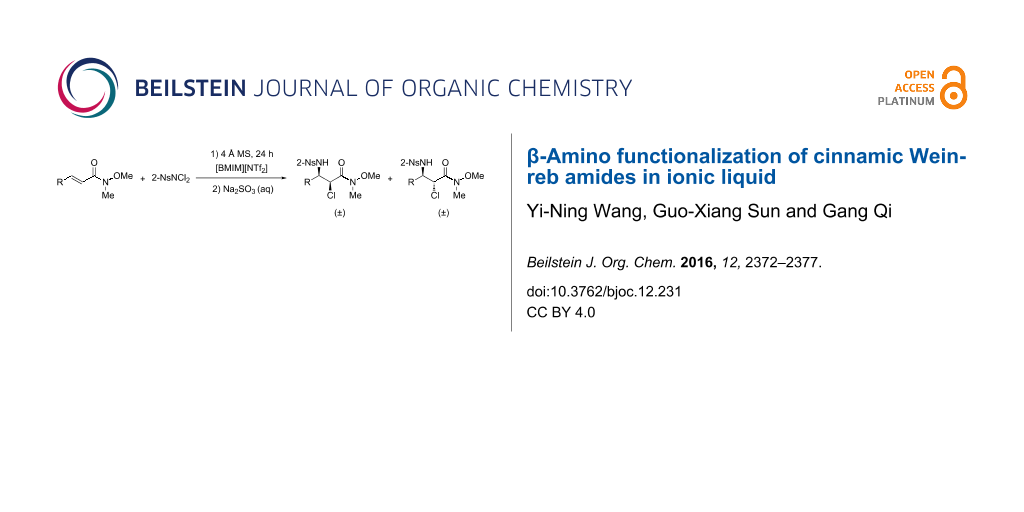
Abstract
2-Ns-Protected β-amino Weinreb amides were synthesized by aminochlorination of α,β-unsaturated Weinreb amides in an ionic liquid, 1-n-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([BMIM][NTf2]). Processed without the use of metal catalysts or the need of an inert gas atmosphere, the presented process can be readily performed as a one-pot synthesis at room temperature. Moreover, the preparation has the distinct advantages of the use of 2-NsNCl2 as an inexpensive and stable nitrogen/halogen source and the ionic liquid as a recyclable reaction media. Nine examples were examined, and modest to good isolated chemical yields (40–83%) were obtained.

Graphical Abstract
Introduction
Unlike N-protected α-amino carbonyl compounds, their analogues, the β-amino carbonyl compounds, have drawn relatively little attention. However, the β-amino carbonyl moieties are not only found in natural products [1-3], e.g., in the side chain of Taxol, but also in important building blocks widely used in the organic syntheses. They are especially used in the synthesis of β-amino acids [4,5], which are precursors for many biological and pharmacological active compounds, such as β-lactams [6] and β-peptides [7]. The most straightforward approach towards the preparation of β-amino carbonyl compounds is the Mannich reaction [8,9]. However, there are few reported methods specifically focused on this target, which include oxidation of γ-amino alcohols [10], hydrolysis of 1,3-oxazines [11], rearrangement of 2,3-aziridinio alcohols [12], etc.
N-Methoxy-N-methylamides, known as Weinreb amides, were first reported by Nahm and Weinreb in 1981 [13]. Since then, the Weinreb amides were found to be useful precursors for the synthesis of their carbonyl equivalents in organic chemistry [14]. By treating with various nucleophiles, such as hydrides, Grignard and organolithium reagents or ester enolates, the Weinreb amides can be easily converted into aldehydes, ketones, and β-keto esters, respectively [13-20]. Therefore, the transformation of β-amino Weinreb amides can provide a promising pathway towards the achievement of β-amino carbonyl derivatives. Surprisingly, there have been only a few successful preparations for β-amino Weinreb amides to date. One of which is the addition of the enolate of commercially available N-methoxy-N-methylacetamide to sulfinimines to generate the corresponding N-sulfinyl β-amino Weinreb amides [21-26]. Another approach was the direct amination of α,β-unsaturated Weinreb amides by using lithium (S)-N-benzyl-N-α-methylbenzylamide as the nitrogen source [27-29]. Recently, a new chiral N-phosphonylimine chemistry was developed by the Li group. By reacting N-phosphonylimines 1 with Weinreb amides, N-phosphonyl-β-amino Weinreb amides 2 can be obtained (Scheme 1) [30]. Although great progress has already been achieved, there were still some limitations in the existing syntheses of β-amino Weinreb amides, such as the difficult syntheses of starting materials or the harsh reaction conditions required for the removal of the N-protecting groups.
