of Pd-catalyzed Buchwald–Hartwig amination/cyclization reactions of brominated thiophene-based precursors and amines [11,12], the series of S,N-heteroacenes has been systematically extended from pentamer SN5 [13,14] up to a stable and still soluble S,N-heterotridecacene SN13 [7,15-17]. Correlation of, e.g., absorption or redox potentials vs conjugated chain length led to interesting structure–property relationships showing the influence of the number and sequence of pyrrole rings. The highly planar π-conjugated systems revealed interesting structural features such as nearly complete bond length equalization and S–S and S–π dipolar interactions in the solid state. Due to tunable optoelectronic properties, a variety of functionalized derivatives, mainly trimeric DTP (SN3) [18], pentameric SN5 [19-23], and hexameric SN6 [24-28], have been successfully implemented as active semiconducting components in organic solar cells [18-21,24-26], perovskite solar cells [22,27], organic field-effect transistors [28], or sensors [23]. The interesting structure–property relationships of the S,N-heteroacene series inspired as well theoretical work. In this respect, De Simone et al. computed electronic spectra of the neutral, charged radical cationic, and dicationic species [29], whereas Sarkar et al. undertook quantum chemical investigations on the photovoltaic performance revealing an odd-even relation of the charge separation [30]. Furthermore, they calculated the single molecule transport behavior of various S,N-heteroacenes with different conjugation length between nanocontacts [31].
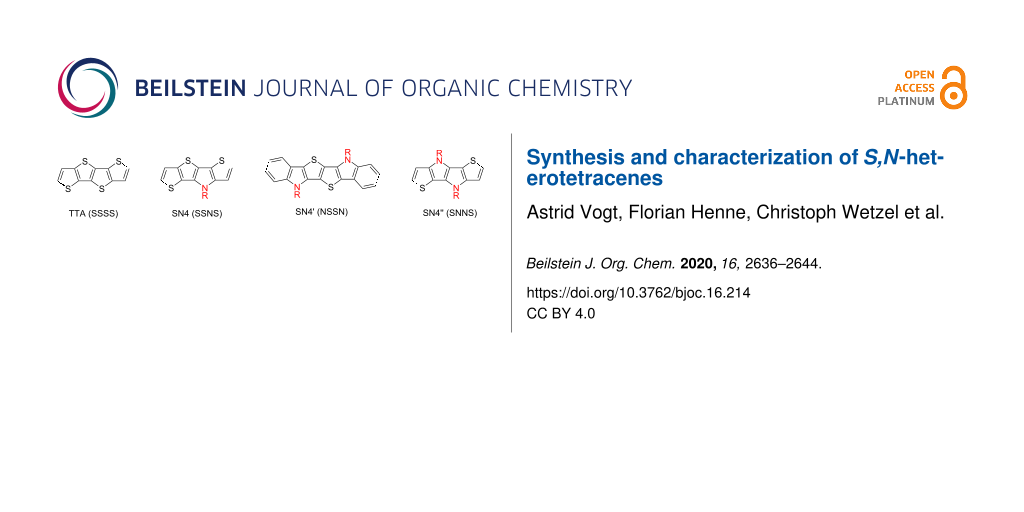
The obvious missing link in the series of S,N-heteroacenes is represented by heterotetrameric SN4, which is very rarely described in the literature. SN4 is derived from well-known parent tetrathienoacene (TTA, ‘SSSS’, Figure 1) by replacing one “S” by “N” or in other words one fused thiophene unit by a pyrrole ring. For TTA various synthetic methods have been developed and it has been broadly implemented as building block into materials for organic electronic devices [32,33]. We have disclosed the first SN4 derivative with the heteroatom sequence ‘SSNS’ in the context of complementing the series, but did not yet describe synthetic details [7]. Xue et al. published a donor–acceptor photosensitizer for dye-sensitized solar cells, in which the N-phenylated SN4 unit serves as bridge between donor and acceptor moieties [34]. Further replacement of sulfur by nitrogen can lead to different isomeric SN4 structures with possible sequences ‘NSSN’, which we denominate as SN4', and ‘SNNS’ as SN4'' (Figure 1). In this respect, the only known structures were prepared by Gryko et al., who synthesized heterohexacenes with N-phenylated SN4'' cores [35]. The same group furthermore prepared heterotetracenes comprising four pyrrole rings (‘NNNN’) as electron-rich, ladder-type fluorophores by a two-step synthesis [36]. The possible asymmetric SN4 isomer ‘SNSN’ and heterotetracenes comprising three pyrrole rings are unknown so far.
![[1860-5397-16-214-1]](/bjoc/content/figures/1860-5397-16-214-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Heteroacenes: tetrathienoacene (TTA), S,N-heteroacenes SN4, SN4', and SN4''.
Figure 1: Heteroacenes: tetrathienoacene (TTA), S,N-heteroacenes SN4, SN4', and SN4''.
In continuation of our work on heteroacenes, we now report in detail the syntheses and characterization of fused S,N-heterotetracenes, SN4 (‘SSNS’), benzannulated SN4' (‘NSSN’), and SN4'' (‘SNNS’) by application of various cyclization methods to build up pyrrole rings in the tetraheterocyclic systems. Among them, a Buchwald–Hartwig amination of brominated precursors, thermolysis of azide precursors, and a Cadogan cyclization of nitro-substituted precursors were applied to prepare various unknown derivatives. These represent novel core molecules, which by further derivatisation, for example with terminal acceptor groups, would lead to interesting π-conjugated materials for application in organic electronics.
Results and Discussion
Synthesis of N-alkylated S,N-heterotetracene 9 by Pd-catalyzed amination. Asymmetric S,N-heterotetracene Hex-SN4 9 comprises the sequence of heteroatoms ‘SSNS’ in the tetracyclic conjugated π-system [37]. In a multistep synthesis, 2,6-dibromothienothiophene 2 [39], which was obtained from thieno[3,2-b]thiophene (1) [38], was triisopropylsilyl (TIPS)-protected by lithium-halogen exchange with n-BuLi and triisopropylsilyl chloride to give thienothiophene 3 in 69% yield. A halogen dance reaction of 3, induced by lithium diisopropylamide (LDA) at −78 °C, gave the corresponding β-brominated thienothiophene 4 in 91% yield [6]. Compound 4 was selectively brominated in α-position by using NBS in DMF at room temperature to give dibrominated thienothiophene 5 in 92% yield which was coupled in a Negishi-type cross-coupling reaction with zincated 2,3-dibromothiophene 6 and Pd(dppf)Cl2 as catalyst to give the corresponding dibromide 7 in a yield of 70%. Organozinc species 6 was obtained from 2,3-dibromothiophene by lithiation with n-BuLi and reaction with zink dichloride. Annulation to TIPS-protected SN4-Hex 8 was achieved in 87% yield by a tandem Buchwald–Hartwig amination of dibromide 7 with n-hexylamine in the presence of sodium tert-butoxide as base and Pd(dba)2/dppf as the catalytic system. The TIPS group was removed from Hex-SN4 8 upon treatment with tetra-n-butylammonium fluoride (TBAF) and the parent system Hex-SN4 9 was obtained in 80% yield (Scheme 1).