![[1860-5397-12-231-i1]](/bjoc/content/inline/1860-5397-12-231-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of N-phosphonyl-β-amino Weinreb amides.
Scheme 1: Synthesis of N-phosphonyl-β-amino Weinreb amides.
With the aminochlorination developed in the past few years [31], we found that when the aminochlorination reactions of α,β-unsaturated ketones 3 were carried out in ionic liquid, [BMIM][NTf2] (1-n-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide, Tf = SO2CF3), a series of α-chloro-β-amino ketone derivatives 4 could be obtained with excellent regioselectivities (Scheme 2) [32]. Encouraged by these promising results, we thought that the α,β-unsaturated Weinreb amides could be potential substrates for the preparation of β-amino Weinreb amides. Compared with the reported methods, there are several advantages for the proposed aminochlorination reactions: 1) it is much easier to achieve the starting materials, including the α,β-unsaturated Weinreb amides and the nitrogen sources, such as 4-TsNCl2 and 2-NsNCl2; 2) the removal of the N-protecting groups is relatively easier than those shown above; and 3) the ionic liquids are environmentally friendly and can be readily recycled.
![[1860-5397-12-231-i2]](/bjoc/content/inline/1860-5397-12-231-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Aminochlorination of α,β-unsaturated ketones in ionic liquid.
Scheme 2: Aminochlorination of α,β-unsaturated ketones in ionic liquid.
Results and Discussion
The first attempt was to conduct the aminochlorination reaction of N-methoxy-N-methylcinnamoylamide (5a) in acetonitrile by using 2-NsNCl2 as the nitrogen source and Cu(I)OTf as the metal catalyst [33,34]. However, there no aminochlorination products were observed. Next, we turned our attention to the use of ionic liquids, which have shown many significant advantages in the reported aminochlorination reactions [35-37]. However, when the ionic liquid [BMIM][BF4] was utilized as the reaction media, no anticipated haloamine products were detected, which was similar to the aminochlorination of α,β-unsaturated ketones [32]. Fortunately, when we switched the ionic liquid to [BMIM][NTf2], it was found that the starting material, N-methoxy-N-methylcinnamoylamide (5a), was consumed completely after 24 h without utilizing any metal catalysts. Furthermore, under the optimized reaction conditions, the isolated chemical yield of the β-amino product could be increased up to 81% (Table 1). A series of common α,β-unsaturated Weinreb amides were systematically investigated as well. Similarly, the aminochlorination reactions proceeded smoothly, and the anticipated α-chloro-β-amino products were obtained in modest to good yields and excellent regioselectivities (Table 2).
Table 1: Optimization of aminochlorination of N-methoxy-N-methylcinnamoylamide (5a).
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-231-i7.svg?max-width=637&scale=1.0)
|
|||
| Entry | Temp (°C) | Cu(I)OTf | Yield (%)a |
|---|---|---|---|
| 1 | 25 | – | 81 |
| 2 | 25 | 10 mol % | 71 |
| 3 | 80 | – | 60 |
| 4 | 80 | 10 mol % | 61 |
aCombined yields of two isomers purified by column chromatography.