![[1860-5397-16-214-i1]](/bjoc/content/inline/1860-5397-16-214-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of fused S,N-heterotetracene SN4 9 starting from thieno[3,2-b]thiophene (1).
Scheme 1: Synthesis of fused S,N-heterotetracene SN4 9 starting from thieno[3,2-b]thiophene (1).
Synthesis of S,N-heterotetracene H-SN4 13 by thermolysis of azide precursors. Smaller parent heterotriacene dithienopyrrole (H-DTP) was first synthesized by Zanirato et al. by thermolysis of 3-azido-2,2’-bithiophene as the key step [9]. We therefore tried to build up tetracyclic H-SN4 13 via the azide route. The synthesis started from the above mentioned brominated thieno[3,2-b]thiophene 3, which was coupled with zincated 2,3-dibromothiophene 6 in a Negishi-type cross-coupling reaction with Pd(dppf)Cl2 as catalyst to give thiophene-substituted thienothiophene 10 in 72% yield. In the next step, an azide group was introduced to 10 by lithiation with n-BuLi followed by the reaction with tosyl azide giving azide 11 in an excellent yield of 92%. Thermally induced ring-closure under release of nitrogen and formation of a nitrene intermediate was achieved by refluxing azide 11 in chlorobenzene for 30 min to yield TIPS-protected H-SN4 12 in 46% yield. Finally, the TIPS group of S,N-heterotetracene 12 was smoothly removed using TBAF in THF solution to yield the basic structure H-SN4 13 in almost quantitative yield (Scheme 2).
![[1860-5397-16-214-i2]](/bjoc/content/inline/1860-5397-16-214-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of parent H-SN4 13 via the azide route.
Scheme 2: Synthesis of parent H-SN4 13 via the azide route.
Synthesis of S,N-heterotetracene H-SN4 13 and related derivatives by Cadogan reaction of nitro-substituted precursors. Another possibility for the preparation of fused pyrrole rings is established by the Cadogan reaction [40]. Well-known examples are the reductive cyclization of 2-nitro-1,1’-biphenyls with triethyl phosphite or phosphanes as reducing agent to provide carbazoles covering a wide substrate and functional group spectrum [41]. Moreover, penta- and heptafused heteroacenes were prepared by the Cadogan reaction by annulation of nitrophenyl or nitrobenzothienyl precursors [42-45]. In this respect, we recently reported a Cadogan cyclization of 3-nitro-2,2’-bithiophene with triethyl phosphite under microwave irradiation and surprisingly obtained targeted heterotriacene H-DTP (vide supra) with only 11% yield [45]. This result prompted us to have a closer look on the applicability of the Cadogan reaction/cyclization in order to provide S,N-heterotetracenes and related derivatives from nitro-substituted thiophene-based precursors. First of all, the reductive cyclization of nitrothienyl-substituted thienothiophene 16 to H-SN4 13 was investigated. For this purpose, 2-stannylthienothiophene 14, which was obtained from thienothiophene 1, was converted in a Pd-catalyzed Stille-type cross-coupling with 2-bromo-3-nitrothiophene (15) to precursor 2-(3-nitrothien-2-yl)thieno[3,2-b]thiophene (16) in 64% yield. Cadogan cyclization of nitro derivative 16 with triethyl phosphite at reflux gave 4H-thieno[3,2-b]thieno[2’, 3’:4,5]thieno[2,3-d]pyrrole (H-SN4, 13) in a similar low yield (16%) as before H-DTP (vide supra) (Scheme 3). The change of the reducing agent to triphenyl phosphite did not lead to a reaction at all.
![[1860-5397-16-214-i3]](/bjoc/content/inline/1860-5397-16-214-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of tetracyclic H-SN4 13 via the Cadogan route.
Scheme 3: Synthesis of tetracyclic H-SN4 13 via the Cadogan route.
We turned then to related thienothiophene 18, which bears an o-nitrophenyl substituent instead of a 3-nitrothienyl residue in 16. 2-(2-Nitrophenyl)thieno[3,2-b]thiophene (18) was similarly prepared by Pd-catalyzed Stille-type reaction of monostannylated thienothiophene 14 and o-iodonitrobenzene (17) in 86% yield. Successive Cadogan cyclization of 18 with triethyl phosphite under reflux gave tetracyclic 9H-thieno[2’,3’:4,5]thieno[3,2-b]indole (19) in an increased yield of 58% (Scheme 4).
![[1860-5397-16-214-i4]](/bjoc/content/inline/1860-5397-16-214-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of tetracyclic indole derivative 19 via the Cadogan route.
Scheme 4: Synthesis of tetracyclic indole derivative 19 via the Cadogan route.
Comparison of the reactions indicated that o-nitrophenyl substituents as in 18 give higher cyclization yields than the corresponding 3-nitrothienyls as in 16. We rationalize this finding by a higher reactivity of the intermediate thienyl nitrene, which is formed during the cyclization reaction of 16 compared to phenyl nitrene [42]. Besides H-SN4 13, which is formed by insertion of the nitrene N-atom into the C3–H σ-bond of the thienothiophene in 16 and simultaneous migration of the H-atom to the nitrogen, mostly undefined black oligomeric or polymeric products were obtained.
We further tested a twofold nitrophenyl-substituted thienothiophene in a Cadogan cyclization. Thus, distannylated thienothiophene 20 was obtained in 68% yield from thienothiophene 1 with two equivalents each of n-BuLi at −78 °C and trimethylstannyl chloride in THF. The stannyl reacted with o-iodonitrobenzene (17) and Pd[PPh3]4 as catalyst in DMF to give 2,5-bis(2-nitrophenyl)thieno[3,2-b]thiophene (21) in 86% yield. Subsequent Cadogan cyclization with triethyl phosphite under reflux gave hexacyclic S,N-heteroacene 22 in 38% yield which reflects a benzannulated SN4' system with the heteroatom sequence ‘NSSN’ (Scheme 5). The use of triphenyl phosphite as reducing agent gave a decreased yield of 24%. The N-alkylated derivative of hexacyclic SN4' 22 was prepared by Wong et al., who applied the Cadogan reaction to cyclize a dibrominated precursor similar to 21. Without isolation, the SN4’-intermediate was directly alkylated with 2-ethylhexyl bromide and the corresponding dialkylated SN4' system was subsequently obtained by removal of the bromines by lithium–halogen exchange and acidic work-up [28].
![[1860-5397-16-214-i5]](/bjoc/content/inline/1860-5397-16-214-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of hexacyclic heteroacene SN4' 22 via the Cadogan route.
Scheme 5: Synthesis of hexacyclic heteroacene SN4' 22 via the Cadogan route.