Table 2: Results of aminochlorination of α,β-unsaturated Weinreb amides.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-231-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | R | Product | Yielda (%) | Stereoselectivityb (A:B) |
|---|---|---|---|---|---|
| 1 | 5a | H | 6a | 81 | 1.0:1.3 |
| 2 | 5b | 2-fluoro | 6b | 83 | 1.0:1.5 |
| 3 | 5c | 4-chloro | 6c | 70 | 1.0:1.4 |
| 4 | 5d | 2,4-dichloro | 6d | 79 | 1.0:1.8 |
| 5 | 5e | 4-bromo | 6e | 79 | 1.0:1.3 |
| 6 | 5f | 4-methyl | 6f | 78 | 1.0:1.2 |
| 7 | 5g | 4-methoxy | 6g | 63 | 1.0:1.2 |
| 8 | 5h | 4-phenyl | 6h | 69 | 1.0:0.7 |
| 9 | 5i | 4-naphthyl | 6i | 40 | 1.0:0.7 |
aCombined yields of two isomers separated by column chromatography. bDetermined after column chromatography.
Different from the similar aminochlorination reactions of α,β-unsaturated ketones mediated in the same ionic liquid, two different isomers were formed nearly equally. However, these diastereomers can be easily separated by flash column chromatography. The regioselectivities of the isomers were determined by mass spectrometric analyses, both of which prominent peaks corresponding to [PhCHNHNs]+ (m/z = 291) could be clearly observed. The results indicated that the isolated products were both α-chloro-β-amino Weinreb amide derivatives. The assignments for the stereoselectivities were determined by the conversions of the diastereomers to their corresponding aziridines (Scheme 3), and diastereomers 6a-A and 6a-B were chosen as representative substrates. Based on the vicinal Karplus correlation diagram, the vicinal proton coupling constants in the 1H NMR spectra of cis-aziridines are larger than those of trans-azridines [38,39]. In the 1H NMR spectra of the resulting aziridines derived from compounds 6a-A and 6a-B, the vicinal proton coupling constants were 7.5 Hz and 4.5 Hz, respectively. Therefore, compound 6a-A was assigned as syn-stereoisomer, and compound 6a-B was assigned as anti-stereoisomer.
![[1860-5397-12-231-i3]](/bjoc/content/inline/1860-5397-12-231-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Conversion of β-amino Weinreb amides to aziridines.
Scheme 3: Conversion of β-amino Weinreb amides to aziridines.
In the 1H NMR spectra of the isolated diastereomers, an obvious and interesting phenomenon can be seen; namely, the difference of the proton chemical shifts of the hydrogen atoms of the nitrobenzenesulfonylamines. Compared with the chemical shifts of these protons belonging to the syn-stereoisomers, the signals of the same kind of protons of the anti-stereoisomers move to much lower fields (e.g., 8.25 ppm vs 6.33 ppm for compounds 6a-B vs 6a-A). The reason for this phenomenon is probably due to hydrogen bonding between the protons of the nitrobenzenesulfonyl amines and the neighboring carbonyl groups, which form stable six-membered rings as shown in Figure 1. In addition, because of the resonance structures (Scheme 4), the hydrogen bonding can be further strengthened by the oxygen which is more electronegative. As a result, the chemical shifts of these protons belonging to the anti-stereoisomers can move to much lower fields than those of the similar hydrogen atoms of not only the syn-stereoisomers but also the β-amino compounds derived from α,β-unsaturated ketones. This hypothesis can be supported by an unexpected transformation. When aziridine 7-B was purified by flash column chromatography, part of the aziridine ring was opened by a chlorine anion to form the α-amino-β-chloro Weinreb amide derivative 8 (Scheme 5). This could be confirmed by mass spectroscopic analysis as well. In its mass spectrum, a prominent peak denoting [NsNHCHC(O)NMe(OMe)]+ (m/z = 302) was exclusively observed. Because compound 8 was derived from trans-aziridine, it should be the trans-regioisomer of compound 6a-B. As expected, its proton chemical shift of nitrobenzenesulfonylamine was only 6.20 ppm.
![[1860-5397-12-231-1]](/bjoc/content/figures/1860-5397-12-231-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Possible hydrogen bonding conformation.
Figure 1: Possible hydrogen bonding conformation.
![[1860-5397-12-231-i4]](/bjoc/content/inline/1860-5397-12-231-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Resonance structures of Weinreb amides.
Scheme 4: Resonance structures of Weinreb amides.