Synthesis of S,N-heterotetracenes SN4''. In contrast to SN4’, S,N-heterotetracene system SN4'' comprising the heteroatom sequence ‘SNNS’ is built-up by annulation of two ‘outer’ thiophene and two ‘inner’ pyrrole rings resulting in the symmetric sequence of heteroatoms in the tetracyclic conjugated π-system. The multistep synthesis of Pr-SN4'' 33 started from 2-thienylcarbaldehyde (23), which was put to reaction with methyl 2-azidoactetate [46] and sodium methanolate in a Knoevenagel condensation to give azide 24 in 81% yield. A solution of the azide 24 was added to boiling toluene and cyclization to thienopyrrole 25 occurred via a nitrene intermediate in nearly quantitative yield [47]. Single crystals of intermediate thienopyrrole 25 were obtained by slow evaporation of a chloroform solution and an X-ray structure analysis was performed. Structural details and packing motifs of the molecule are described in Supporting Information File 1. Alkylation to N-propyl-substituted thienopyrrole 26, saponification to carbonic acid 27, and subsequent Cu-mediated decarboxylation in quinoline resulted in thienopyrrole 28 in more than 80% yield over three steps. Lithiation of 28 with n-BuLi and reaction with TIPS chloride selectively occurred at the thiophene α-position to give TIPS-protected thienopyrrole 29 in 92% yield which was further brominated with N-bromosuccinimide (NBS) to dibrominated thienopyrrole 30 in 99% yield. Pd-catalyzed Neghishi-type coupling of 30 with 3-bromo-2-thienylzinc chloride (6) resulted in thienyl-substituted thienopyrrole 31 in moderate 37% yield. Cyclization to form the second pyrrole ring was achieved by selective Buchwald–Hartwig amination of dibromide 31 with n-propylamine to give TIPS-protected SN4'' 32 in 61% yield. Final removal of the TIPS-protecting group by TBAF gave the novel SN4''-system 33 in 98% yield (Scheme 6).
![[1860-5397-16-214-i6]](/bjoc/content/inline/1860-5397-16-214-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of heterotetracene SN4'' 33 via the azide and Buchwald–Hartwig amination route.
Scheme 6: Synthesis of heterotetracene SN4'' 33 via the azide and Buchwald–Hartwig amination route.
All prepared S,N-heterotetracenes and -hexacene were clearly identified, fully characterized, and their structures confirmed by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS) via matrix-assisted laser desorption/ionization (MALDI) (Supporting Information File 1, Figures S1–S5). In the 1H NMR spectra, the S,N-heterotetracenes typically showed two doublets with 3J-coupling constants of 5.2–5.3 Hz for the α- and β-protons of the terminal thiophene units in the fused skeleton. Thereby the α-protons typically occur slightly downfield-shifted relative to the ß-protons [20]. If one compares the shifts of these protons of the basic systems TTA (Hα 7.52, Hß 7.41 ppm) with those of SN4 13 (Hα 7.39, Hß 7.33 ppm) and SN4'' 33 (Hα 7.06, Hß 7.02 ppm), both protons gradually shift up-field the more electron-rich pyrrole rings are present in the system. In contrast, for benzofused heteroacene 19 these protons are shifted down-field (Hα 7.67, Hß 7.49 ppm) compared to SN4 13. MALDI–HRMS of all synthesized S,N-heteroacenes showed only a single peak for the molecular ions accounting for their purity.
Optical and redox properties of S,N-heteroacenes 9, 13, 19, 22, and 33 in comparison to tetrathienoacene (TTA). Optical spectroscopy of the various SN4-heteroacenes was performed in THF solutions. UV–vis absorption spectra of TTA, SN4 9, and SN4'' 33 are shown in Figure 2, left, those of heteroacenes 13, 19, and 22 in Figure S6 (Supporting Information File 1) and data is listed in Table 1. In general, the heteroacenes exhibited one vibronically well-resolved strong absorption band in the UV-regime of 250–360 nm which we address to the π–π* transition corresponding to the HOMO–LUMO energy gap. The absorption maximum is gradually red-shifted from 318 nm for TTA and with increasing number of pyrrole rings to 330 nm for SN4'' 33 including molar extinction coefficients in the range from 30 500 M−1 cm−1 for TTA to 33 700 M−1 cm−1 for SN4'' derivative 33. In comparison to H-SN4 13, the influence of the hexyl side chain in Hex-SN4 9 on the optical properties is as expected marginal and the absorption maximum is only slightly red-shifted. The effect of benzannulation in 19 compared to H-SN4 13 as well results only in small differences, which, however, become larger in comparison to hexacyclic heteroacene 22 showing the largest bathochromic shift in the series (Figure S6, Supporting Information File 1). The absorption maximum (378 nm) and the shape of the band are comparable to those of SN6 (379 nm) [17]. The pronounced vibronic fine splitting of the main absorption bands is typical for the planar and stiff-fused backbones of the heteroacenes [20]. The optical gap, which is determined from the absorption onset, gradually decreases with increasing number of pyrrole rings in the heteroacenes from 3.62 eV for TTA to 3.50 eV for 33 (Figure 2, left).
![[1860-5397-16-214-2]](/bjoc/content/figures/1860-5397-16-214-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis absorption spectra of TTA, Hex-SN4 9, Pr-SN4'' 33 and fluorescence spectrum of 33 in THF at rt (left) and corresponding cyclic voltammograms in dichloromethane/tetrabutylammonium hexafluorophosphate (0.1 M), 100 mV/s (right).
Figure 2: UV–vis absorption spectra of TTA, Hex-SN4 9, Pr-SN4'' 33 and fluorescence spectrum of 33 in THF at ...
The SN4-heteroacenes fluoresce, if at all, only very weakly and emission maxima lay in between 356 nm for TTA and 388 nm for 22 with the most extended conjugated π-system (Table 1). Fluorescence quantum yields were far below 1% except for hexacene 22, which showed a very moderate value of 1.9%. This result is in line with the series of S,N-heteroacenes where noticeable fluorescence started with heptacene SN7 (ϕem = 19%) reaching 93% for the longest tridecamer SN13 [17,37]. The Stokes shifts, which were determined from the 0,0-transitions, lay in between 682 cm−1 for hexacene 22 and 1433 cm−1 for SN4'-derivative 9. These relatively small values indicate small geometric changes and structural deformation between the ground and excited states which is typical for such planar and conformationally restricted systems [48].
Table 1: Thermal, optical, and electrochemical data of S,N-heterotetracenes 9, 13, and 33 in comparison to tetrathienoacene (TTA) and benzofused S,N-heteroacenes 19 and 22.
| Heteroacene |
Mp
[°C] |
λabs
[nm]a |
ε
[M−1 cm−1] |
Egopt
[eV]b |
λem
[nm]a |
ϕem
[%]c |
Stokes shift
[cm−1] |
EpOx1
[V] |
HOMO
[eV]d |
LUMO
[eV]e |
| TTA | 222.0 | 332, 318, 305 | 30 500 | 3.62 | 356 | <<1 | 965f | 0.81 | −5.76 | −2.14 |
|
H-SN4
(13) |
186.3 | 333, 320, 305 | 29 300 | 3.57 | n.d.g | n.d.g | n.d.g | 0.37 | −5.37 | −1.80 |
|
Hex-SN4
(9) |
66.3 | 336, 322, 310 | 30 700 | 3.55 | 364 | <<1 | 1433f | 0.39 | −5.39 | −1.84 |
|
Pr-SN4’’
(33) |
141.0 | 344, 330, 317 | 33 700 | 3.50 | 355 | <<1 | 861 | 0.07 | −5.10 | −1.60 |
|
H-SN3B
(19) |
225–226 | 339, 325, 313 | 29 500 | 3.55 | n.d.g | n.d.g | n.d.g | 0.56 | −5.56 | −2.01 |
|
H-BSN4’B
(22) |
452 | 378, 358, 336 | 52 500 | 3.20 | 388 | 1.9 | 682 | 0.33 | −5.27 | −2.07 |
aAbsorption and emission measured in THF. bCalculated by Eg = 1240/λonset. cQuantum yields referred to anthracene in ethanol (ϕem = 0.27). dCalculated from the onset value of the oxidation wave; Fc/Fc+ was set to −5.1 eV vs vacuum. eCalculated with EHOMO and Egopt sol. fValue calculated through Gaussian deconvolution of the unstructured emission spectrum. gn.d. = not detectable.
In order to get information about the redox properties and energetics of the frontier orbitals, the S,N-heteroacenes were studied by cyclic voltammetry and all potentials were referenced against the ferrocene/ferricenium couple (Table 1). In the series TTA, SN4 9, and SN4'' 33 irreversible oxidation waves were obtained due to follow-up reactions of the formed radical cations and peak potentials gradually decrease from 0.81 V for TTA to 0.07 V for SN4'' 33 (Figure 2, right). As expected, the oxidation potential is substantially influenced by the increasing number of electron-rich pyrrole rings and drops continuously. As expected, if an electron-rich fused thiophene ring in H-SN4 13 is replaced by a phenyl ring in 19, the oxidation potential is increased by 190 mV. The value for hexacyclic 22 (Epox1 = 0.33 V) is in the same regime than for H-SN4 13, but substantially higher than this for SN6 (Epox1 = 0.06 V) [7,37] clearly showing the effect of the two-fold benzannulation.
From the optoelectronic data a schematic energy level diagram of the frontier orbitals and the optical transitions in the series can be derived (Figure 3). The HOMO energy levels were determined from the onset of the oxidation wave and are continuously destabilized in the series TTA, SN4 13 and 9, and SN4'' 33. Due to the absence of reduction waves in the cyclic voltammograms, the LUMO energy levels were calculated from the optical energy gaps Egopt and the HOMO energy levels. As well the LUMOs are destabilized with increasing number of pyrrole rings.
![[1860-5397-16-214-3]](/bjoc/content/figures/1860-5397-16-214-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Energy diagram of the frontier molecular orbitals of heterotetracenes TTA, 9, 13, 19, 22, and 33, and heterohexacene 22.
Figure 3: Energy diagram of the frontier molecular orbitals of heterotetracenes TTA, 9, 13, 19, 22, and 33, a...
Conclusion
In summary, we have presented the synthesis and characterization of novel S,N-heterotetracenes and -hexacene with varying the number and sequence of the sulfur and nitrogen heteroatoms which complement the general series of S,N-heteroacenes. Various cyclization methods to eventually build up pyrrole rings in the tetraheterocyclic systems have been tested and used to prepare SN4, SN4'', and benzannulated SN4' derivatives. Among them, the Buchwald–Hartwig amination of brominated precursors, the thermolysis of azide precursors, and a Cadogan cyclization of nitro-substituted precursors were successfully applied. The various heteroatom sequences ‘SSNS’, ‘SNNS’, and benzannulated ‘NSSN’ allowed for evaluation of structure–property relationships relative to sulfur analogue tetrathienoacene (‘SSSS’). In line with the results for the whole series of S,N-heteroacenes, we found that replacement of sulfur by nitrogen atoms in the tetracyclic systems leads to a red-shift in absorption, a decrease in oxidation potential and energy gap. On the other hand, the replacement of a thiophene ring by benzene leads to the opposite effects. The basic SN4 and SN4'' frameworks represent novel core molecules for further functionalization, for example with terminal acceptor groups, leading to highly absorbing dye molecules for application in organic solar cells.
References
-
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Wu, W.; Liu, Y.; Zhu, D. Chem. Soc. Rev. 2010, 39, 1489–1502. doi:10.1039/b813123f
Return to citation in text: [1] -
Mori, T.; Nishimura, T.; Yamamoto, T.; Doi, I.; Miyazaki, E.; Osaka, I.; Takimiya, K. J. Am. Chem. Soc. 2013, 135, 13900–13913. doi:10.1021/ja406257u
Return to citation in text: [1] -
Aragó, J.; Viruela, P. M.; Ortí, E.; Malavé Osuna, R.; Vercelli, B.; Zotti, G.; Hernández, V.; López Navarrete, J. T.; Henssler, J. T.; Matzger, A. J.; Suzuki, Y.; Yamaguchi, S. Chem. – Eur. J. 2010, 16, 5481–5491. doi:10.1002/chem.200903343
Return to citation in text: [1] -
Okamoto, T.; Kudoh, K.; Wakamiya, A.; Yamaguchi, S. Chem. – Eur. J. 2007, 13, 548–556. doi:10.1002/chem.200601064
Return to citation in text: [1] -
Zhang, X.; Côté, A. P.; Matzger, A. J. J. Am. Chem. Soc. 2005, 127, 10502–10503. doi:10.1021/ja053326m
Return to citation in text: [1] [2] -
Wetzel, C.; Brier, E.; Vogt, A.; Mishra, A.; Mena-Osteritz, E.; Bäuerle, P. Angew. Chem., Int. Ed. 2015, 54, 12334–12338. doi:10.1002/anie.201502840
Return to citation in text: [1] [2] [3] [4] -
Snyder, H. R.; Carpino, L. A.; Zack, J. F., Jr.; Mills, J. F. J. Am. Chem. Soc. 1957, 79, 2556–2559. doi:10.1021/ja01567a053
Return to citation in text: [1] -
Zanirato, P.; Spagnolo, P.; Zanardi, G. J. Chem. Soc., Perkin Trans. 1 1983, 2551–2554. doi:10.1039/p19830002551
Return to citation in text: [1] [2] -
Bulumulla, C.; Gunawardhana, R.; Gamage, P. L.; Miller, J. T.; Kularatne, R. N.; Biewer, M. C.; Stefan, M. C. ACS Appl. Mater. Interfaces 2020, 12, 32209–32232. doi:10.1021/acsami.0c07161
Return to citation in text: [1] -
Ogawa, K.; Rasmussen, S. C. J. Org. Chem. 2003, 68, 2921–2928. doi:10.1021/jo034078k
Return to citation in text: [1] -
Nozaki, K.; Takahashi, K.; Nakano, K.; Hiyama, T.; Tang, H.-Z.; Fujiki, M.; Yamaguchi, S.; Tamao, K. Angew. Chem., Int. Ed. 2003, 42, 2051–2053. doi:10.1002/anie.200250648
Return to citation in text: [1] -
Mitsudo, K.; Shimohara, S.; Mizoguchi, J.; Mandai, H.; Suga, S. Org. Lett. 2012, 14, 2702–2705. doi:10.1021/ol300887t
Return to citation in text: [1] -
Extended S,N-heteroacenes were named according the number (n) of fused five-membered rings and the heteroatoms (S, N) to SNn.