![[1860-5397-12-231-i5]](/bjoc/content/inline/1860-5397-12-231-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Transformation of trans-aziridine.
Scheme 5: Transformation of trans-aziridine.
As shown in Table 2, nearly 1:1 stereoselectivities were observed in all cases. This indicated that instead of a chlorinium or aziridinium ion intermediate, as hypothesized in the previous analogous reactions, a relatively stable carbocation intermediate was more likely to have formed in this aminochlorination reaction (Scheme 6). Moreover, when the carbocation intermediate was formed, strong electron-withdrawing groups at the phenyl ring in the 3-position of the carbocation are highly disfavored for the aminochlorination reaction. For example, when the nitro group was attached to the phenyl ring, no aminochlorination reaction took place, and only starting material remained even after stirring for a long time.
![[1860-5397-12-231-i6]](/bjoc/content/inline/1860-5397-12-231-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed mechanism of aminochlorination of α,β-unsaturated Weinreb amides.
Scheme 6: Proposed mechanism of aminochlorination of α,β-unsaturated Weinreb amides.
Conclusion
A new approach to afford β-amino Weinreb amides was achieved by the aminochlorination of α,β-unsaturated Weinreb amides. The new process has the advantages that the starting materials can be easily achieved and the N-protecting groups simply removed. Additionally, the reactions can be readily performed in the ionic liquid [BMIM][NTf2] at room temperature without the use of metal catalysts or the need for inert gases protection, and the final products can be easily isolated by flash column chromatography. Compared with the existing methods, the aminochlorination reactions of α,β-unsaturated Weinreb amides provided a promising direction towards the preparation of β-amino Weinreb amides, which have shown great importance in not only organic synthesis, but also in medicinal and pharmaceutical chemistry.
Experimental
General procedure: α,β-Unsaturated Weinreb amides were prepared following literature methods [40]. The ionic liquid [BMIM][NTf2] was readily prepared by reacting 1-methylimidazole with n-butyl bromide [41-43], followed by anion metathesis using N-lithiotrifluoromethanesulfonimide in acetone solution. The resulting ionic liquid, [BMIM][NTf2], was carefully dried by heating at 60 °C in vacuum, then confirmed by 1H NMR analysis [44]. All aminochlorination reactions were performed in oven-dried vials. Flash column chromatography was performed using silica gel (Merck 60, 230–400 mesh). 1H and 13C NMR spectra were obtained with Varian Inova 500 MHz and Varian Mercury Plus 300 MHz spectrometers using deuterated chloroform as a solvent. Internal TMS (δ = 0.0 ppm) was used as the reference for 1H NMR, while the deuterated chloroform peak (δ = 77.0 ppm) was used as the reference for 13C NMR.
Typical procedure for the aminochlorination of α,β-unsaturated Weinreb amides 5a–i: Analogous to the procedure described in [32], N-methoxy-N-methylcinnamoylamide (5a, 96 mg, 0.5 mmol, 1.0 equiv), 4 Å molecular sieves (100 mg), 2-NsNCl2 (163 mg, 0.6 mmol, 1.2 equiv) and [BMIM][NTf2] (500 mg) were loaded into an oven-dried vial. The resulting mixture was stirred at room temperature for 24 h. The reaction was finally quenched with saturated aqueous Na2SO3solution. The product was extracted with ethyl acetate (5 mL × 3) and the combined organic phases were washed with brine and dried over anhydrous Na2SO4. The crude product was subjected to flash column chromatography (EtOAc/dichloromethane/hexane, v/v/v = 1:3:2) to yield 174 mg of product as a white solid (81%).
Typical procedure for the preparation of aziridines 7A and 7B: The β-amino Weinreb amide (6a-A or 6a-B, 70 mg, 0.16 mmol, 1.0 equiv), dichloromethane (5 mL) and triethylamine (0.1 mL) were loaded into an oven-dried vial. After stirring at room temperature for 24 h, the reaction mixture was concentrated to dryness and purified by flash column chromatography (EtOAc/hexane, v/v = 1:3) to yield the product as a white solid.
Supporting Information
| Supporting Information File 1: Full compound characterization data for products 6a–i, 7A, 7B and 8. | ||
| Format: PDF | Size: 165.1 KB | Download |
References
-
Davis, F. A.; Szewczyk, J. M. Tetrahedron Lett. 1998, 39, 5951–5954. doi:10.1016/S0040-4039(98)01223-4
Return to citation in text: [1] -
Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957–5958. doi:10.1016/S0040-4020(02)00584-7
Return to citation in text: [1] -
Seebach, D.; Kimmerlin, T.; Šebesta, R.; Campo, M. A.; Beck, A. K. Tetrahedron 2004, 60, 7455–7506. doi:10.1016/j.tet.2004.06.043
Return to citation in text: [1] -
Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2
Return to citation in text: [1] -
Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Rev. 2007, 107, 4437–4492. doi:10.1021/cr0307300
Return to citation in text: [1] -
Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219–3232. doi:10.1021/cr000045i
Return to citation in text: [1] -
Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 5797–5815. doi:10.1002/ejoc.200700409
Return to citation in text: [1] -
Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29–41. doi:10.1039/b713885g
Return to citation in text: [1] -
Davies, S. B.; McKervey, M. A. Tetrahedron Lett. 1999, 40, 1229–1232. doi:10.1016/S0040-4039(98)02573-8
Return to citation in text: [1] -
Gizecki, P.; Dhal, R.; Toupet, L.; Dujardin, G. Org. Lett. 2000, 2, 585–588. doi:10.1021/ol991326j
Return to citation in text: [1] -
Wang, B. M.; Song, Z. L.; Fan, C. A.; Tu, Y. Q.; Shi, Y. Org. Lett. 2002, 4, 363–366. doi:10.1021/ol0170410
Return to citation in text: [1] -
Nahm, S.; Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815–3818. doi:10.1016/S0040-4039(01)91316-4
Return to citation in text: [1] [2] -
Sibi, M. P. Org. Prep. Proced. Int. 1993, 25, 15–40. doi:10.1080/00304949309457931
Return to citation in text: [1] [2] -
Turner, J. A.; Jacks, W. S. J. Org. Chem. 1989, 54, 4229–4231. doi:10.1021/jo00278a047
Return to citation in text: [1] -
Murphy, J. A.; Commeureuc, A. G. J.; Snaddon, T. N.; McGuire, T. M.; Khan, T. A.; Hisler, K.; Dewis, M. L.; Carling, R. Org. Lett. 2005, 7, 1427–1429. doi:10.1021/ol050337b
Return to citation in text: [1] -
Kojima, S.; Hidaka, T.; Yamakawa, A. Chem. Lett. 2005, 34, 470–471. doi:10.1246/cl.2005.470
Return to citation in text: [1] -
Qu, B.; Collum, D. B. J. Org. Chem. 2006, 71, 7117–7119. doi:10.1021/jo061223w
Return to citation in text: [1] -
Persson, T.; Nielsen, J. Org. Lett. 2006, 8, 3219–3222. doi:10.1021/ol0611088
Return to citation in text: [1] -
Rudzinski, D. M.; Kelly, C. B.; Leadbeater, N. E. Chem. Commun. 2012, 48, 9610–9612. doi:10.1039/C2CC35037H
Return to citation in text: [1] -
Davis, F. A.; Prasad, K. R.; Brad Nolt, M.; Wu, Y. Org. Lett. 2003, 5, 925–927. doi:10.1021/ol034119z
Return to citation in text: [1] -