Return to citation in text: [1] -
Wetzel, C.; Vogt, A.; Rudnick, A.; Mena-Osteritz, E.; Köhler, A.; Bäuerle, P. Org. Chem. Front. 2017, 4, 1629–1635. doi:10.1039/c7qo00294g
Return to citation in text: [1] -
Rudnick, A.; Wetzel, C.; Tscheuschner, S.; Schmalz, H.; Vogt, A.; Greiner, A.; Bässler, H.; Mena-Osteritz, E.; Bäuerle, P.; Köhler, A. J. Phys. Chem. B 2017, 121, 7492–7501. doi:10.1021/acs.jpcb.7b02935
Return to citation in text: [1] -
Brier, E.; Wetzel, C.; Bauer, M.; Mena-Osteritz, E.; Wunderlin, M.; Bäuerle, P. Chem. Mater. 2019, 31, 7007–7023. doi:10.1021/acs.chemmater.9b01652
Return to citation in text: [1] [2] [3] -
Ata, I.; Dkhil, S. B.; Pfannmöller, M.; Bals, S.; Duché, D.; Simon, J.-J.; Koganezawa, T.; Yoshimoto, N.; Videlot-Ackermann, C.; Margeat, O.; Ackermann, J.; Bäuerle, P. Org. Chem. Front. 2017, 4, 1561–1573. doi:10.1039/c7qo00222j
Return to citation in text: [1] [2] -
Mishra, A.; Popovic, D.; Vogt, A.; Kast, H.; Leitner, T.; Walzer, K.; Pfeiffer, M.; Mena-Osteritz, E.; Bäuerle, P. Adv. Mater. (Weinheim, Ger.) 2014, 26, 7217–7223. doi:10.1002/adma.201402448
Return to citation in text: [1] [2] -
Leitner, T. D.; Vogt, A.; Popović, D.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. Mater. Chem. Front. 2018, 2, 959–968. doi:10.1039/c7qm00542c
Return to citation in text: [1] [2] [3] [4] -
Chen, X.; Liu, H.; Xia, L.; Hayat, T.; Alsaedi, A.; Tan, Z. Chem. Commun. 2019, 55, 7057–7060. doi:10.1039/c9cc03536b
Return to citation in text: [1] [2] -
Steck, C.; Franckevičius, M.; Zakeeruddin, S. M.; Mishra, A.; Bäuerle, P.; Grätzel, M. J. Mater. Chem. A 2015, 3, 17738–17746. doi:10.1039/c5ta03865k
Return to citation in text: [1] [2] -
Meng, L.; Xia, D.; Liu, S.; Yi, X.; Ding, F.; Fan, R.; Yang, Y. Chem. Lett. 2020, 49, 947–951. doi:10.1246/cl.200290
Return to citation in text: [1] [2] -
Wetzel, C.; Mishra, A.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. J. Mater. Chem. C 2016, 4, 3715–3725. doi:10.1039/c5tc03539b
Return to citation in text: [1] [2] -
Wang, C.-K.; Jiang, B.-H.; Lu, J.-H.; Cheng, M.-T.; Jeng, R.-J.; Lu, Y.-W.; Chen, C.-P.; Wong, K.-T. ChemSusChem 2020, 13, 903–913. doi:10.1002/cssc.201903087
Return to citation in text: [1] [2] -
Huang, C.; Liao, X.; Gao, K.; Zuo, L.; Lin, F.; Shi, X.; Li, C.-Z.; Liu, H.; Li, X.; Liu, F.; Chen, Y.; Chen, H.; Jen, A. K.-Y. Chem. Mater. 2018, 30, 5429–5434. doi:10.1021/acs.chemmater.8b02276
Return to citation in text: [1] [2] -
Arora, N.; Wetzel, C.; Dar, M. I.; Mishra, A.; Yadav, P.; Steck, C.; Zakeeruddin, S. M.; Bäuerle, P.; Grätzel, M. ACS Appl. Mater. Interfaces 2017, 9, 44423–44428. doi:10.1021/acsami.7b10039
Return to citation in text: [1] [2] -
Huang, Y.-F.; Wang, C.-K.; Lai, B.-H.; Chung, C.-L.; Chen, C.-Y.; Ciou, G.-T.; Wong, K.-T.; Wang, C.-L. ACS Appl. Mater. Interfaces 2019, 11, 21756–21765. doi:10.1021/acsami.9b04284
Return to citation in text: [1] [2] [3] -
De Simone, B. C.; Marino, T.; Prejanò, M.; Russo, N. Theor. Chem. Acc. 2016, 135, 238. doi:10.1007/s00214-016-1994-6
Return to citation in text: [1] -
Biswas, S.; Pramanik, A.; Pal, S.; Sarkar, P. J. Phys. Chem. C 2017, 121, 2574–2587. doi:10.1021/acs.jpcc.6b11471
Return to citation in text: [1] -
Roy, P.; Biswas, S.; Pramanik, A.; Sarkar, P. Chem. Phys. Lett. 2018, 708, 87–93. doi:10.1016/j.cplett.2018.08.007
Return to citation in text: [1] -
Mazaki, Y.; Kobayashi, K. Tetrahedron Lett. 1989, 30, 3315–3318. doi:10.1016/s0040-4039(00)99231-1
Return to citation in text: [1] -
Youn, J.; Huang, P.-Y.; Huang, Y.-W.; Chen, M.-C.; Lin, Y.-J.; Huang, H.; Ortiz, R. P.; Stern, C.; Chung, M.-C.; Feng, C.-Y.; Chen, L.-H.; Facchetti, A.; Marks, T. J. Adv. Funct. Mater. 2012, 22, 48–60. doi:10.1002/adfm.201101053
Return to citation in text: [1] -
Wang, Z.; Liang, M.; Dong, H.; Gao, P.; Su, Y.; Cai, P.; Ding, S.; Chen, J.; Xue, S. Org. Lett. 2017, 19, 3711–3714. doi:10.1021/acs.orglett.7b01465
Return to citation in text: [1] -
Tasior, M.; Czichy, M.; Łapkowski, M.; Gryko, D. T. Chem. – Asian J. 2018, 13, 449–456. doi:10.1002/asia.201701639
Return to citation in text: [1] -
Janiga, A.; Krzeszewski, M.; Gryko, D. T. Chem. – Asian J. 2015, 10, 212–218. doi:10.1002/asia.201402925
Return to citation in text: [1] -
Wetzel, C. Novel Thiophene-Pyrrole-Containing S,N-Heteroacenes: Synthesis, Characterization, and Application in Organic Solar Cells. Ph.D. Thesis, University of Ulm, Ulm, Germany, 2016.
Return to citation in text: [1] [2] [3] -
Fuller, L. S.; Iddon, B.; Smith, K. A. J. Chem. Soc., Perkin Trans. 1 1997, 3465–3470. doi:10.1039/a701877k
Return to citation in text: [1] -
Dogru, M.; Handloser, M.; Auras, F.; Kunz, T.; Medina, D.; Hartschuh, A.; Knochel, P.; Bein, T. Angew. Chem., Int. Ed. 2013, 52, 2920–2924. doi:10.1002/anie.201208514
Return to citation in text: [1] -
Kaur, M.; Kumar, R. ChemistrySelect 2018, 3, 5330–5340. doi:10.1002/slct.201800779
Return to citation in text: [1] -
Freeman, A. W.; Urvoy, M.; Criswell, M. E. J. Org. Chem. 2005, 70, 5014–5019. doi:10.1021/jo0503299
Return to citation in text: [1] -
Balaji, G.; Phua, D. I.; Shim, W. L.; Valiyaveettil, S. Org. Lett. 2010, 12, 232–235. doi:10.1021/ol902528b
Return to citation in text: [1] [2] -
Qi, T.; Guo, Y.; Liu, Y.; Xi, H.; Zhang, H.; Gao, X.; Liu, Y.; Lu, K.; Du, C.; Yu, G.; Zhu, D. Chem. Commun. 2008, 6227. doi:10.1039/b813683a
Return to citation in text: [1] -
Balaji, G.; Della Pelle, A. M.; Popere, B. C.; Chandrasekaran, A.; Thayumanavan, S. Org. Biomol. Chem. 2012, 10, 3455–3462. doi:10.1039/c2ob25087j
Return to citation in text: [1] -
Förtsch, S.; Vogt, A.; Bäuerle, P. J. Phys. Org. Chem. 2017, 30, e3743. doi:10.1002/poc.3743
Return to citation in text: [1] [2] -
O'Brien, A. G.; Lévesque, F.; Seeberger, P. H. Chem. Commun. 2011, 47, 2688–2690. doi:10.1039/c0cc04481d
Return to citation in text: [1] -
Srinivasan, S.; Schuster, G. B. Org. Lett. 2008, 10, 3657–3660. doi:10.1021/ol801137t
Return to citation in text: [1] -
Zhou, J.; Tang, R.; Wang, X.; Zhang, W.; Zhuang, X.; Zhang, F. J. Mater. Chem. C 2016, 4, 1159–1164. doi:10.1039/c5tc04139b
Return to citation in text: [1]
| 6. | Zhang, X.; Côté, A. P.; Matzger, A. J. J. Am. Chem. Soc. 2005, 127, 10502–10503. doi:10.1021/ja053326m |
| 9. | Zanirato, P.; Spagnolo, P.; Zanardi, G. J. Chem. Soc., Perkin Trans. 1 1983, 2551–2554. doi:10.1039/p19830002551 |
| 40. | Kaur, M.; Kumar, R. ChemistrySelect 2018, 3, 5330–5340. doi:10.1002/slct.201800779 |
| 1. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 2. | Wu, W.; Liu, Y.; Zhu, D. Chem. Soc. Rev. 2010, 39, 1489–1502. doi:10.1039/b813123f |
| 3. | Mori, T.; Nishimura, T.; Yamamoto, T.; Doi, I.; Miyazaki, E.; Osaka, I.; Takimiya, K. J. Am. Chem. Soc. 2013, 135, 13900–13913. doi:10.1021/ja406257u |
| 9. | Zanirato, P.; Spagnolo, P.; Zanardi, G. J. Chem. Soc., Perkin Trans. 1 1983, 2551–2554. doi:10.1039/p19830002551 |
| 28. | Huang, Y.-F.; Wang, C.-K.; Lai, B.-H.; Chung, C.-L.; Chen, C.-Y.; Ciou, G.-T.; Wong, K.-T.; Wang, C.-L. ACS Appl. Mater. Interfaces 2019, 11, 21756–21765. doi:10.1021/acsami.9b04284 |
| 47. | Srinivasan, S.; Schuster, G. B. Org. Lett. 2008, 10, 3657–3660. doi:10.1021/ol801137t |
| 8. | Snyder, H. R.; Carpino, L. A.; Zack, J. F., Jr.; Mills, J. F. J. Am. Chem. Soc. 1957, 79, 2556–2559. doi:10.1021/ja01567a053 |
| 23. | Meng, L.; Xia, D.; Liu, S.; Yi, X.; Ding, F.; Fan, R.; Yang, Y. Chem. Lett. 2020, 49, 947–951. doi:10.1246/cl.200290 |
| 20. | Leitner, T. D.; Vogt, A.; Popović, D.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. Mater. Chem. Front. 2018, 2, 959–968. doi:10.1039/c7qm00542c |
| 7. | Wetzel, C.; Brier, E.; Vogt, A.; Mishra, A.; Mena-Osteritz, E.; Bäuerle, P. Angew. Chem., Int. Ed. 2015, 54, 12334–12338. doi:10.1002/anie.201502840 |
| 18. | Ata, I.; Dkhil, S. B.; Pfannmöller, M.; Bals, S.; Duché, D.; Simon, J.-J.; Koganezawa, T.; Yoshimoto, N.; Videlot-Ackermann, C.; Margeat, O.; Ackermann, J.; Bäuerle, P. Org. Chem. Front. 2017, 4, 1561–1573. doi:10.1039/c7qo00222j |
| 19. | Mishra, A.; Popovic, D.; Vogt, A.; Kast, H.; Leitner, T.; Walzer, K.; Pfeiffer, M.; Mena-Osteritz, E.; Bäuerle, P. Adv. Mater. (Weinheim, Ger.) 2014, 26, 7217–7223. doi:10.1002/adma.201402448 |
| 20. | Leitner, T. D.; Vogt, A.; Popović, D.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. Mater. Chem. Front. 2018, 2, 959–968. doi:10.1039/c7qm00542c |
| 21. | Chen, X.; Liu, H.; Xia, L.; Hayat, T.; Alsaedi, A.; Tan, Z. Chem. Commun. 2019, 55, 7057–7060. doi:10.1039/c9cc03536b |
| 24. | Wetzel, C.; Mishra, A.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. J. Mater. Chem. C 2016, 4, 3715–3725. doi:10.1039/c5tc03539b |
| 25. | Wang, C.-K.; Jiang, B.-H.; Lu, J.-H.; Cheng, M.-T.; Jeng, R.-J.; Lu, Y.-W.; Chen, C.-P.; Wong, K.-T. ChemSusChem 2020, 13, 903–913. doi:10.1002/cssc.201903087 |