Davis, F. A.; Yang, B. Org. Lett. 2003, 5, 5011–5014. doi:10.1021/ol035981+
Return to citation in text: [1] -
Davis, F. A.; Brad Nolt, M.; Wu, Y.; Prasad, K. R.; Li, D.; Yang, B.; Bowen, K.; Lee, S. H.; Eardley, J. H. J. Org. Chem. 2005, 70, 2184–2190. doi:10.1021/jo0402780
Return to citation in text: [1] -
Davis, F. A.; Song, M. Org. Lett. 2007, 9, 2413–2416. doi:10.1021/ol0708166
Return to citation in text: [1] -
Davis, F. A.; Theddu, N. J. Org. Chem. 2010, 75, 3814–3820. doi:10.1021/jo100680b
Return to citation in text: [1] -
Davis, F. A.; Xu, P. J. Org. Chem. 2011, 76, 3329–3337. doi:10.1021/jo2002352
Return to citation in text: [1] -
Davies, S. G.; McCarthy, T. D. Synlett 1995, 700–702. doi:10.1055/s-1995-5059
Return to citation in text: [1] -
Davies, S. G.; Iwamoto, K.; Smethurst, C. A. P.; Smith, A. D.; Rodriguez-Solla, H. Synlett 2002, 1146–1148. doi:10.1055/s-2002-32580
Return to citation in text: [1] -
Burke, A. J.; Davies, S. G.; Garner, A. C.; McCarthy, T. D.; Roberts, P. M.; Smith, A. D.; Rodriguez-Solla, H.; Vickers, R. J. Org. Biomol. Chem. 2004, 2, 1387–1394. doi:10.1039/B402531H
Return to citation in text: [1] -
Kaur, P.; Nguyen, T.; Li, G. Eur. J. Org. Chem. 2009, 912–916. doi:10.1002/ejoc.200801061
Return to citation in text: [1] -
Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076–1091. doi:10.1021/cs400138b
Return to citation in text: [1] -
Wang, Y.-N.; Ni, B.; Headley, A. D.; Li, G. Adv. Synth. Catal. 2007, 349, 319–322. doi:10.1002/adsc.200600277
Return to citation in text: [1] [2] [3] -
Li, G.; Wei, H.-X.; Kim, S. H.; Neighbors, M. Org. Lett. 1999, 1, 395–398. doi:10.1021/ol990059e
Return to citation in text: [1] -
Li, G.; Wei, H.-X.; Kim, S. H. Org. Lett. 2000, 2, 2249–2252. doi:10.1021/ol000120b
Return to citation in text: [1] -
Kotti, S. R. S. S.; Xu, X.; Wang, Y.; Headley, A. D.; Li, G. Tetrahedron Lett. 2004, 45, 7209–7212. doi:10.1016/j.tetlet.2004.08.040
Return to citation in text: [1] -
Xu, X.; Kotti, S. R. S. S.; Liu, J.; Cannon, J. F.; Headley, A. D.; Li, G. Org. Lett. 2004, 6, 4881–4884. doi:10.1021/ol048045i
Return to citation in text: [1] -
Wang, Y.-N.; Kattuboina, A.; Ai, T.; Banerjee, D.; Li, G. Tetrahedron Lett. 2007, 48, 7894–7898. doi:10.1016/j.tetlet.2007.08.099
Return to citation in text: [1] -
Ploux, O.; Caruso, M.; Chassaing, G.; Marquet, A. J. Org. Chem. 1988, 53, 3154–3158. doi:10.1021/jo00249a006
Return to citation in text: [1] -
Davis, F. A.; Zhou, P.; Reddy, G. V. J. Org. Chem. 1994, 59, 3243–3245. doi:10.1021/jo00091a001
Return to citation in text: [1] -
De Luca, L.; Giacomelli, G.; Taddei, M. J. Org. Chem. 2001, 66, 2534–2537. doi:10.1021/jo015524b
Return to citation in text: [1] -
Earle, M. J.; McCormac, P. B.; Seddon, K. R. Green Chem. 1999, 23–25. doi:10.1039/A808052F
Return to citation in text: [1] -
Sheldon, R. Chem. Commun. 2001, 2399–2407. doi:10.1039/B107270F