| 26. | Huang, C.; Liao, X.; Gao, K.; Zuo, L.; Lin, F.; Shi, X.; Li, C.-Z.; Liu, H.; Li, X.; Liu, F.; Chen, Y.; Chen, H.; Jen, A. K.-Y. Chem. Mater. 2018, 30, 5429–5434. doi:10.1021/acs.chemmater.8b02276 |
| 28. | Huang, Y.-F.; Wang, C.-K.; Lai, B.-H.; Chung, C.-L.; Chen, C.-Y.; Ciou, G.-T.; Wong, K.-T.; Wang, C.-L. ACS Appl. Mater. Interfaces 2019, 11, 21756–21765. doi:10.1021/acsami.9b04284 |
| 4. | Aragó, J.; Viruela, P. M.; Ortí, E.; Malavé Osuna, R.; Vercelli, B.; Zotti, G.; Hernández, V.; López Navarrete, J. T.; Henssler, J. T.; Matzger, A. J.; Suzuki, Y.; Yamaguchi, S. Chem. – Eur. J. 2010, 16, 5481–5491. doi:10.1002/chem.200903343 |
| 5. | Okamoto, T.; Kudoh, K.; Wakamiya, A.; Yamaguchi, S. Chem. – Eur. J. 2007, 13, 548–556. doi:10.1002/chem.200601064 |
| 6. | Zhang, X.; Côté, A. P.; Matzger, A. J. J. Am. Chem. Soc. 2005, 127, 10502–10503. doi:10.1021/ja053326m |
| 22. | Steck, C.; Franckevičius, M.; Zakeeruddin, S. M.; Mishra, A.; Bäuerle, P.; Grätzel, M. J. Mater. Chem. A 2015, 3, 17738–17746. doi:10.1039/c5ta03865k |
| 27. | Arora, N.; Wetzel, C.; Dar, M. I.; Mishra, A.; Yadav, P.; Steck, C.; Zakeeruddin, S. M.; Bäuerle, P.; Grätzel, M. ACS Appl. Mater. Interfaces 2017, 9, 44423–44428. doi:10.1021/acsami.7b10039 |
| 46. | O'Brien, A. G.; Lévesque, F.; Seeberger, P. H. Chem. Commun. 2011, 47, 2688–2690. doi:10.1039/c0cc04481d |
| 7. | Wetzel, C.; Brier, E.; Vogt, A.; Mishra, A.; Mena-Osteritz, E.; Bäuerle, P. Angew. Chem., Int. Ed. 2015, 54, 12334–12338. doi:10.1002/anie.201502840 |
| 15. | Wetzel, C.; Vogt, A.; Rudnick, A.; Mena-Osteritz, E.; Köhler, A.; Bäuerle, P. Org. Chem. Front. 2017, 4, 1629–1635. doi:10.1039/c7qo00294g |
| 16. | Rudnick, A.; Wetzel, C.; Tscheuschner, S.; Schmalz, H.; Vogt, A.; Greiner, A.; Bässler, H.; Mena-Osteritz, E.; Bäuerle, P.; Köhler, A. J. Phys. Chem. B 2017, 121, 7492–7501. doi:10.1021/acs.jpcb.7b02935 |
| 17. | Brier, E.; Wetzel, C.; Bauer, M.; Mena-Osteritz, E.; Wunderlin, M.; Bäuerle, P. Chem. Mater. 2019, 31, 7007–7023. doi:10.1021/acs.chemmater.9b01652 |
| 19. | Mishra, A.; Popovic, D.; Vogt, A.; Kast, H.; Leitner, T.; Walzer, K.; Pfeiffer, M.; Mena-Osteritz, E.; Bäuerle, P. Adv. Mater. (Weinheim, Ger.) 2014, 26, 7217–7223. doi:10.1002/adma.201402448 |
| 20. | Leitner, T. D.; Vogt, A.; Popović, D.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. Mater. Chem. Front. 2018, 2, 959–968. doi:10.1039/c7qm00542c |
| 21. | Chen, X.; Liu, H.; Xia, L.; Hayat, T.; Alsaedi, A.; Tan, Z. Chem. Commun. 2019, 55, 7057–7060. doi:10.1039/c9cc03536b |
| 22. | Steck, C.; Franckevičius, M.; Zakeeruddin, S. M.; Mishra, A.; Bäuerle, P.; Grätzel, M. J. Mater. Chem. A 2015, 3, 17738–17746. doi:10.1039/c5ta03865k |
| 23. | Meng, L.; Xia, D.; Liu, S.; Yi, X.; Ding, F.; Fan, R.; Yang, Y. Chem. Lett. 2020, 49, 947–951. doi:10.1246/cl.200290 |
| 45. | Förtsch, S.; Vogt, A.; Bäuerle, P. J. Phys. Org. Chem. 2017, 30, e3743. doi:10.1002/poc.3743 |
| 13. | Mitsudo, K.; Shimohara, S.; Mizoguchi, J.; Mandai, H.; Suga, S. Org. Lett. 2012, 14, 2702–2705. doi:10.1021/ol300887t |
| 14. | Extended S,N-heteroacenes were named according the number (n) of fused five-membered rings and the heteroatoms (S, N) to SNn. |
| 24. | Wetzel, C.; Mishra, A.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. J. Mater. Chem. C 2016, 4, 3715–3725. doi:10.1039/c5tc03539b |
| 25. | Wang, C.-K.; Jiang, B.-H.; Lu, J.-H.; Cheng, M.-T.; Jeng, R.-J.; Lu, Y.-W.; Chen, C.-P.; Wong, K.-T. ChemSusChem 2020, 13, 903–913. doi:10.1002/cssc.201903087 |
| 26. | Huang, C.; Liao, X.; Gao, K.; Zuo, L.; Lin, F.; Shi, X.; Li, C.-Z.; Liu, H.; Li, X.; Liu, F.; Chen, Y.; Chen, H.; Jen, A. K.-Y. Chem. Mater. 2018, 30, 5429–5434. doi:10.1021/acs.chemmater.8b02276 |
| 27. | Arora, N.; Wetzel, C.; Dar, M. I.; Mishra, A.; Yadav, P.; Steck, C.; Zakeeruddin, S. M.; Bäuerle, P.; Grätzel, M. ACS Appl. Mater. Interfaces 2017, 9, 44423–44428. doi:10.1021/acsami.7b10039 |
| 28. | Huang, Y.-F.; Wang, C.-K.; Lai, B.-H.; Chung, C.-L.; Chen, C.-Y.; Ciou, G.-T.; Wong, K.-T.; Wang, C.-L. ACS Appl. Mater. Interfaces 2019, 11, 21756–21765. doi:10.1021/acsami.9b04284 |
| 42. | Balaji, G.; Phua, D. I.; Shim, W. L.; Valiyaveettil, S. Org. Lett. 2010, 12, 232–235. doi:10.1021/ol902528b |
| 11. | Ogawa, K.; Rasmussen, S. C. J. Org. Chem. 2003, 68, 2921–2928. doi:10.1021/jo034078k |