Return to citation in text: [1] -
Wu, J. X.; Beck, B.; Ren, R. X. Tetrahedron Lett. 2002, 43, 387–389. doi:10.1016/S0040-4039(01)02168-2
Return to citation in text: [1] -
Lucas, P.; El Mehdi, N.; Anh Ho, H.; Bélanger, D.; Breau, L. Synthesis 2000, 1253–1258. doi:10.1055/s-2000-6416
Return to citation in text: [1]
| 32. | Wang, Y.-N.; Ni, B.; Headley, A. D.; Li, G. Adv. Synth. Catal. 2007, 349, 319–322. doi:10.1002/adsc.200600277 |
| 33. | Li, G.; Wei, H.-X.; Kim, S. H.; Neighbors, M. Org. Lett. 1999, 1, 395–398. doi:10.1021/ol990059e |
| 34. | Li, G.; Wei, H.-X.; Kim, S. H. Org. Lett. 2000, 2, 2249–2252. doi:10.1021/ol000120b |
| 35. | Kotti, S. R. S. S.; Xu, X.; Wang, Y.; Headley, A. D.; Li, G. Tetrahedron Lett. 2004, 45, 7209–7212. doi:10.1016/j.tetlet.2004.08.040 |
| 36. | Xu, X.; Kotti, S. R. S. S.; Liu, J.; Cannon, J. F.; Headley, A. D.; Li, G. Org. Lett. 2004, 6, 4881–4884. doi:10.1021/ol048045i |
| 37. | Wang, Y.-N.; Kattuboina, A.; Ai, T.; Banerjee, D.; Li, G. Tetrahedron Lett. 2007, 48, 7894–7898. doi:10.1016/j.tetlet.2007.08.099 |
| 1. | Davis, F. A.; Szewczyk, J. M. Tetrahedron Lett. 1998, 39, 5951–5954. doi:10.1016/S0040-4039(98)01223-4 |
| 2. | Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957–5958. doi:10.1016/S0040-4020(02)00584-7 |
| 3. | Seebach, D.; Kimmerlin, T.; Šebesta, R.; Campo, M. A.; Beck, A. K. Tetrahedron 2004, 60, 7455–7506. doi:10.1016/j.tet.2004.06.043 |
| 8. | Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 5797–5815. doi:10.1002/ejoc.200700409 |
| 9. | Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29–41. doi:10.1039/b713885g |
| 31. | Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076–1091. doi:10.1021/cs400138b |
| 7. | Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219–3232. doi:10.1021/cr000045i |
| 32. | Wang, Y.-N.; Ni, B.; Headley, A. D.; Li, G. Adv. Synth. Catal. 2007, 349, 319–322. doi:10.1002/adsc.200600277 |
| 6. | Alcaide, B.; Almendros, P.; Aragoncillo, C. Chem. Rev. 2007, 107, 4437–4492. doi:10.1021/cr0307300 |
| 27. | Davies, S. G.; McCarthy, T. D. Synlett 1995, 700–702. doi:10.1055/s-1995-5059 |
| 28. | Davies, S. G.; Iwamoto, K.; Smethurst, C. A. P.; Smith, A. D.; Rodriguez-Solla, H. Synlett 2002, 1146–1148. doi:10.1055/s-2002-32580 |
| 29. | Burke, A. J.; Davies, S. G.; Garner, A. C.; McCarthy, T. D.; Roberts, P. M.; Smith, A. D.; Rodriguez-Solla, H.; Vickers, R. J. Org. Biomol. Chem. 2004, 2, 1387–1394. doi:10.1039/B402531H |
| 32. | Wang, Y.-N.; Ni, B.; Headley, A. D.; Li, G. Adv. Synth. Catal. 2007, 349, 319–322. doi:10.1002/adsc.200600277 |
| 4. | Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2 |
| 5. | Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h |
| 30. | Kaur, P.; Nguyen, T.; Li, G. Eur. J. Org. Chem. 2009, 912–916. doi:10.1002/ejoc.200801061 |