| 12. | Nozaki, K.; Takahashi, K.; Nakano, K.; Hiyama, T.; Tang, H.-Z.; Fujiki, M.; Yamaguchi, S.; Tamao, K. Angew. Chem., Int. Ed. 2003, 42, 2051–2053. doi:10.1002/anie.200250648 |
| 41. | Freeman, A. W.; Urvoy, M.; Criswell, M. E. J. Org. Chem. 2005, 70, 5014–5019. doi:10.1021/jo0503299 |
| 10. | Bulumulla, C.; Gunawardhana, R.; Gamage, P. L.; Miller, J. T.; Kularatne, R. N.; Biewer, M. C.; Stefan, M. C. ACS Appl. Mater. Interfaces 2020, 12, 32209–32232. doi:10.1021/acsami.0c07161 |
| 18. | Ata, I.; Dkhil, S. B.; Pfannmöller, M.; Bals, S.; Duché, D.; Simon, J.-J.; Koganezawa, T.; Yoshimoto, N.; Videlot-Ackermann, C.; Margeat, O.; Ackermann, J.; Bäuerle, P. Org. Chem. Front. 2017, 4, 1561–1573. doi:10.1039/c7qo00222j |
| 42. | Balaji, G.; Phua, D. I.; Shim, W. L.; Valiyaveettil, S. Org. Lett. 2010, 12, 232–235. doi:10.1021/ol902528b |
| 43. | Qi, T.; Guo, Y.; Liu, Y.; Xi, H.; Zhang, H.; Gao, X.; Liu, Y.; Lu, K.; Du, C.; Yu, G.; Zhu, D. Chem. Commun. 2008, 6227. doi:10.1039/b813683a |
| 44. | Balaji, G.; Della Pelle, A. M.; Popere, B. C.; Chandrasekaran, A.; Thayumanavan, S. Org. Biomol. Chem. 2012, 10, 3455–3462. doi:10.1039/c2ob25087j |
| 45. | Förtsch, S.; Vogt, A.; Bäuerle, P. J. Phys. Org. Chem. 2017, 30, e3743. doi:10.1002/poc.3743 |
| 31. | Roy, P.; Biswas, S.; Pramanik, A.; Sarkar, P. Chem. Phys. Lett. 2018, 708, 87–93. doi:10.1016/j.cplett.2018.08.007 |
| 29. | De Simone, B. C.; Marino, T.; Prejanò, M.; Russo, N. Theor. Chem. Acc. 2016, 135, 238. doi:10.1007/s00214-016-1994-6 |
| 17. | Brier, E.; Wetzel, C.; Bauer, M.; Mena-Osteritz, E.; Wunderlin, M.; Bäuerle, P. Chem. Mater. 2019, 31, 7007–7023. doi:10.1021/acs.chemmater.9b01652 |
| 30. | Biswas, S.; Pramanik, A.; Pal, S.; Sarkar, P. J. Phys. Chem. C 2017, 121, 2574–2587. doi:10.1021/acs.jpcc.6b11471 |
| 20. | Leitner, T. D.; Vogt, A.; Popović, D.; Mena-Osteritz, E.; Walzer, K.; Pfeiffer, M.; Bäuerle, P. Mater. Chem. Front. 2018, 2, 959–968. doi:10.1039/c7qm00542c |
| 17. | Brier, E.; Wetzel, C.; Bauer, M.; Mena-Osteritz, E.; Wunderlin, M.; Bäuerle, P. Chem. Mater. 2019, 31, 7007–7023. doi:10.1021/acs.chemmater.9b01652 |
| 37. | Wetzel, C. Novel Thiophene-Pyrrole-Containing S,N-Heteroacenes: Synthesis, Characterization, and Application in Organic Solar Cells. Ph.D. Thesis, University of Ulm, Ulm, Germany, 2016. |
| 39. | Dogru, M.; Handloser, M.; Auras, F.; Kunz, T.; Medina, D.; Hartschuh, A.; Knochel, P.; Bein, T. Angew. Chem., Int. Ed. 2013, 52, 2920–2924. doi:10.1002/anie.201208514 |
| 38. | Fuller, L. S.; Iddon, B.; Smith, K. A. J. Chem. Soc., Perkin Trans. 1 1997, 3465–3470. doi:10.1039/a701877k |
| 36. | Janiga, A.; Krzeszewski, M.; Gryko, D. T. Chem. – Asian J. 2015, 10, 212–218. doi:10.1002/asia.201402925 |
| 37. | Wetzel, C. Novel Thiophene-Pyrrole-Containing S,N-Heteroacenes: Synthesis, Characterization, and Application in Organic Solar Cells. Ph.D. Thesis, University of Ulm, Ulm, Germany, 2016. |
| 34. | Wang, Z.; Liang, M.; Dong, H.; Gao, P.; Su, Y.; Cai, P.; Ding, S.; Chen, J.; Xue, S. Org. Lett. 2017, 19, 3711–3714. doi:10.1021/acs.orglett.7b01465 |
| 35. | Tasior, M.; Czichy, M.; Łapkowski, M.; Gryko, D. T. Chem. – Asian J. 2018, 13, 449–456. doi:10.1002/asia.201701639 |
| 32. | Mazaki, Y.; Kobayashi, K. Tetrahedron Lett. 1989, 30, 3315–3318. doi:10.1016/s0040-4039(00)99231-1 |
| 33. | Youn, J.; Huang, P.-Y.; Huang, Y.-W.; Chen, M.-C.; Lin, Y.-J.; Huang, H.; Ortiz, R. P.; Stern, C.; Chung, M.-C.; Feng, C.-Y.; Chen, L.-H.; Facchetti, A.; Marks, T. J. Adv. Funct. Mater. 2012, 22, 48–60. doi:10.1002/adfm.201101053 |
| 48. | Zhou, J.; Tang, R.; Wang, X.; Zhang, W.; Zhuang, X.; Zhang, F. J. Mater. Chem. C 2016, 4, 1159–1164. doi:10.1039/c5tc04139b |
| 7. | Wetzel, C.; Brier, E.; Vogt, A.; Mishra, A.; Mena-Osteritz, E.; Bäuerle, P. Angew. Chem., Int. Ed. 2015, 54, 12334–12338. doi:10.1002/anie.201502840 |
| 7. | Wetzel, C.; Brier, E.; Vogt, A.; Mishra, A.; Mena-Osteritz, E.; Bäuerle, P. Angew. Chem., Int. Ed. 2015, 54, 12334–12338. doi:10.1002/anie.201502840 |
| 37. | Wetzel, C. Novel Thiophene-Pyrrole-Containing S,N-Heteroacenes: Synthesis, Characterization, and Application in Organic Solar Cells. Ph.D. Thesis, University of Ulm, Ulm, Germany, 2016. |
© 2020 Vogt et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)