| 13. | Nahm, S.; Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815–3818. doi:10.1016/S0040-4039(01)91316-4 |
| 13. | Nahm, S.; Weinreb, S. M. Tetrahedron Lett. 1981, 22, 3815–3818. doi:10.1016/S0040-4039(01)91316-4 |
| 14. | Sibi, M. P. Org. Prep. Proced. Int. 1993, 25, 15–40. doi:10.1080/00304949309457931 |
| 15. | Turner, J. A.; Jacks, W. S. J. Org. Chem. 1989, 54, 4229–4231. doi:10.1021/jo00278a047 |
| 16. | Murphy, J. A.; Commeureuc, A. G. J.; Snaddon, T. N.; McGuire, T. M.; Khan, T. A.; Hisler, K.; Dewis, M. L.; Carling, R. Org. Lett. 2005, 7, 1427–1429. doi:10.1021/ol050337b |
| 17. | Kojima, S.; Hidaka, T.; Yamakawa, A. Chem. Lett. 2005, 34, 470–471. doi:10.1246/cl.2005.470 |
| 18. | Qu, B.; Collum, D. B. J. Org. Chem. 2006, 71, 7117–7119. doi:10.1021/jo061223w |
| 19. | Persson, T.; Nielsen, J. Org. Lett. 2006, 8, 3219–3222. doi:10.1021/ol0611088 |
| 20. | Rudzinski, D. M.; Kelly, C. B.; Leadbeater, N. E. Chem. Commun. 2012, 48, 9610–9612. doi:10.1039/C2CC35037H |
| 41. | Earle, M. J.; McCormac, P. B.; Seddon, K. R. Green Chem. 1999, 23–25. doi:10.1039/A808052F |
| 42. | Sheldon, R. Chem. Commun. 2001, 2399–2407. doi:10.1039/B107270F |
| 43. | Wu, J. X.; Beck, B.; Ren, R. X. Tetrahedron Lett. 2002, 43, 387–389. doi:10.1016/S0040-4039(01)02168-2 |
| 12. | Wang, B. M.; Song, Z. L.; Fan, C. A.; Tu, Y. Q.; Shi, Y. Org. Lett. 2002, 4, 363–366. doi:10.1021/ol0170410 |
| 21. | Davis, F. A.; Prasad, K. R.; Brad Nolt, M.; Wu, Y. Org. Lett. 2003, 5, 925–927. doi:10.1021/ol034119z |
| 22. | Davis, F. A.; Yang, B. Org. Lett. 2003, 5, 5011–5014. doi:10.1021/ol035981+ |
| 23. | Davis, F. A.; Brad Nolt, M.; Wu, Y.; Prasad, K. R.; Li, D.; Yang, B.; Bowen, K.; Lee, S. H.; Eardley, J. H. J. Org. Chem. 2005, 70, 2184–2190. doi:10.1021/jo0402780 |
| 24. | Davis, F. A.; Song, M. Org. Lett. 2007, 9, 2413–2416. doi:10.1021/ol0708166 |
| 25. | Davis, F. A.; Theddu, N. J. Org. Chem. 2010, 75, 3814–3820. doi:10.1021/jo100680b |
| 26. | Davis, F. A.; Xu, P. J. Org. Chem. 2011, 76, 3329–3337. doi:10.1021/jo2002352 |
| 44. | Lucas, P.; El Mehdi, N.; Anh Ho, H.; Bélanger, D.; Breau, L. Synthesis 2000, 1253–1258. doi:10.1055/s-2000-6416 |
| 11. | Gizecki, P.; Dhal, R.; Toupet, L.; Dujardin, G. Org. Lett. 2000, 2, 585–588. doi:10.1021/ol991326j |
| 38. | Ploux, O.; Caruso, M.; Chassaing, G.; Marquet, A. J. Org. Chem. 1988, 53, 3154–3158. doi:10.1021/jo00249a006 |
| 39. | Davis, F. A.; Zhou, P.; Reddy, G. V. J. Org. Chem. 1994, 59, 3243–3245. doi:10.1021/jo00091a001 |
| 10. | Davies, S. B.; McKervey, M. A. Tetrahedron Lett. 1999, 40, 1229–1232. doi:10.1016/S0040-4039(98)02573-8 |
| 14. | Sibi, M. P. Org. Prep. Proced. Int. 1993, 25, 15–40. doi:10.1080/00304949309457931 |
| 40. | De Luca, L.; Giacomelli, G.; Taddei, M. J. Org. Chem. 2001, 66, 2534–2537. doi:10.1021/jo015524b |
© 2016 Wang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)